
Kinase-Targeted Therapies for Glioblastoma


 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Kinase Inhibitors: Mechanisms and Therapeutic Approaches
2.1. Mechanisms of Action of Kinases
2.2. Multi-Kinase Inhibitors
2.2.1. Inhibitors of EGFR Kinase Activity
2.2.2. Inhibitors of Platelet-Derived Growth Factor (PDGF) Kinase Activity
2.3. Promising Multi-Kinase Inhibitors
3. Mechanisms of Resistance to Kinase Inhibitors
4. Possible Strategies to Escape Resistance to TKIs
5. Discussion
6. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| KI | Kinase inhibitors |
| MKI | Multi kinase inhibitor |
| RTK | Receptor tyrosine kinase |
| VEGF | Vascular Endothelial Growth Factor |
| TTFields | Tumor Treatment Fields |
| PKA | Cyclic-AMP-dependent protein kinase |
| EGFR | Epidermal Growth Factor Receptor |
| FGFR | Fibroblast Growth Factor Receptor |
| EGFR | Epidermal Growth Factor Receptor |
| NSCLS | Non small cell lung cancer |
| TGF-α | Transforming Growth Factor alpha |
| HB-EGF | Heparin-binding EGF-like Growth Factor |
| MAPK | Mitogen-Activated Protein Kinase |
| NSCLC | Non Small Cell Lung Cancer |
| OS | Overall Survival |
| BBB | Blood–Brain Barrier |
| PDGF | Platelet-Derived Growth Factor |
| PDGFR | Platelet-Derived Growth Factor Receptor |
| VEGFR1-3 | Vascular Endothelial Growth Factor Receptors 1, 2 and 3 |
| TIS | Therapy-Induced Senescence |
| GSC | Glioma Stem Cell |
| ROS | Reactive Oxygen Species |
| ATM | Ataxia Telangectasia Mutated |
| HIF 1 α | Hypoxia inducible factor 1α |
| CDKN1A | Cyclin dependent kinase inhibitor 1A |
| FFPE | Formalin fixed paraffin embedded |
| PSAT1 | Phosphoserine aminotransferase 1 |
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T.; Scott, J.D. Protein phosphorylation in signaling—50 years and counting. Trends Biochem. Sci. 2005, 30, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.D.; Krebs, E.G. Protein phosphorylation and signal transduction. Pharmacol. Ther. 1999, 82, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef]
- Miller, M.L.; Jensen, L.J.; Diella, F.; Jørgensen, C.; Tinti, M.; Li, L.; Hsiung, M.; Parker, S.A.; Bordeaux, J.; Sicheritz-Ponten, T.; et al. Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal 2008, 1, ra2. [Google Scholar] [CrossRef]
- Hunter, T. Tyrosine phosphorylation: Thirty years and counting. Curr. Opin. Cell Biol. 2009, 21, 140–146. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Kim, G.; Ko, Y.T. Small molecule tyrosine kinase inhibitors in glioblastoma. Arch. Pharm. Res. 2020, 43, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Qiu, W.; Sun, T.; Wang, L.; Du, C.; Hu, Y.; Liu, W.; Feng, F.; Chen, Y.; Sun, H. Therapeutic strategies of glioblastoma (GBM): The current advances in the molecular targets and bioactive small molecule compounds. Acta Pharm. Sin. B 2022, 12, 1781–1804. [Google Scholar] [CrossRef]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Miljković, F.; Bajorath, J. Exploring Selectivity of Multikinase Inhibitors across the Human Kinome. ACS Omega 2018, 3, 1147–1153. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. J. Med. Chem. 2008, 51, 7921–7932. [Google Scholar] [CrossRef]
- Eglen, R.; Reisine, T. Drug discovery and the human kinome: Recent trends. Pharmacol. Ther. 2011, 130, 144–156. [Google Scholar] [CrossRef]
- Das, R.; Choithramani, A.; Shard, A. A molecular perspective for the use of type IV tyrosine kinase inhibitors as anticancer therapeutics. Drug Discov. Today 2022, 27, 808–821. [Google Scholar] [CrossRef]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013, 20, 146–159. [Google Scholar] [CrossRef]
- Cox, K.J.; Shomin, C.D.; Ghosh, I. Tinkering outside the kinase ATP box: Allosteric (type IV) and bivalent (type V) inhibitors of protein kinases. Future Med. Chem. 2011, 3, 29–43. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2024 update. Pharmacol. Res. 2024, 200, 107059. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Broekman, F.; Giovannetti, E.; Peters, G.J. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J. Clin. Oncol. 2011, 2, 80–93. [Google Scholar] [CrossRef]
- Miljković, F.; Bajorath, J. Evaluation of Kinase Inhibitor Selectivity Using Cell-based Profiling Data. Mol. Inform. 2018, 37, e1800024. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
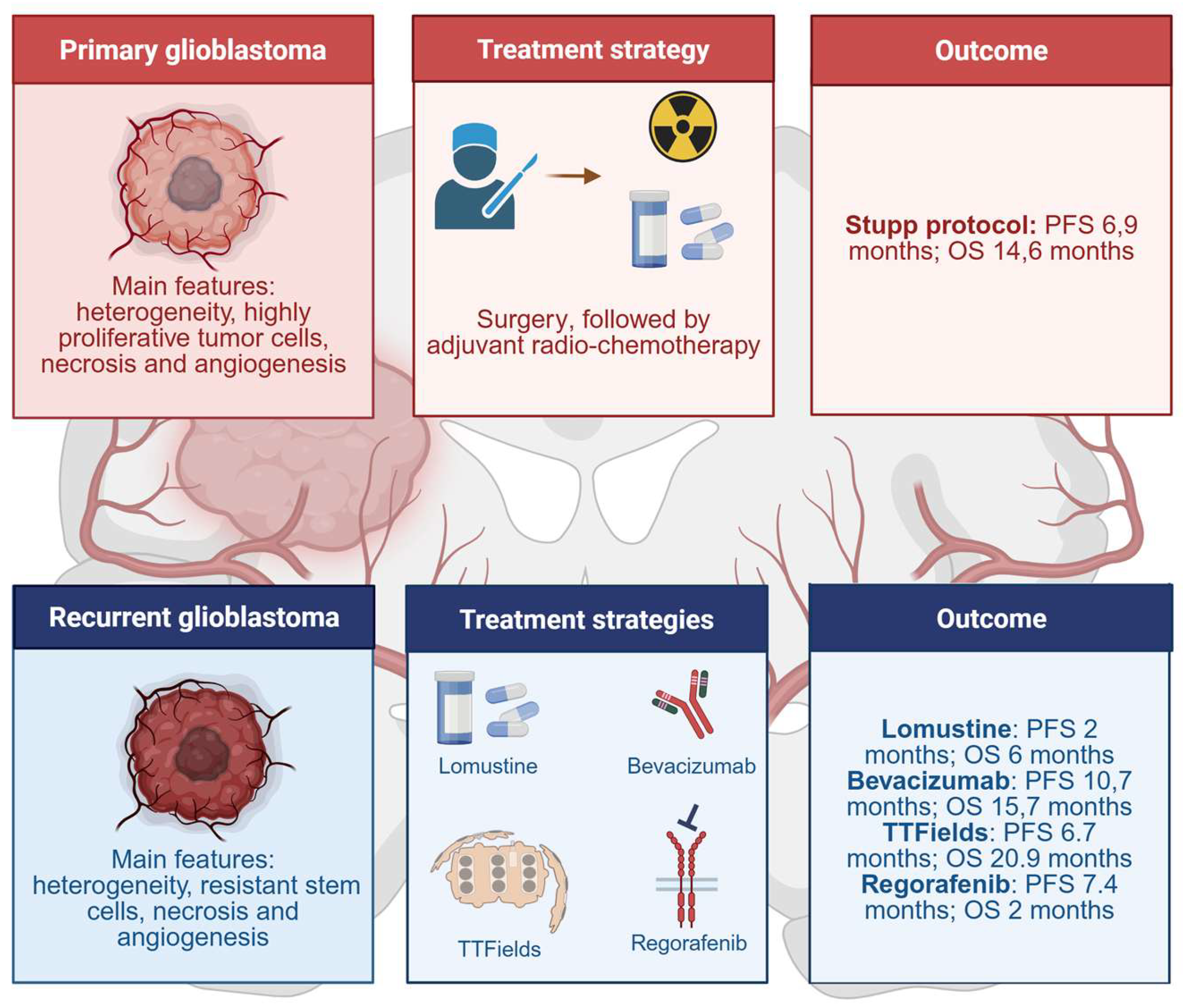
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef]
- de Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor invasion after treatment of glioblastoma with bevacizumab: Radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010, 12, 233–242. [Google Scholar] [CrossRef]
- Gomez-Manzano, C.; Holash, J.; Fueyo, J.; Xu, J.; Conrad, C.A.; Aldape, K.D.; de Groot, J.F.; Bekele, B.N.; Yung, W.K. VEGF Trap induces antiglioma effect at different stages of disease. Neuro Oncol. 2008, 10, 940–945. [Google Scholar] [CrossRef]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Rominiyi, O.; Vanderlinden, A.; Clenton, S.J.; Bridgewater, C.; Al-Tamimi, Y.; Collis, S.J. Tumour treating fields therapy for glioblastoma: Current advances and future directions. Br. J. Cancer 2021, 124, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Kirson, E.D.; Dbalý, V.; Tovarys, F.; Vymazal, J.; Soustiel, J.F.; Itzhaki, A.; Mordechovich, D.; Steinberg-Shapira, S.; Gurvich, Z.; Schneiderman, R.; et al. Alternating electric fields arrest cell proliferation in animal tumor models and human brain tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 10152–10157. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Wen, P.; Nishikawa, R.; Reardon, D.; Peters, K. Critical review of the addition of tumor treating fields (TTFields) to the existing standard of care for newly diagnosed glioblastoma patients. Crit. Rev. Oncol. Hematol. 2017, 111, 60–65. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Knighton, D.R.; Zheng, J.H.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef]
- Rahban, M.; Joushi, S.; Bashiri, H.; Saso, L.; Sheibani, V. Characterization of prevalent tyrosine kinase inhibitors and their challenges in glioblastoma treatment. Front. Chem. 2023, 11, 1325214. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Wang, Z.; Peet, N.P.; Zhang, P.; Jiang, Y.; Rong, L. Current Development of Glioblastoma Therapeutic Agents. Mol. Cancer Ther. 2021, 20, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Burgess, A.W. EGFR family: Structure physiology signalling and therapeutic targets. Growth Factors 2008, 26, 263–274. [Google Scholar] [CrossRef]
- Quesnelle, K.M.; Boehm, A.L.; Grandis, J.R. STAT-mediated EGFR signaling in cancer. J. Cell Biochem. 2007, 102, 311–319. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification and overexpression of the EGF receptor gene in primary human glioblastomas. J. Cell Sci. Suppl. 1985, 3, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.; et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar]
- Cataldo, V.D.; Gibbons, D.L.; Pérez-Soler, R.; Quintás-Cardama, A. Treatment of non-small-cell lung cancer with erlotinib or gefitinib. N. Engl. J. Med. 2011, 364, 947–955. [Google Scholar] [CrossRef]
- Burotto, M.; Manasanch, E.E.; Wilkerson, J.; Fojo, T. Gefitinib and erlotinib in metastatic non-small cell lung cancer: A meta-analysis of toxicity and efficacy of randomized clinical trials. Oncologist 2015, 20, 400–410. [Google Scholar] [CrossRef]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Vega-González, M.T.; López-Macías, D.; Martínez-Hernández, J.N.; Bacon-Fonseca, L.; Macedo-Pérez, E.O.; Ramírez-Tirado, L.A.; Flores-Estrada, D.; de la Garza-Salazar, J. Randomized, open-label trial evaluating the preventive effect of tetracycline on afatinib induced-skin toxicities in non-small cell lung cancer patients. Lung Cancer 2015, 88, 282–288. [Google Scholar] [CrossRef]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Robins, H.I.; Rohle, D.; Campos, C.; Grommes, C.; Nghiemphu, P.L.; Kubek, S.; Oldrini, B.; Chheda, M.G.; Yannuzzi, N.; et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Mukasa, A.; Wykosky, J.; Ligon, K.L.; Chin, L.; Cavenee, W.K.; Furnari, F. Mutant EGFR is required for maintenance of glioma growth in vivo, and its ablation leads to escape from receptor dependence. Proc. Natl. Acad. Sci. USA 2010, 107, 2616–2621. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Ahluwalia, M.S.; Ye, X.; Supko, J.G.; Hilderbrand, S.L.; Phuphanich, S.; Nabors, L.B.; Rosenfeld, M.R.; Mikkelsen, T.; Grossman, S.A.; et al. NABTT 0502: A phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol. 2013, 15, 490–496. [Google Scholar] [CrossRef]
- Sathornsumetee, S.; Desjardins, A.; Vredenburgh, J.J.; McLendon, R.E.; Marcello, J.; Herndon, J.E.; Mathe, A.; Hamilton, M.; Rich, J.N.; Norfleet, J.A.; et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro Oncol. 2010, 12, 1300–1310. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Bady, P.; Kamoshima, Y.; Kouwenhoven, M.C.; Delorenzi, M.; Lambiv, W.L.; Hamou, M.F.; Matter, M.S.; Koch, A.; et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib—A phase II trial. Mol. Cancer Ther. 2011, 10, 1102–1112. [Google Scholar] [CrossRef]
- Thiessen, B.; Stewart, C.; Tsao, M.; Kamel-Reid, S.; Schaiquevich, P.; Mason, W.; Easaw, J.; Belanger, K.; Forsyth, P.; McIntosh, L.; et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2010, 65, 353–361. [Google Scholar] [CrossRef]
- Eller, J.L.; Longo, S.L.; Kyle, M.M.; Bassano, D.; Hicklin, D.J.; Canute, G.W. Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery 2005, 56, 155–162; discussion 162. [Google Scholar] [CrossRef]
- Buendia Duque, M.; Pinheiro, K.V.; Thomaz, A.; da Silva, C.A.; Freire, N.H.; Brunetto, A.T.; Schwartsmann, G.; Jaeger, M.; de Farias, C.B.; Roesler, R. Combined Inhibition of HDAC and EGFR Reduces Viability and Proliferation and Enhances STAT3 mRNA Expression in Glioblastoma Cells. J. Mol. Neurosci. 2019, 68, 49–57. [Google Scholar] [CrossRef]
- Xu, W.; Bi, Y.; Kong, J.; Zhang, J.; Wang, B.; Li, K.; Tian, M.; Pan, X.; Shi, B.; Gu, J.; et al. Combination of an anti-EGFRvIII antibody CH12 with Rapamycin synergistically inhibits the growth of EGFRvIII+PTEN-glioblastoma in vivo. Oncotarget 2016, 7, 24752–24765. [Google Scholar] [CrossRef]
- Hasselbalch, B.; Lassen, U.; Hansen, S.; Holmberg, M.; Sørensen, M.; Kosteljanetz, M.; Broholm, H.; Stockhausen, M.T.; Poulsen, H.S. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: A phase II trial. Neuro Oncol. 2010, 12, 508–516. [Google Scholar] [CrossRef]
- Tian, L.; Xu, B.; Chen, Y.; Li, Z.; Wang, J.; Zhang, J.; Ma, R.; Cao, S.; Hu, W.; Chiocca, E.A.; et al. Specific targeting of glioblastoma with an oncolytic virus expressing a cetuximab-CCL5 fusion protein via innate and adaptive immunity. Nat. Cancer 2022, 3, 1318–1335. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Paulsson, J.; Lindh, M.B.; Jarvius, M.; Puputti, M.; Nistér, M.; Nupponen, N.N.; Paulus, W.; Söderberg, O.; Dresemann, G.; von Deimling, A.; et al. Prognostic but not predictive role of platelet-derived growth factor receptors in patients with recurrent glioblastoma. Int. J. Cancer 2011, 128, 1981–1988. [Google Scholar] [CrossRef]
- Lassman, A.B.; Pugh, S.L.; Gilbert, M.R.; Aldape, K.D.; Geinoz, S.; Beumer, J.H.; Christner, S.M.; Komaki, R.; DeAngelis, L.M.; Gaur, R.; et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro Oncol. 2015, 17, 992–998. [Google Scholar] [CrossRef]
- Hu-Lowe, D.D.; Zou, H.Y.; Grazzini, M.L.; Hallin, M.E.; Wickman, G.R.; Amundson, K.; Chen, J.H.; Rewolinski, D.A.; Yamazaki, S.; Wu, E.Y.; et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin. Cancer Res. 2008, 14, 7272–7283. [Google Scholar] [CrossRef] [PubMed]
- Bellesoeur, A.; Carton, E.; Alexandre, J.; Goldwasser, F.; Huillard, O. Axitinib in the treatment of renal cell carcinoma: Design, development, and place in therapy. Drug Des. Dev. Ther. 2017, 11, 2801–2811. [Google Scholar] [CrossRef]
- Infante, J.R.; Reid, T.R.; Cohn, A.L.; Edenfield, W.J.; Cescon, T.P.; Hamm, J.T.; Malik, I.A.; Rado, T.A.; McGee, P.J.; Richards, D.A.; et al. Axitinib and/or bevacizumab with modified FOLFOX-6 as first-line therapy for metastatic colorectal cancer: A randomized phase 2 study. Cancer 2013, 119, 2555–2563. [Google Scholar] [CrossRef]
- Lu, L.; Saha, D.; Martuza, R.L.; Rabkin, S.D.; Wakimoto, H. Single agent efficacy of the VEGFR kinase inhibitor axitinib in preclinical models of glioblastoma. J. Neurooncol. 2015, 121, 91–100. [Google Scholar] [CrossRef]
- Duerinck, J.; Du Four, S.; Bouttens, F.; Andre, C.; Verschaeve, V.; Van Fraeyenhove, F.; Chaskis, C.; D’Haene, N.; Le Mercier, M.; Rogiers, A.; et al. Randomized phase II trial comparing axitinib with the combination of axitinib and lomustine in patients with recurrent glioblastoma. J. Neurooncol. 2018, 136, 115–125. [Google Scholar] [CrossRef]
- Duerinck, J.; Du Four, S.; Vandervorst, F.; D’Haene, N.; Le Mercier, M.; Michotte, A.; Van Binst, A.M.; Everaert, H.; Salmon, I.; Bouttens, F.; et al. Randomized phase II study of axitinib versus physicians best alternative choice of therapy in patients with recurrent glioblastoma. J. Neurooncol. 2016, 128, 147–155. [Google Scholar] [CrossRef]
- Du Four, S.; Maenhout, S.K.; Benteyn, D.; De Keersmaecker, B.; Duerinck, J.; Thielemans, K.; Neyns, B.; Aerts, J.L. Disease progression in recurrent glioblastoma patients treated with the VEGFR inhibitor axitinib is associated with increased regulatory T cell numbers and T cell exhaustion. Cancer Immunol. Immunother. 2016, 65, 727–740. [Google Scholar] [CrossRef]
- Chryplewicz, A.; Scotton, J.; Tichet, M.; Zomer, A.; Shchors, K.; Joyce, J.A.; Homicsko, K.; Hanahan, D. Cancer cell autophagy, reprogrammed macrophages, and remodeled vasculature in glioblastoma triggers tumor immunity. Cancer Cell 2022, 40, 1111–1127.e9. [Google Scholar] [CrossRef]
- Morelli, M.B.; Amantini, C.; Santoni, M.; Soriani, A.; Nabissi, M.; Cardinali, C.; Santoni, A.; Santoni, G. Axitinib induces DNA damage response leading to senescence, mitotic catastrophe, and increased NK cell recognition in human renal carcinoma cells. Oncotarget 2015, 6, 36245–36259. [Google Scholar] [CrossRef]
- Morelli, M.B.; Amantini, C.; Nabissi, M.; Cardinali, C.; Santoni, M.; Bernardini, G.; Santoni, A.; Santoni, G. Axitinib induces senescence-associated cell death and necrosis in glioma cell lines: The proteasome inhibitor, bortezomib, potentiates axitinib-induced cytotoxicity in a p21(Waf/Cip1) dependent manner. Oncotarget 2017, 8, 3380–3395. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Radice, G.; Piras, M.; Stagni, V.; Pacioni, S.; Re, A.; Putti, S.; Ferrè, F.; Farsetti, A.; Pallini, R.; et al. Axitinib exposure triggers endothelial cells senescence through ROS accumulation and ATM activation. Oncogene 2019, 38, 5413–5424. [Google Scholar] [CrossRef]
- Formato, A.; Salbini, M.; Orecchini, E.; Pellegrini, M.; Buccarelli, M.; Vitiani, L.R.; Giannetti, S.; Pallini, R.; D’Alessandris, Q.G.; Lauretti, L.; et al. N-Acetyl-L-Cysteine (NAC) Blunts Axitinib-Related Adverse Effects in Preclinical Models of Glioblastoma. Cancer Med. 2024, 13, e70279. [Google Scholar] [CrossRef]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Santangelo, A.; Rossato, M.; Lombardi, G.; Benfatto, S.; Lavezzari, D.; De Salvo, G.L.; Indraccolo, S.; Dechecchi, M.C.; Prandini, P.; Gambari, R.; et al. A molecular signature associated with prolonged survival in glioblastoma patients treated with regorafenib. Neuro Oncol. 2021, 23, 264–276. [Google Scholar] [CrossRef]
- Chiesa, S.; Mangraviti, A.; Martini, M.; Cenci, T.; Mazzarella, C.; Gaudino, S.; Bracci, S.; Martino, A.; Della Pepa, G.M.; Offi, M.; et al. Clinical and NGS predictors of response to regorafenib in recurrent glioblastoma. Sci. Rep. 2022, 12, 16265. [Google Scholar] [CrossRef]
- Indraccolo, S.; De Salvo, G.L.; Verza, M.; Caccese, M.; Esposito, G.; Piga, I.; Del Bianco, P.; Pizzi, M.; Gardiman, M.P.; Eoli, M.; et al. Phosphorylated Acetyl-CoA Carboxylase Is Associated with Clinical Benefit with Regorafenib in Relapsed Glioblastoma: REGOMA Trial Biomarker Analysis. Clin. Cancer Res. 2020, 26, 4478–4484. [Google Scholar] [CrossRef]
- Chiang, I.T.; Liu, Y.C.; Liu, H.S.; Ali, A.A.A.; Chou, S.Y.; Hsu, T.I.; Hsu, F.T. Regorafenib Reverses Temozolomide-Induced CXCL12/CXCR4 Signaling and Triggers Apoptosis Mechanism in Glioblastoma. Neurotherapeutics 2022, 19, 616–634. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Buccarelli, M.; Formato, A.; Orecchini, E.; Salbini, M.; Ricci, V.; Orsini, T.; Putti, S.; Chiesa, S.; Ricci-Vitiani, L.; et al. Characterization of Glioblastoma Cells Response to Regorafenib. Cancers 2022, 14, 6193. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, L.; Chen, H.; Lei, Y.; Zhang, T.; Wang, Y.; Jin, P.; Lan, J.; Zhou, L.; Huang, Z.; et al. Regorafenib induces lethal autophagy arrest by stabilizing PSAT1 in glioblastoma. Autophagy 2020, 16, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Haeusser, L.A.; Becker, H.; Kuhlburger, L.; Zago, M.; Walter, B.; Tsiami, F.; Erdmann, S.; Trampert, J.; Surender, S.; Stahl, A.; et al. Genome-wide CRISPR-Cas 9 screens identify BCL family members as modulators of response to regorafenib in experimental glioma. Neuro Oncol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Cao, R.; Yang, Y.; Chen, X.; Liu, L.; Ren, B.; Wang, L.; Goh, B.C. Blood-Brain Barrier Conquest in Glioblastoma Nanomedicine: Strategies, Clinical Advances, and Emerging Challenges. Cancers 2024, 16, 3300. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Focosi, D. Three paths to better tyrosine kinase inhibition behind the blood-brain barrier in treating chronic myelogenous leukemia and glioblastoma with imatinib. Transl. Oncol. 2010, 3, 13–15. [Google Scholar] [CrossRef]
- Beccaria, K.; Canney, M.; Bouchoux, G.; Desseaux, C.; Grill, J.; Heimberger, A.B.; Carpentier, A. Ultrasound-induced blood-brain barrier disruption for the treatment of gliomas and other primary CNS tumors. Cancer Lett. 2020, 479, 13–22. [Google Scholar] [CrossRef]
- Krchniakova, M.; Skoda, J.; Neradil, J.; Chlapek, P.; Veselska, R. Repurposing Tyrosine Kinase Inhibitors to Overcome Multidrug Resistance in Cancer: A Focus on Transporters and Lysosomal Sequestration. Int. J. Mol. Sci. 2020, 21, 3157. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [Google Scholar] [CrossRef]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014, 343, 72–76. [Google Scholar] [CrossRef]
- Day, E.K.; Sosale, N.G.; Xiao, A.; Zhong, Q.; Purow, B.; Lazzara, M.J. Glioblastoma Cell Resistance to EGFR and MET Inhibition Can Be Overcome via Blockade of FGFR-SPRY2 Bypass Signaling. Cell Rep. 2020, 30, 3383–3396.e7. [Google Scholar] [CrossRef]
- Ma, Y.; Tang, N.; Thompson, R.C.; Mobley, B.C.; Clark, S.W.; Sarkaria, J.N.; Wang, J. InsR/IGF1R Pathway Mediates Resistance to EGFR Inhibitors in Glioblastoma. Clin. Cancer Res. 2016, 22, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ma, X.; Gao, P.; Han, X.; Zhao, P.; Xie, F.; Liu, M. Advancing glioblastoma treatment by targeting metabolism. Neoplasia 2024, 51, 100985. [Google Scholar] [CrossRef] [PubMed]
- Aldaz, P.; Olias-Arjona, A.; Lasheras-Otero, I.; Ausin, K.; Redondo-Muñoz, M.; Wellbrock, C.; Santamaria, E.; Fernandez-Irigoyen, J.; Arozarena, I. Drug-Induced Reorganisation of Lipid Metabolism Limits the Therapeutic Efficacy of Ponatinib in Glioma Stem Cells. Pharmaceutics 2024, 16, 728. [Google Scholar] [CrossRef]
- Bausart, M.; Préat, V.; Malfanti, A. Immunotherapy for glioblastoma: The promise of combination strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35. [Google Scholar] [CrossRef]
- Ellenbogen, Y.; Zadeh, G. A new paradigm for immunotherapy in glioblastoma. Nat. Med. 2025. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, S.; Wang, Y.; Zhao, Y.; Li, Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: Molecular mechanisms and future perspective. Signal Transduct. Target. Ther. 2022, 7, 329. [Google Scholar] [CrossRef]
- Dymova, M.A.; Kuligina, E.V.; Richter, V.A. Molecular Mechanisms of Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 6358. [Google Scholar] [CrossRef]
- Pinto-Fraga, J.; García-Chico, C.; Lista, S.; Lacal, P.M.; Carpenzano, G.; Salvati, M.; Santos-Lozano, A.; Graziani, G.; Ceci, C. Protein kinase inhibitors as targeted therapy for glioblastoma: A meta-analysis of randomized controlled clinical trials. Pharmacol. Res. 2025, 212, 107528. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Mitchell, K.; Troike, K.; Silver, D.J.; Lathia, J.D. The evolution of the cancer stem cell state in glioblastoma: Emerging insights into the next generation of functional interactions. Neuro Oncol. 2021, 23, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, G.; Wang, X.; Hong, H.; Li, T.; Li, L.; Wang, H.; Xie, J.; Li, B.; Lu, D.; et al. Glioblastoma stem cell-specific histamine secretion drives pro-angiogenic tumor microenvironment remodeling. Cell Stem. Cell 2022, 29, 1531–1546.e1537. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef]
- Xie, X.P.; Laks, D.R.; Sun, D.; Ganbold, M.; Wang, Z.; Pedraza, A.M.; Bale, T.; Tabar, V.; Brennan, C.; Zhou, X.; et al. Quiescent human glioblastoma cancer stem cells drive tumor initiation, expansion, and recurrence following chemotherapy. Dev. Cell 2022, 57, 32–46.e8. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Barria, A. Dangerous liaisons as tumour cells form synapses with neurons. Nature 2019, 573, 499–501. [Google Scholar] [CrossRef]
- Monje, M.; Borniger, J.C.; D’Silva, N.J.; Deneen, B.; Dirks, P.B.; Fattahi, F.; Frenette, P.S.; Garzia, L.; Gutmann, D.H.; Hanahan, D.; et al. Roadmap for the Emerging Field of Cancer Neuroscience. Cell 2020, 181, 219–222. [Google Scholar] [CrossRef]
- Winkler, F.; Venkatesh, H.S.; Amit, M.; Batchelor, T.; Demir, I.E.; Deneen, B.; Gutmann, D.H.; Hervey-Jumper, S.; Kuner, T.; Mabbott, D.; et al. Cancer neuroscience: State of the field, emerging directions. Cell 2023, 186, 1689–1707. [Google Scholar] [CrossRef]
- Kim, K.H.; Migliozzi, S.; Koo, H.; Hong, J.H.; Park, S.M.; Kim, S.; Kwon, H.J.; Ha, S.; Garofano, L.; Oh, Y.T.; et al. Integrated proteogenomic characterization of glioblastoma evolution. Cancer Cell 2024, 42, 358–377.e8. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, X.; Zhang, D.Y.; Zhang, Z.; Bhattarai, J.P.; Wang, Y.; Park, K.H.; Dong, W.; Hung, Y.F.; Yang, Q.; et al. Brain-wide neuronal circuit connectome of human glioblastoma. Nature 2025. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, S.K.; Reyhan, E.; Layer, N.; Bengtson, C.P.; Heuer, A.; Schroers, J.; Faymonville, A.J.; Langeroudi, A.P.; Drewa, N.; Keifert, E.; et al. Characterizing and targeting glioblastoma neuron-tumor networks with retrograde tracing. Cell 2025, 188, 390–411.e36. [Google Scholar] [CrossRef] [PubMed]
- Correia, C.D.; Calado, S.M.; Matos, A.; Esteves, F.; De Sousa-Coelho, A.L.; Campinho, M.A.; Fernandes, M.T. Advancing Glioblastoma Research with Innovative Brain Organoid-Based Models. Cells 2025, 14, 292. [Google Scholar] [CrossRef]
- Peng, T.; Ma, X.; Hua, W.; Wang, C.; Chu, Y.; Sun, M.; Fermi, V.; Hamelmann, S.; Lindner, K.; Shao, C.; et al. Individualized patient tumor organoids faithfully preserve human brain tumor ecosystems and predict patient response to therapy. Cell Stem Cell 2025, 32, 652–669.e11. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salbini, M.; Formato, A.; Mongiardi, M.P.; Levi, A.; Falchetti, M.L. Kinase-Targeted Therapies for Glioblastoma. Int. J. Mol. Sci. 2025, 26, 3737. https://doi.org/10.3390/ijms26083737
Salbini M, Formato A, Mongiardi MP, Levi A, Falchetti ML. Kinase-Targeted Therapies for Glioblastoma. International Journal of Molecular Sciences. 2025; 26(8):3737. https://doi.org/10.3390/ijms26083737
Chicago/Turabian StyleSalbini, Maria, Alessia Formato, Maria Patrizia Mongiardi, Andrea Levi, and Maria Laura Falchetti. 2025. "Kinase-Targeted Therapies for Glioblastoma" International Journal of Molecular Sciences 26, no. 8: 3737. https://doi.org/10.3390/ijms26083737
APA StyleSalbini, M., Formato, A., Mongiardi, M. P., Levi, A., & Falchetti, M. L. (2025). Kinase-Targeted Therapies for Glioblastoma. International Journal of Molecular Sciences, 26(8), 3737. https://doi.org/10.3390/ijms26083737