
Induction of Cell Death and Regulation of Autocrine Vitamin D Metabolism in Cervical Cancer by Physiological and GI20 Doses of 25-Hydroxycholecalciferol

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
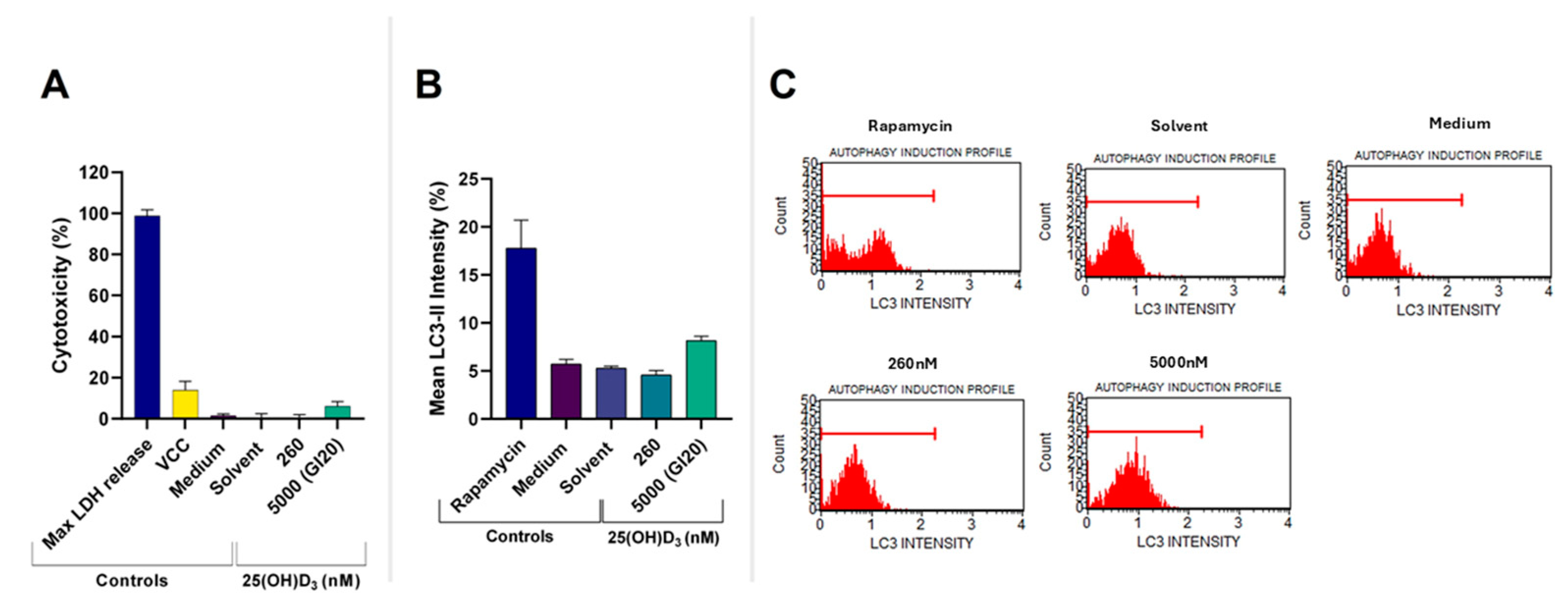
2.1. Assessment of GI20 and Cell Health Parameters
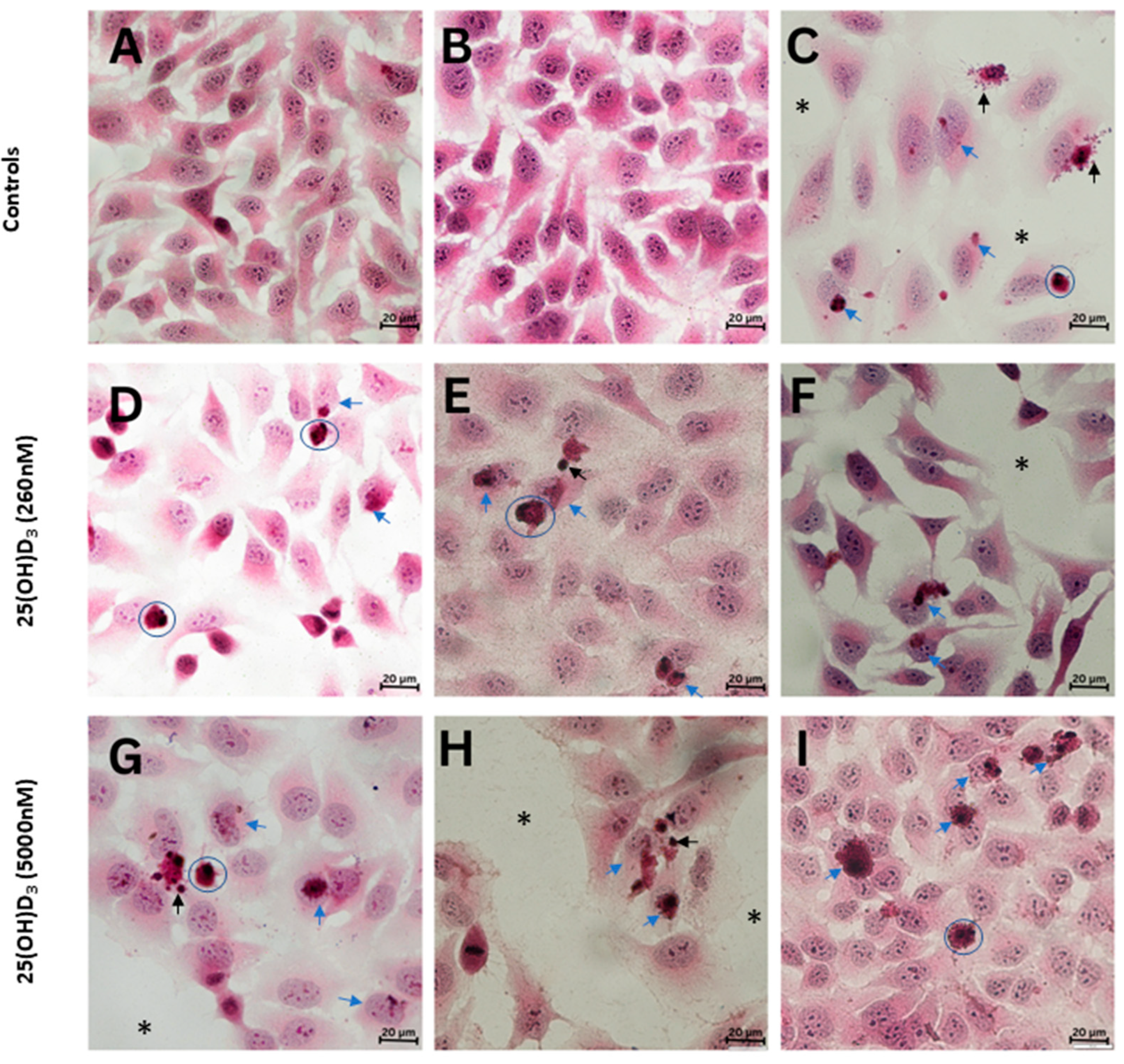
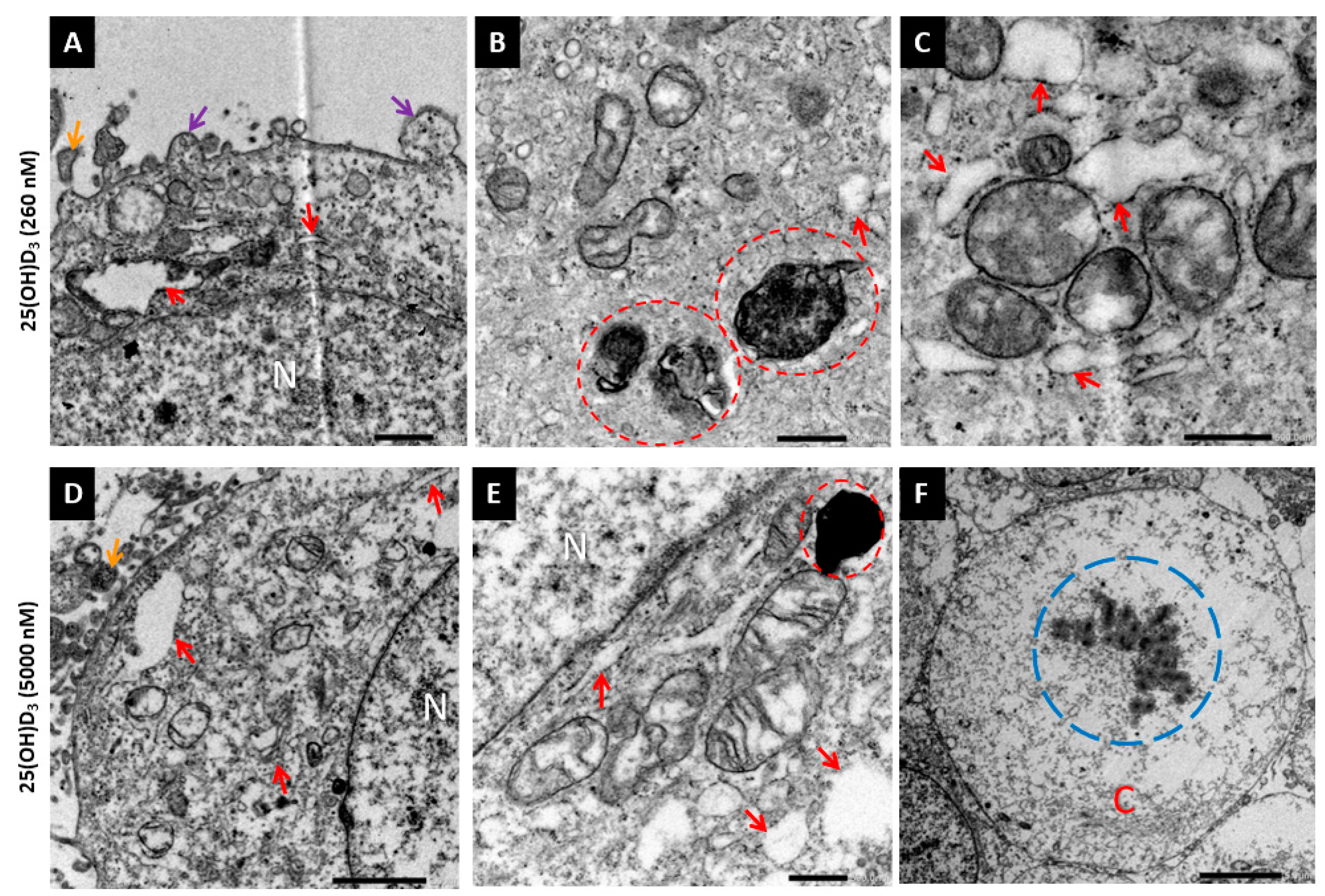
2.2. 25(OH)D3 Induced Apoptotic Cell Death
2.3. 25(OH)D3 Regulates VDMS in HeLa Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Crystal Violet Assay
4.3. AlamarBlue Assay
4.4. Cell Cycle Distribution by Flow Cytometric Analysis
4.5. Mitochondrial Membrane Disruption Analysis with Flow Cytometry
4.6. Annexin V Staining by Flow Cytometry
4.7. Caspase-3/7 Assay with Flow Cytometry
4.8. Autophagy LC3-II Assay with Flow Cytometry
4.9. Lactate Dehydrogenase (LDH) Assay
4.10. Brightfield Microscopy
4.11. Transmission Electron Microscopy (TEM)
4.12. RNA Isolation
4.13. RT-qPCR
4.14. Western Blot
4.15. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1,25(OH)2D3 | 1,25-dihydroxycholecalciferol |
| 25(OH)D3 | 25-hydroxycholecalciferol |
| ANOVA | Analysis of variance |
| BSA | Bovine serum albumin |
| CYP24A1 | Cytochrome P450 25-hydroxyvitamin D-24-hydroxylase |
| CYP27B1 | Cytochrome P450 25-hydroxyvitamin D-1α-hydroxylase |
| ER | Endoplasmic reticulum |
| HRP | Horseradish peroxidase |
| PBS | Phosphate buffered saline |
| PVDF | Polyvinylidene fluoride |
| SEM | Standard error of mean |
| VDMS | Vitamin D metabolizing system |
| VDR | Vitamin D receptor |
Appendix A
References
- Holick, M.F. Vitamin D Deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.M.; Mason, R.S. Vitamin D. Int. J. Biochem. Cell Biol. 2009, 41, 982–985. [Google Scholar] [CrossRef]
- Berridge, M.J. Vitamin D cell signalling in health and disease. Biochem. Biophys. Res. Commun. 2015, 460, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Deuster, E.; Jeschke, U.; Ye, Y.; Mahner, S.; Czogalla, B. Vitamin D and VDR in Gynecological Cancers—A Systematic Review. Int. J. Mol. Sci. 2017, 18, 2328. [Google Scholar] [CrossRef]
- Moukayed, M.; Grant, W.B. Molecular link between vitamin D and cancer prevention. Nutrients 2013, 5, 3993–4021. [Google Scholar] [CrossRef]
- Rosen, C.J.; Adams, J.S.; Bikle, D.D.; Black, D.M.; Demay, M.B.; Manson, J.E.; Murad, M.H.; Kovacs, C. The nonskeletal effects of vitamin D: An Endocrine Society scientific statement. Endocr. Rev. 2012, 33, 456–492. [Google Scholar] [CrossRef]
- St-Arnold, R.; Jones, G. CYP24A1: Structure, Function, and Physiological Role. In Vitamin D, Biochemistry, Physiology and Diagnostics, 4th ed.; Feldman, D., Pike, J.W., Bouillon, R., Eds.; Academic Press (Elsevier): London, UK, 2018; pp. 81–95. [Google Scholar]
- Jones, G.; Prosser, D.E.; Kaufmann, M. The Activating Enzymes of Vitamin D Metabolism (25- and 1α-Hydroxylases). In Vitamin D, Biochemistry, Physiology and Diagnostics, 4th ed.; Feldman, D., Pike, J.W., Bouillon, R., Eds.; Academic Press (Elsevier): London, UK, 2018; pp. 57–79. [Google Scholar]
- Seraphin, G.; Rieger, S.; Hewison, M.; Capobianco, E.; Lisse, T.S. The impact of vitamin D on cancer: A mini review. J. Steroid Biochem. Mol. Biol. 2023, 231, 106308. [Google Scholar] [CrossRef]
- Mathiasen, I.S.; Lademann, U.; Jäättelä, M. Apoptosis induced by vitamin D compounds in breast cancer cells is inhibited by Bcl-2 but does not involve known caspases or p53. Cancer Res. 1999, 59, 48–56. [Google Scholar]
- Swami, S.; Raghavachari, N.; Muller, U.R.; Bao, Y.P.; Feldman, D. Vitamin D growth inhibition of breast cancer cells: Gene expression patterns assessed by cDNA microarray. Breast Cancer Res. Treat. 2003, 80, 49–62. [Google Scholar] [CrossRef]
- Karlsson, S.; Diaz Cruz, M.A.; Faresjö, M.; Khamou, A.P.; Larsson, D. Inhibition of CYP27B1 and CYP24 Increases the Anti-prolifeative Effects of 25-Hydroxyvitamin D(3) in LNCaP Cells. Anticancer. Res. 2021, 41, 4733–4740. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Ramshaw, H.; Zhang, A.; Cook, R.; Byford, V.; White, J.; Petkovich, M. Expression and Activity of Vitamin D-Metabolizing Cytochrome P450s (CYP1α and CYP24) in Human Nonsmall Cell Lung Carcinomas. Endocrinology 1999, 140, 3303–3310. [Google Scholar] [CrossRef] [PubMed]
- Reichrath, J.; Rech, M.; Moeini, M.; Meese, E.; Tilgen, W.; Seifert, M. In vitro comparison of the vitamin D endocrine system in 1,25(OH)2D3-responsive and -resistant melanoma cells. Cancer Biol. Ther. 2007, 6, 48–55. [Google Scholar] [CrossRef]
- Grant, W.B. 25-Hydroxyvitamin D and Breast Cancer, Colorectal Cancer, and Colorectal Adenomas: Case–Control versus Nested Case–Control Studies. Anticancer Res. 2015, 35, 1153. [Google Scholar] [PubMed]
- González-Sancho, J.M.; Larriba, M.J.; Ordóñez-Morán, P.; Pálmer, H.G.; Muñoz, A. Effects of 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Anticancer Res. 2006, 26, 2669–2681. [Google Scholar]
- Friedrich, M.; Rafi, L.; Mitschele, T.; Tilgen, W.; Schmidt, W.; Reichrath, J. Analysis of the vitamin D system in cervical carcinomas, breast cancer and ovarian cancer. Recent. Results Cancer Res. 2003, 164, 239–246. [Google Scholar] [CrossRef]
- Bhoora, S.; Pillay, T.S.; Punchoo, R. Cholecalciferol Mediates Apoptosis in Siha Cervical Cancer Line via Autocrine Mechanisms. J. Endocr. Soc. 2021, 5, A1013. [Google Scholar] [CrossRef]
- Punchoo, R.; Dreyer, G.; Pillay, T.S. 25-Hydroxycholecalciferol Inhibits Cell Growth and Induces Apoptosis in SiHa Cervical Cells via Autocrine Vitamin D Metabolism. Biomedicines 2023, 11, 871. [Google Scholar] [CrossRef]
- Bhoora, S.; Pather, Y.; Marais, S.; Punchoo, R. Cholecalciferol Inhibits Cell Growth and Induces Apoptosis in the CaSki Cell Line. Med. Sci. 2020, 8, 12. [Google Scholar] [CrossRef]
- Bhoora, S.; Pillay, T.S.; Punchoo, R. Cholecalciferol induces apoptosis via autocrine metabolism in epidermoid cervical cancer cells. Biochem. Cell Biol. 2022, 100, 387–402. [Google Scholar] [CrossRef]
- Punchoo, R.; Bhoora, S. Autocrine Vitamin D Metabolism Mediates Anticancer Actions in Uterine Cervical Cancer. In Interdisciplinary Cancer Research; Springer: Cham, Switzerland, 2024. [Google Scholar] [CrossRef]
- Dong, H.; Chen, S.; Liang, X.; Cai, Q.; Zhang, X.; Xiel, J.; Sun, Z. Vitamin D and Its Receptors in Cervical Cancer. J. Cancer 2024, 4, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Trump, D.L.; Johnson, C.S.; Feldman, D. The role of vitamin D in cancer prevention and treatment. Endocrinol. Metab. Clin. N. Am. 2010, 39, 401–418. [Google Scholar] [CrossRef]
- Welsh, J. Cellular and molecular effects of vitamin D on carcinogenesis. Arch. Biochem. Biophys. 2012, 523, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M.; Shin, E.A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fleet, J.C.; Desmet, M.; Johnson, R.; Li, Y. Vitamin D and cancer: A review of molecular mechanisms. Biochem. J. 2012, 441, 61–76. [Google Scholar] [CrossRef]
- Wang, J.; Lian, H.; Zhao, Y.; Kauss, M.A.; Spindel, S. Vitamin D3 Induces Autophagy of Human Myeloid Leukemia Cells. J. Biol. Chem. 2008, 283, 25596–25605. [Google Scholar] [CrossRef]
- Avila, E.; Noriega-Mejía, B.J.; González-Macías, J.; Cortes-Hernández, U.; García-Quiroz, J.; García-Becerra, R.; Díaz, L. The Preventive Role of the Vitamin D Endocrine System in Cervical Cancer. Int. J. Mol. Sci. 2023, 10, 8665. [Google Scholar] [CrossRef]
- Gholamalizadeh, M.; Ardekanizadeh, N.H.; Aghakhaninejad, Z.; Mohammadi, S.; Majidi, N.; Masoumvand, M.; Bakhshimoghaddam, F.; Ahmadzadeh, M.; Tajadod, S.; Askarpour, S.A.; et al. The effects of dietary supplements in patients with cervical cancer: A comprehensive systematic review. Eur. J. Obstet. Gynecol. Reprod. Biol. X 2023, 19, 100217. [Google Scholar] [CrossRef]
- World Health Organisation. Cervical Cancer. Available online: https://www.who.int/cancer/prevention/diagnosis-screening/cervical-cancer/en/ (accessed on 1 May 2020).
- Burmeister, C.A.; Khan, S.F.; Schafer, G.; Mbatani, N.; Adams, T.; Moodley, J.; Prince, S. Cervical cancer therapies: Current challenges and future perspectives. Tumour Virus Res. 2022, 13, 200238. [Google Scholar] [CrossRef]
- StatPearls. Available online: https://www.ncbi.nlm.nih.gov/books/NBK431093 (accessed on 31 March 2025).
- Holick, M.F. Vitamin D: Importance in the prevention of cancers, type 1 diabetes, heart disease, and osteoporosis. Am. J. Clin. Nutr. 2004, 79, 362–371. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D status: Measurement, interpretation, and clinical application. Ann. Epidemiol. 2009, 2, 73–78. [Google Scholar] [CrossRef]
- Ramasamy, I. Vitamin D Metabolism and Guidelines for Vitamin D Supplementation. Clin. Biochem. Rev. 2020, 3, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Shruthi, N.K.; Prashanthkumar, M.; Venugopalreddy, B.; Suma, M.; Subba Rao, V. Analysis of the Cytotoxic Effects of Vitamin D3 on Colorectal, Breast and Cervical Carcinoma Cell Lines. Biochem. Anal. Biochem. 2017, 6, 1000318. [Google Scholar] [CrossRef]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Galluzzi, L.; Linkermann, A.; Keep, O.; Kroemer, G. Pathophysiology of Cancer Cell Death. In Abeloff’s Clinical Oncology, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 74–83. [Google Scholar] [CrossRef]
- Pather, Y.M.; Bhoora, S.; Joubert, A.M.; Mercier, A.E.; Punchoo, R. Cholecalciferol induces cell cycle arrest and apoptosis in HeLa cell line. In Proceedings of the 56th Annual Conference of the Microscopy Society of Southern Africa, Langebaan, South Africa, 1–5 December 2019; Volume 48. [Google Scholar]
- Kara, M.; Oztas, E. Endoplasmic Reticulum Stress-Mediated Cell Death. In Programmed Cell Death; Gali-Muhtasib, H., Rahal, O.N., Eds.; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Shimizu, T.; Kamel, W.A.; Yamaguchi-Iwai, S.; Fukuchi, Y.; Muto, A.; Saya, H. Calcitriol exerts an anti-tumor effect in osteosarcoma by inducing the endoplasmic reticulum stress response. Cancer Sci. 2017, 108, 1793–1802. [Google Scholar] [CrossRef]
- Haddur, E.; Ozkaya, A.B.; Ak, H.; Aydin, H.H. The effect of calcitriol on endoplasmic reticulum stress response. Biochem. Cell Biol. 2015, 93, 268–271. [Google Scholar] [CrossRef]
- Kloss, M.; Fischer, D.; Thill, M.; Friedrich, M.; Cordes, T.; Salehin, D.; Diedrich, K.; Köster, F. Vitamin D, calcidiol and calcitriol regulate vitamin D metabolizing enzymes in cervical and ovarian cancer cells. Anticancer Res. 2010, 30, 4429–4434. [Google Scholar]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- Zeng, R.; Li, H.; Jia, L.; Lee, S.H.; Jiang, R.; Zhang, Y.; Hu, X.; Ye, T.; Wang, X.; Yan, X.; et al. Association of CYP24A1 with survival and drug resistance in clinical cancer patients: A meta-analysis. BMC Cancer 2022, 22, 1317. [Google Scholar] [CrossRef]
- Shiratsuchi, H.; Wang, Z.; Chen, G.; Ray, P.; Lin, J.; Zhang, Z.; Zhao, L.; Beer, D.; Ray, D.; Ramnath, N. Oncogenic Potential of CYP24A1 in Lung Adenocarcinoma. J. Thorac. Oncol. 2017, 12, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Horváth, H.C.; Lakatos, P.; Kósa, J.P.; Bácsi, K.; Borka, K.; Bises, G.; Nittke, T.; Hershberger, P.A.; Speer, G.; Kállay, E. The candidate oncogene CYP24A1: A potential biomarker for colorectal tumorigenesis. J. Histochem. Cytochem. 2010, 58, 277–285. [Google Scholar] [CrossRef]
- Peixtoto, R.D.; de Carvalho Oliveira, L.J.; de Melo Passarini, T.; Andrade, A.C.; Diniz, P.H.; Prolla, G.; Amorim, L.C.; Gil, M.; Lino, F.; Garicochea, B.; et al. Vitamin D and colorectal cancer—A practical review of the literature. Cancer Treat. Res. Commun. 2022, 32, 100616. [Google Scholar] [CrossRef] [PubMed]
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Renal Physiol. 2005, 289, F8–F28. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, prot087379. [Google Scholar] [CrossRef] [PubMed]
- Punchoo, R.; Zhou, E.; Bhoora, S. Flow Cytometric Analysis of Apoptotic Biomarkers in Actinomycin D-treated SiHa Cervical Cancer Cells. J. Vis. Exp. 2021, 174, e62663. [Google Scholar] [CrossRef]
- Weakley, B.S. A Beginner’s Handbook in Biological Transmission Electron Microscopy, 2nd ed.; Churchill Livingstone: Edinburgh, UK, 1981. [Google Scholar]
- Gounden, S.; Phulukdaree, A.; Moodley, D.; Chuturgoon, A. Increased SIRT3 Expression and Antioxidant Defense under Hyperglycemic Conditions in HepG2 Cells. Metab. Syndr. Relat. Disord. 2015, 13, 255–263. [Google Scholar] [CrossRef]
- Blomberg Jensen, M.; Andersen, C.B.; Nielsen, J.E.; Bagi, P.; Jørgensen, A.; Juul, A.; Leffers, H. Expression of the vitamin D receptor, 25-hydroxylases, 1alpha-hydroxylase and 24-hydroxylase in the human kidney and renal clear cell cancer. J. Steroid Biochem. Mol. Biol. 2010, 121, 376–382. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, E.; Bhoora, S.; Pillay, T.S.; Punchoo, R. Induction of Cell Death and Regulation of Autocrine Vitamin D Metabolism in Cervical Cancer by Physiological and GI20 Doses of 25-Hydroxycholecalciferol. Int. J. Mol. Sci. 2025, 26, 4008. https://doi.org/10.3390/ijms26094008
Zhou E, Bhoora S, Pillay TS, Punchoo R. Induction of Cell Death and Regulation of Autocrine Vitamin D Metabolism in Cervical Cancer by Physiological and GI20 Doses of 25-Hydroxycholecalciferol. International Journal of Molecular Sciences. 2025; 26(9):4008. https://doi.org/10.3390/ijms26094008
Chicago/Turabian StyleZhou, Esther, Sachin Bhoora, Tahir S. Pillay, and Rivak Punchoo. 2025. "Induction of Cell Death and Regulation of Autocrine Vitamin D Metabolism in Cervical Cancer by Physiological and GI20 Doses of 25-Hydroxycholecalciferol" International Journal of Molecular Sciences 26, no. 9: 4008. https://doi.org/10.3390/ijms26094008
APA StyleZhou, E., Bhoora, S., Pillay, T. S., & Punchoo, R. (2025). Induction of Cell Death and Regulation of Autocrine Vitamin D Metabolism in Cervical Cancer by Physiological and GI20 Doses of 25-Hydroxycholecalciferol. International Journal of Molecular Sciences, 26(9), 4008. https://doi.org/10.3390/ijms26094008