

Upregulation of MMP3 Promotes Cisplatin Resistance in Ovarian Cancer

 , and
, and 
Abstract
1. Introduction
2. Results
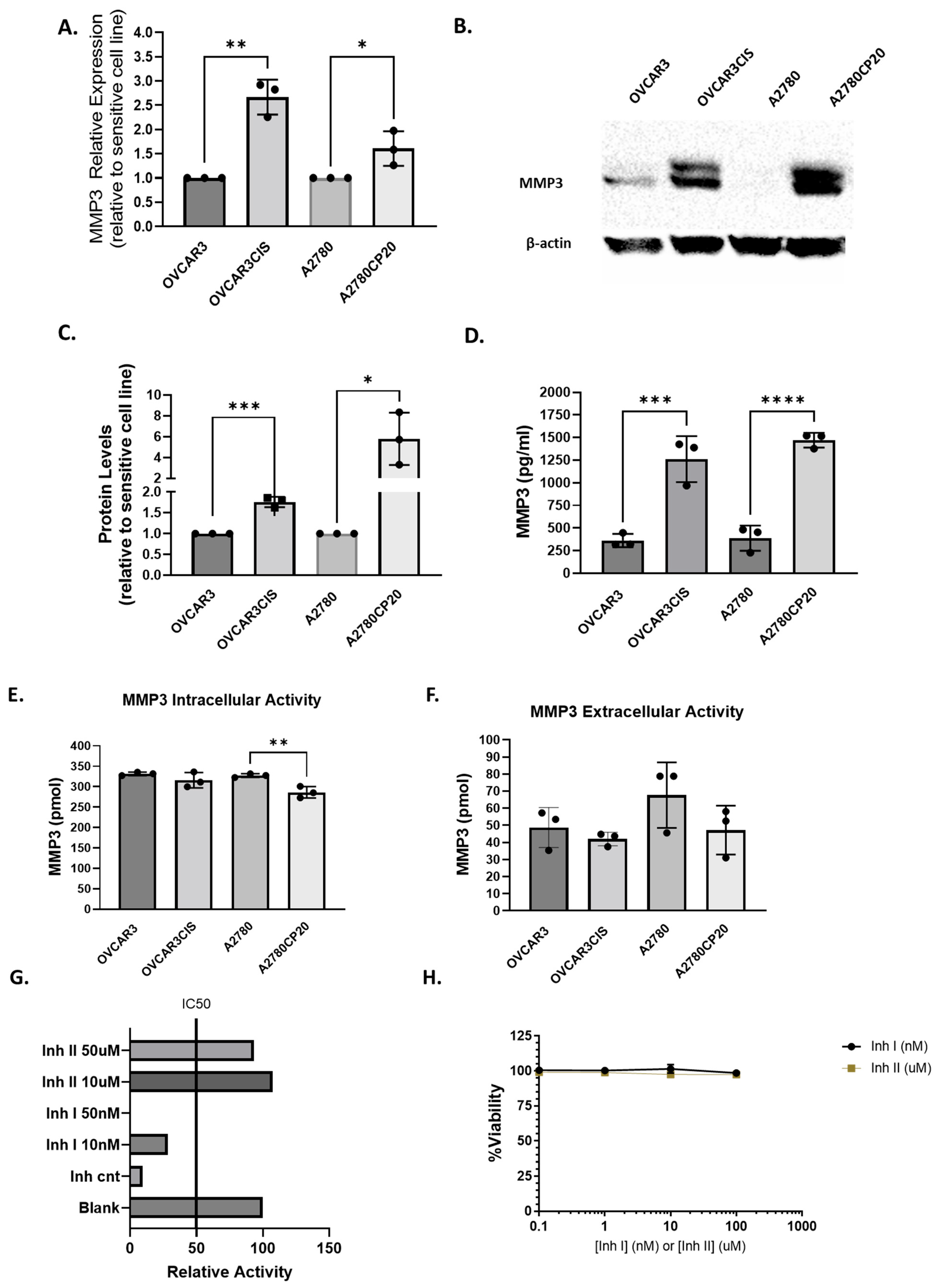
2.1. MMP3 Is Upregulated in Cisplatin-Resistant OC Cells Compared with Their Cisplatin-Sensitive Counterparts
2.1.1. MMP3 Activity Is Not Correlated with the Cisplatin Sensitivity of OC Cells
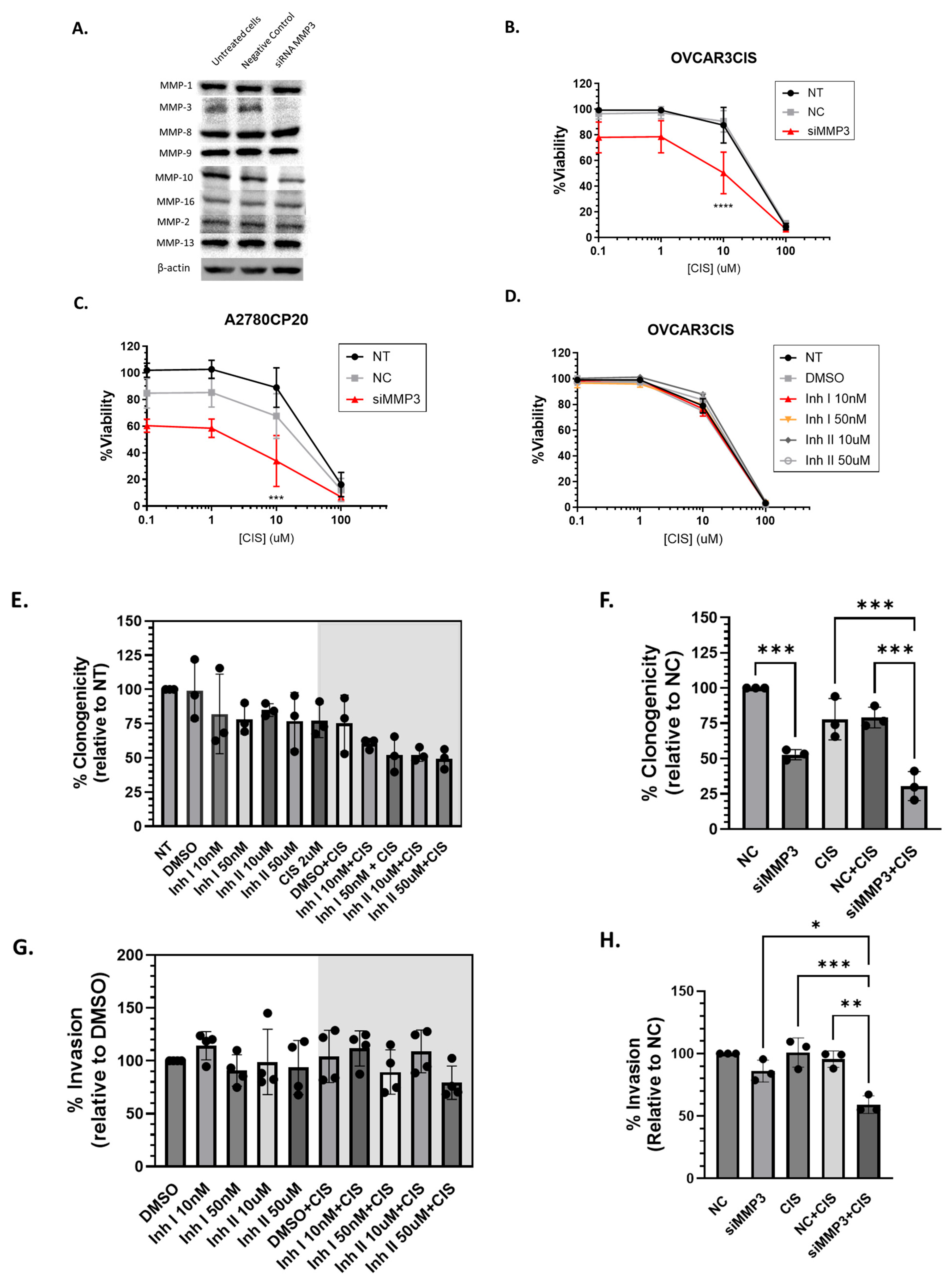
2.1.2. siRNA-Mediated Knockdown of MMP3 and Small-Molecule Inhibitors Have Different Effects on Viability, Proliferation, and Invasion of OC Cells
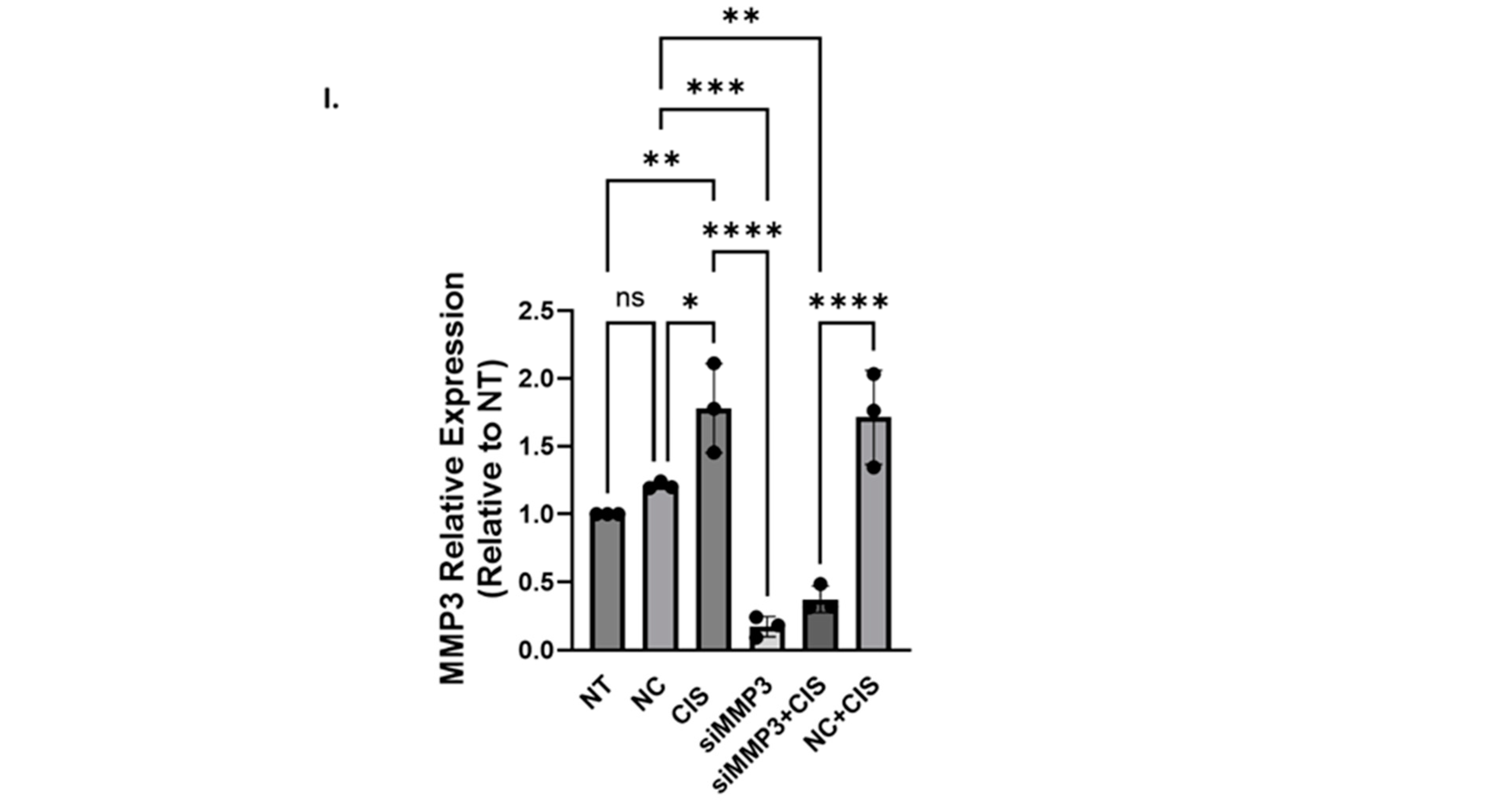
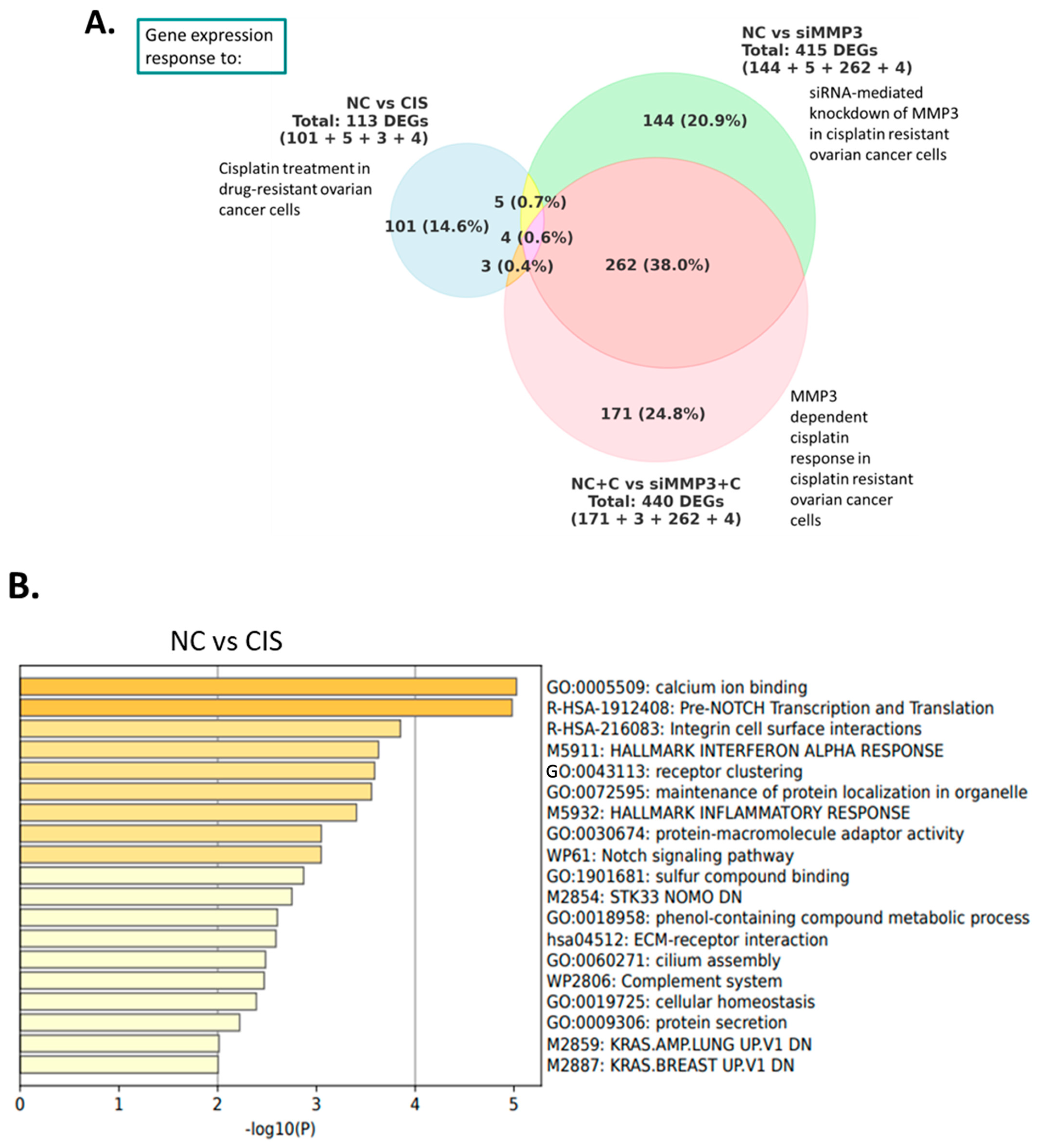
2.2. Downstream Effectors of MMP3 in HGSOC Cells
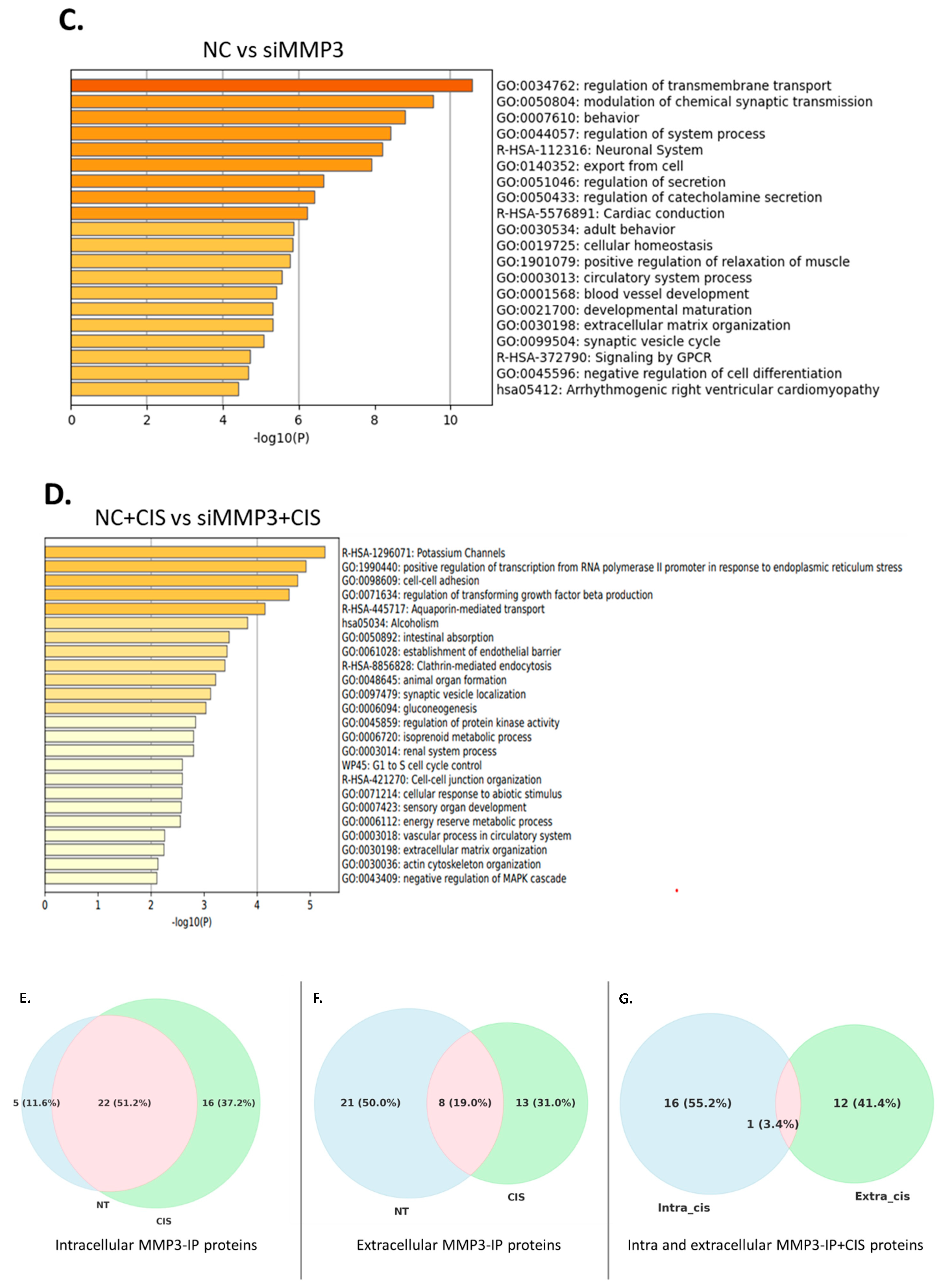
2.3. MMP3 Is Associated with Different Proteins in Untreated and Cisplatin-Treated OVCAR3CIS Cells
2.4. Effect of Multiple Injections of Liposomal-siRNA-MMP3 in an OC Mouse Model
2.5. In Vivo Targeting of MMP3 with Liposome-Encapsulated siRNAs Reduces Cell Proliferation and Angiogenesis in an OC Mouse Model
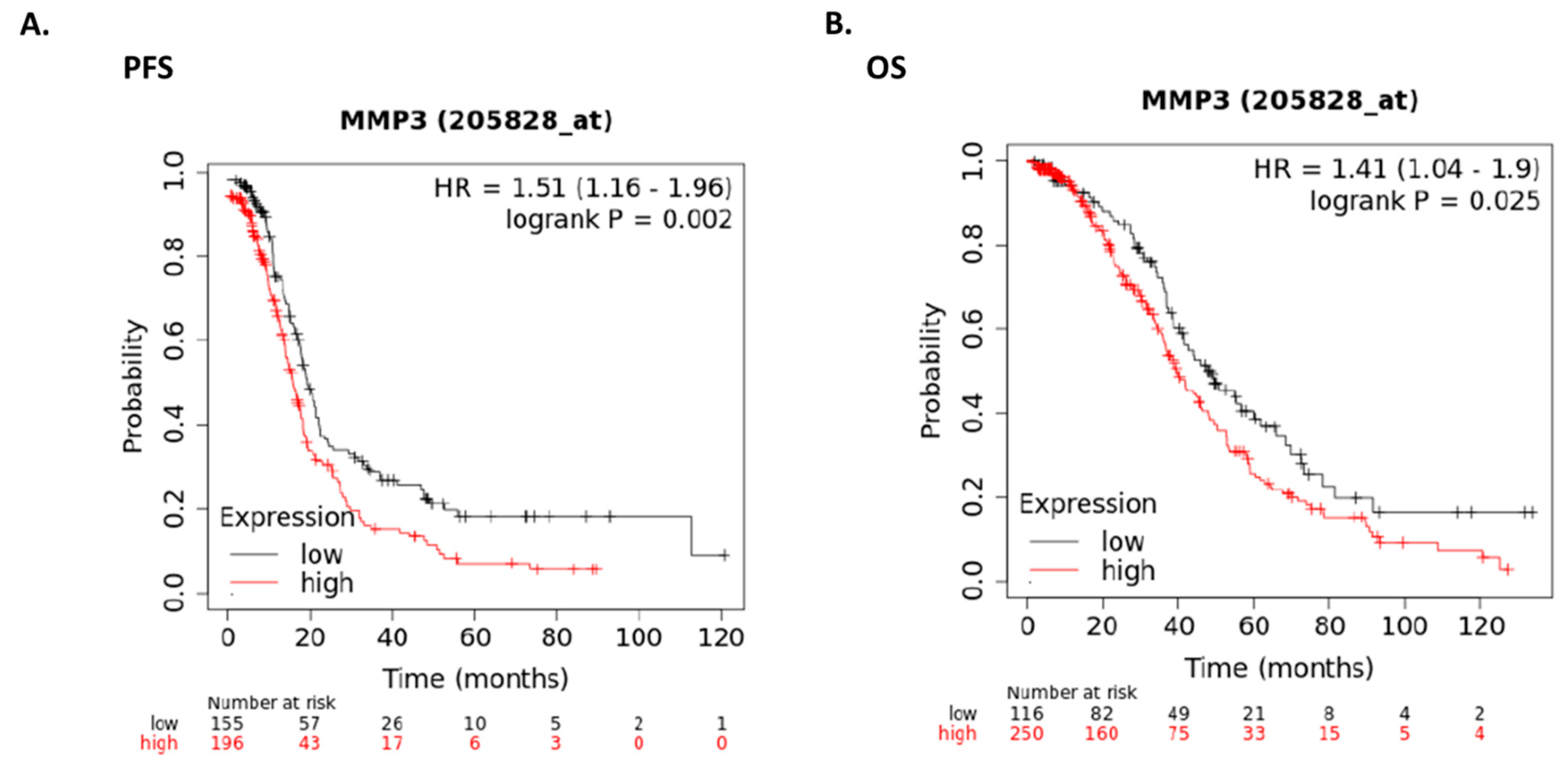
2.6. Expression of MMP3 in Human Ovarian Cancer Patients
3. Discussion
4. Methods
4.1. Cell Lines and Cell Culture Maintenance
4.2. Media Concentration
4.3. Quantitative Real-Time PCR (qRT-PCR)
4.4. Western Blot Analysis
4.5. In Vitro MMP3-siRNA Transfection and Treatment with MMP3 Inhibitors
4.6. MMP3 Activity Assay
4.7. Cell Viability Assays
4.8. Clonogenic Assays
4.9. Migration Assays
4.10. RNA Sequencing of siRNA-Mediated MMP3-Knockdown Cells with and Without Cisplatin
4.11. Kyoto Encyclopedia of Genes and Genomes Pathway Enrichment, Gene Ontology, and Network Analyses
4.12. Immunoprecipitation of the MMP3 Protein
4.13. MS Analyses and Protein Identification
4.14. OVCAR3CIS Tumor Implantation and MMP3-Targeted Treatment with Cisplatin
4.15. Immunohistochemistry
4.16. Survival Analysis
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed]
- Trudu, F.; Amato, F.; Vaňhara, P.; Pivetta, T.; Peña-Méndez, E.M.; Havel, J. Coordination compounds in cancer: Past, present and perspectives. J. Appl. Biomed. 2015, 13, 79–103. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial involvement in cisplatin resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef]
- Dl, R.; Rn, E.; Dm, O. Advances in Ovarian Cancer Care and Unmet Treatment Needs for Patients With Platinum Resistance: A Narrative Review. JAMA Oncol. 2023, 9, 197. [Google Scholar] [CrossRef]
- Song, M.; Cui, M.; Liu, K. Therapeutic strategies to overcome cisplatin resistance in ovarian cancer. Eur. J. Med. Chem. 2022, 232, 114205. [Google Scholar] [CrossRef]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer 2019, 11, 1179299X19860815. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Mendiola, M.; Redondo, A.; Heredia-Soto, V.; Herranz, J.; Berjón, A.; Hernández, A.; Miguel-Martín, M.; Crespo, R.; Barriuso, J.; Cruz, P.; et al. Predicting Response to Standard First-line Treatment in High-grade Serous Ovarian Carcinoma by Angiogenesis-related Genes. Anticancer Res. 2018, 38, 5393–5400. [Google Scholar] [CrossRef]
- Moore, K.N.; Angelergues, A.; Konecny, G.E.; García, Y.; Banerjee, S.; Lorusso, D.; Lee, J.-Y.; Moroney, J.W.; Colombo, N.; Roszak, A.; et al. Mirvetuximab Soravtansine in FRα-Positive, Platinum-Resistant Ovarian Cancer. N. Engl. J. Med. 2023, 389, 2162–2174. [Google Scholar] [CrossRef] [PubMed]
- Piskór, B.M.; Przylipiak, A.; Dąbrowska, E.; Niczyporuk, M.; Ławicki, S. Matrilysins and stromelysins in pathogenesis and diagnostics of cancers. Cancer Manag. Res. 2020, 12, 10949–10964. [Google Scholar] [CrossRef] [PubMed]
- Carey, P.; Low, E.; Harper, E.; Stack, M.S. Metalloproteinases in Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 3403. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271. [Google Scholar] [CrossRef]
- Fingleton, B. MMPs as therapeutic targets—Still a viable option? Semin Cell Dev Biol. 2008, 19, 61–68. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rudek, M.A.; Venitz, J.; Figg, W.D. Matrix metalloproteinase inhibitors: Do they have a place in anticancer therapy? Pharmacotherapy 2002, 22, 705–720. [Google Scholar] [CrossRef]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef]
- Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef]
- Becker, J.W.; Marcy, A.I.; Rokosz, L.L.; Axel, M.G.; Burbaum, J.J.; Fitzgerald, P.M.D.; Cameron, P.M.; Esser, C.K.; Hagmann, W.K.; Hermes, J.D.; et al. Stromelysin-1: Three-dimensional structure of the inhibited catalytic domain and of the C-truncated proenzyme. Protein Sci. 1995, 4, 1966–1976. [Google Scholar] [CrossRef]
- Letra, A.; Silva, R.M.; Rylands, R.J.; Silveira, E.M.; De Souza, A.P.; Wendell, S.K.; Garlet, G.P.; Vieira, A.R. MMP3 and TIMP1 variants contribute to chronic periodontitis and may be implicated in disease progression. J. Clin. Periodontol. 2012, 39, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Beaudeux, J.L.; Giral, P.; Bruckert, E.; Bernard, M.; Foglietti, M.J.; Chapman, M.J. Serum matrix metalloproteinase-3 and tissue inhibitor of metalloproteinases-1 as potential markers of carotid atherosclerosis in infraclinical hyperlipidemia. Atherosclerosis 2003, 169, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Magdalena, K.; Magdalena, K.; Grazyna, S.; Sypniewska, G. The Role of Matrix Metalloproteinase-3 In the Development of Atherosclerosis and Cardiovascular Events. EJIFCC 2006, 17, 2–5. [Google Scholar]
- Huang, J.-F.; Du, W.-X.; Chen, J.-J. Elevated expression of matrix metalloproteinase-3 in human osteosarcoma and its association with tumor metastasis. J. BUON Off. J. Balk. Union Oncol. 2016, 21, 235–243. [Google Scholar]
- Cymbaluk-Płoska, A.; Chudecka-Głaz, A.; Pius-Sadowska, E.; Machaliński, B.; Menkiszak, J.; Sompolska-Rzechuła, A. Suitability assessment of baseline concentration of MMP3, TIMP3, HE4 and CA125 in the serum of patients with ovarian cancer. J. Ovarian Res. 2018, 2018, 1. [Google Scholar] [CrossRef]
- Mehner, C.; Miller, E.; Nassar, A.; Bamlet, W.R.; Radisky, E.S.; Radisky, D.C. Tumor cell expression of MMP3 as a prognostic factor for poor survival in pancreatic, pulmonary, and mammary carcinoma. Genes Cancer 2015, 6, 480. [Google Scholar] [CrossRef]
- Suhaimi, S.A.; Chan, S.C.; Rosli, R. Matrix Metallopeptidase 3 Polymorphisms: Emerging genetic Markers in Human Breast Cancer Metastasis. J. Breast Cancer 2020, 23, 1–9. [Google Scholar] [CrossRef]
- Zheng, J.; Zhou, Y.; Li, X.J.; Hu, J.M. MiR-574-3p exerts as a tumor suppressor in ovarian cancer through inhibiting MMP3 expression. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6839–6848. [Google Scholar] [CrossRef]
- Liang, M.; Wang, J.; Wu, C.; Wu, M.; Hu, J.; Dai, J.; Ruan, H.; Xiong, S.; Dong, C. Targeting matrix metalloproteinase MMP3 greatly enhances oncolytic virus mediated tumor therapy. Transl. Oncol. 2021, 14, 101221. [Google Scholar] [CrossRef]
- Ma, Y.; Cang, S.; Li, G.; Su, Y.; Zhang, H.; Wang, L.; Yang, J.; Shi, X.; Qin, G.; Yuan, H. Integrated analysis of transcriptome data revealed MMP3 and MMP13 as critical genes in anaplastic thyroid cancer progression. J. Cell. Physiol. 2019, 234, 22260–22271. [Google Scholar] [CrossRef]
- Quiñones-Díaz, B.I.; Reyes-González, J.M.; Sánchez-Guzmán, V.; Conde-Del Moral, I.; Valiyeva, F.; Santiago-Sánchez, G.S.; Vivas-Mejía, P.E. MicroRNA-18a-5p Suppresses Tumor Growth via Targeting Matrix Metalloproteinase-3 in Cisplatin-Resistant Ovarian Cancer. Front. Oncol. 2020, 10, 602670. [Google Scholar] [CrossRef] [PubMed]
- Dana, H.; Chalbatani, G.M.; Mahmoodzadeh, H.; Karimloo, R.; Rezaiean, O.; Moradzadeh, A.; Mehmandoost, N.; Moazzen, F.; Mazraeh, A.; Marmari, V.; et al. Molecular Mechanisms and Biological Functions of siRNA. Int. J. Biomed. Sci. IJBS 2017, 13, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Gavrilov, K.; Saltzman, W.M. Therapeutic siRNA: Principles, Challenges, and Strategies. Yale J. Biol. Med. 2012, 85, 187. [Google Scholar]
- Noriega-Rivera, R.; Rivera-Serrano, M.; Rabelo-Fernandez, R.J.; Pérez-Santiago, J.; Valiyeva, F.; Vivas-Mejía, P.E. Upregulation of the Long Noncoding RNA CASC10 Promotes Cisplatin Resistance in High-Grade Serous Ovarian Cancer. Int. J. Mol. Sci. 2022, 23, 7737. [Google Scholar] [CrossRef]
- Li, N.; Yang, L.; Wang, H.; Yi, T.; Jia, X.; Chen, C.; Xu, P. MiR-130a and MiR-374a Function as Novel Regulators of Cisplatin Resistance in Human Ovarian Cancer A2780 Cells. PLoS ONE 2015, 10, e0128886. [Google Scholar] [CrossRef]
- Sørensen, B.H.; Thorsteinsdottir, U.A.; Lambert, I.H. Acquired cisplatin resistance in human ovarian A2780 cancer cells correlates with shift in taurine homeostasis and ability to volume regulate. Am. J. Physiol.-Cell Physiol. 2014, 307, C1071–C1080. [Google Scholar] [CrossRef]
- Parker, R.J.; Eastman, A.; Bostick-Bruton, F.; Reed, E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced repair of cisplatin-DNA lesions and reduced drug accumulation. J. Clin. Investig. 1991, 87, 772–777. [Google Scholar] [CrossRef]
- Gong, T.T.; Liu, F.H.; Xiao, Q.; Li, Y.Z.; Wei, Y.F.; Xu, H.L.; Cao, F.; Sun, M.L.; Jiang, F.L.; Tao, T.; et al. SH3RF2 contributes to cisplatin resistance in ovarian cancer cells by promoting RBPMS degradation. Commun. Biol. 2024, 7, 67. [Google Scholar] [CrossRef]
- Santana-Rivera, Y.; Rabelo-Fernández, R.J.; Quiñones-Díaz, B.I.; Grafals-Ruíz, N.; Santiago-Sánchez, G.; Lozada-Delgado, E.L.; Echevarría-Vargas, I.M.; Apiz, J.; Soto, D.; Rosado, A.; et al. Reduced expression of enolase-1 correlates with high intracellular glucose levels and increased senescence in cisplatin-resistant ovarian cancer cells. Am. J. Transl. Res. 2020, 12, 1275–1292. [Google Scholar]
- Reyes-González, J.M.; Armaiz-Peña, G.N.; Mangala, L.S.; Valiyeva, F.; Ivan, C.; Pradeep, S.; Echevarría-Vargas, I.M.; Rivera-Reyes, A.; Sood, A.K.; Vivas-Mejía, P.E. Targeting c-MYC in Platinum-Resistant Ovarian Cancer. Mol. Cancer Ther. 2015, 14, 2260–2269. [Google Scholar] [CrossRef]
- Celik, Z.B.; Cankara, F.N.; Gunaydin, C. Alterations in the matrix metalloproteinase-3 promoter methylation after common chemotherapeutics: In vitro study of paclitaxel, cisplatin and methotrexate in the MCF-7 and SH-SY5Y cell lines. Mol. Biol. Rep. 2020, 47, 8987–8995. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jia, J.; Liu, D.; Wang, M.; Wang, Z.; Li, X.; Wang, H.; Rui, Y.; Liu, Z.; Guo, W.; et al. Matrix Metalloproteinase Expressions Play Important role in Prediction of Ovarian Cancer Outcome. Sci. Rep. 2019, 9, 11677. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Chen, T.; Yang, M.; Li, N.; Liu, H.; Cao, X. Human SCAMP5, a Novel Secretory Carrier Membrane Protein, Facilitates Calcium-Triggered Cytokine Secretion by Interaction with SNARE Machinery. J. Immunol. 2009, 182, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Aoh, Q.L.; Castle, A.M.; Hubbard, C.H.; Katsumata, O.; Castle, J.D. SCAMP3 Negatively Regulates Epidermal Growth Factor Receptor Degradation and Promotes Receptor Recycling. Mol. Biol. Cell 2009, 20, 1816–1832. [Google Scholar] [CrossRef]
- Li, W.; Webster, K.A.; LeBlanc, M.E.; Tian, H. Secretogranin III: A diabetic retinopathy-selective angiogenic factor. Cell. Mol. Life Sci. CMLS 2017, 75, 635–647. [Google Scholar] [CrossRef]
- Yadav, G.P.; Wang, H.; Ouwendijk, J.; Cross, S.; Wang, Q.; Qin, F.; Verkade, P.; Zhu, M.X.; Jiang, Q.-X. Chromogranin B (CHGB) is dimorphic and responsible for dominant anion channels delivered to cell surface via regulated secretion. Front. Mol. Neurosci. 2023, 16, 1205516. [Google Scholar] [CrossRef]
- Yee, S.W.; Giacomini, K.M. Emerging Roles of the Human Solute Carrier 22 Family. Drug Metab. Dispos. 2022, 50, 1193–1210. [Google Scholar] [CrossRef]
- Shen, T.; Huang, S. The role of Cdc25A in the regulation of cell proliferation and apoptosis. Anticancer. Agents Med. Chem. 2012, 12, 631–639. [Google Scholar] [CrossRef]
- Dufour, A.; Sampson, N.S.; Zucker, S.; Cao, J. Role of the hemopexin domain of matrix metalloproteinases in cell migration. J. Cell. Physiol. 2008, 217, 643–651. [Google Scholar] [CrossRef]
- Piccard, H.; Van den Steen, P.E.; Opdenakker, G. Hemopexin domains as multifunctional liganding modules in matrix metalloproteinases and other proteins. J. Leukoc. Biol. 2007, 81, 870–892. [Google Scholar] [CrossRef]
- García-Pardo, A.; Opdenakker, G. Nonproteolytic functions of matrix metalloproteinases in pathology and insights for the development of novel therapeutic inhibitors. Met. Med. 2015, 2, 19–28. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Dijkgraaf, G.J.P.; Lawson, D.A.; Littlepage, L.E.; Shahi, P.; Pieper, U.; Werb, Z. A Role for matrix metalloproteinases in regulating mammary stem cell function via the Wnt signaling pathway. Cell Stem Cell 2013, 13, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Martin-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Ayala-San Nicolas, M. WNT Signaling in Tumors: The Way to Evade Drugs and Immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef]
- Correia, A.L.; Mori, H.; Chen, E.I.; Schmitt, F.C.; Bissell, M.J. The hemopexin domain of MMP3 is responsible for mammary epithelial invasion and morphogenesis through extracellular interaction with HSP90β. Genes Dev. 2013, 27, 805–817. [Google Scholar] [CrossRef]
- Yin, L.; Yang, Y.; Zhu, W.; Xian, Y.; Han, Z.; Huang, H.; Peng, L.; Zhang, K.; Zhao, Y. Heat Shock Protein 90 Triggers Multi-Drug Resistance of Ovarian Cancer via AKT/GSK3β/β-Catenin Signaling. Front. Oncol. 2021, 11, 558. [Google Scholar] [CrossRef]
- Ivanov, A.I.; Christodoulou, J.; Parkinson, J.A.; Barnham, K.J.; Tucker, A.; Woodrow, J.; Sadler, P.J. Cisplatin Binding Sites on Human Albumin. J. Biol. Chem. 1998, 273, 14721–14730. [Google Scholar] [CrossRef]
- Jia, J.; Han, Z.; Wang, X.; Zheng, X.; Wang, S.; Cui, Y. H2B gene family: A prognostic biomarker and correlates with immune infiltration in glioma. Front. Oncol. 2022, 12, 966817. [Google Scholar] [CrossRef]
- Li, J.; Qi, F.; Su, H.; Zhang, C.; Zhang, Q.; Chen, Y.; Chen, P.; Su, L.; Chen, Y.; Yang, Y.; et al. GRP75-faciliated Mitochondria-associated ER Membrane (MAM) Integrity controls Cisplatin-resistance in Ovarian Cancer Patients. Int. J. Biol. Sci. 2022, 18, 2914–2931. [Google Scholar] [CrossRef]
- Singh, B.; Sundbom, M.F.; Muthukrishnan, U.; Natarajan, B.; Stransky, S.; Görgens, A.; Nordin, J.Z.; Wiklander, O.P.B.; Sandblad, L.; Sidoli, S.; et al. Histones are exosome membrane proteins regulated by cell stress. bioRxiv 2024, 2024, 588575. [Google Scholar] [CrossRef]
- Lee, Y.J.; Shin, K.J.; Chae, Y.C. Regulation of cargo selection in exosome biogenesis and its biomedical applications in cancer. Exp. Mol. Med. 2024, 56, 877–889. [Google Scholar] [CrossRef]
- Starokadomskyy, P.; Gluck, N.; Li, H.; Chen, B.; Wallis, M.; Maine, G.N.; Mao, X.; Zaidi, I.W.; Hein, M.Y.; McDonald, F.J.; et al. CCDC22 deficiency in humans blunts activation of proinflammatory NF-κB signaling. J. Clin. Investig. 2013, 123, 2244–2256. [Google Scholar] [CrossRef]
- Schoeberl, A.; Gutmann, M.; Theiner, S.; Corte-Rodríguez, M.; Braun, G.; Vician, P.; Berger, W.; Koellensperger, G. The copper transporter CTR1 and cisplatin accumulation at the single-cell level by LA-ICP-TOFMS. Front. Mol. Biosci. 2022, 9, 1055356. [Google Scholar] [CrossRef]
- Uptake of the Anticancer Drug Cisplatin Mediated by the Copper Transporter Ctr1 in Yeast and Mammals|PNAS. Available online: https://www.pnas.org/doi/full/10.1073/pnas.162491399 (accessed on 13 January 2025).
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Basak, U.; Sarkar, T.; Mukherjee, S.; Chakraborty, S.; Dutta, A.; Dutta, S.; Nayak, D.; Kaushik, S.; Das, T.; Sa, G. Tumor-associated macrophages: An effective player of the tumor microenvironment. Front. Immunol. 2023, 14, 1295257. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int. J. Mol. Sci. 2022, 23, 146. [Google Scholar] [CrossRef]
- Bohonowych, J.; Hance, M.; Nolan, K.; Defee, M.; Parsons, C.; Isaacs, J. Extracellular Hsp90 mediates an NF-κB dependent inflammatory stromal program: Implications for the prostate tumor microenvironment. Prostate 2014, 74, 395–407. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, J.; Li, W.; Zhang, C. Micro-RNA-21 Regulates Cancer-Associated Fibroblast-Mediated Drug Resistance in Pancreatic Cancer. Oncol. Res. 2018, 26, 827–835. [Google Scholar] [CrossRef]
- PubChem. (2R)-N-[(2S)-3,3-dimethyl-1-oxo-1-[[(1R)-1-phenylethyl]amino]butan-2-yl]-N’-hydroxy-2-[3-(3-methyl-4-phenylphenyl)propyl]butanediamide. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/10370504 (accessed on 13 January 2025).
- PubChem. MMP-3 Inhibitor I. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/99648983 (accessed on 13 January 2025).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | ID | log2FoldChange | p-adj |
|---|---|---|---|
| siRNA-mediated MMP3 knockdown | |||
| SCAMP5 | ENSG00000198794 | 1.060988 | 1.76 × 10−51 |
| NEFH | ENSG00000100285 | 1.139433 | 2.61 × 10−40 |
| BTG2 | ENSG00000159388 | −1.09713 | 4.09 × 10−26 |
| VASP | ENSG00000125753 | −1.05933 | 5.52 × 10−25 |
| ITGA5 | ENSG00000161638 | −1.00827 | 5.68 × 10−24 |
| WWTR1 | ENSG00000018408 | −1.03085 | 2.2 × 10−22 |
| PHF21B | ENSG00000056487 | 1.042219 | 9.14 × 10−22 |
| RPS6KA4 | ENSG00000162302 | −1.02319 | 1.39 × 10−21 |
| DLX2 | ENSG00000115844 | −1.22783 | 1.52 × 10−20 |
| RELL2 | ENSG00000164620 | 1.035967 | 9.42 × 10−16 |
| KCNQ2 | ENSG00000075043 | 1.043291 | 3.8 × 10−15 |
| SERPINB9 | ENSG00000170542 | −1.02407 | 1.95 × 10−11 |
| SNCB | ENSG00000074317 | 1.281406 | 5.91 × 10−11 |
| SVOP | ENSG00000166111 | 4.904506 | 4.58 × 10−10 |
| MPC1 | ENSG00000060762 | 1.037979 | 6.96 × 10−10 |
| GPR3 | ENSG00000181773 | −1.12902 | 7.66 × 10−10 |
| ERO1A | ENSG00000197930 | −1.01424 | 8.17 × 10−10 |
| C3orf14 | ENSG00000114405 | 1.073282 | 9.45 × 10−10 |
| LMLN | ENSG00000185621 | −1.0919 | 2.97 × 10−9 |
| FGF7P3 | ENSG00000204837 | 1.163488 | 9.88 × 10−9 |
| MMP3-dependent cisplatin response | |||
| CHGB | ENSG00000089199 | 3.603976 | 9.84 × 10−154 |
| CHRNB2 | ENSG00000160716 | 4.26102 | 7.07 × 10−137 |
| SCG3 | ENSG00000104112 | 5.04094 | 2.42 × 10−130 |
| AP3B2 | ENSG00000103723 | 2.221219 | 4.52 × 10−122 |
| SYP | ENSG00000102003 | 2.309564 | 1.06 × 10−121 |
| STMN3 | ENSG00000197457 | 2.493578 | 1.14 × 10−110 |
| RUNDC3A | ENSG00000108309 | 3.001584 | 2.17 × 10−102 |
| ACTL6B | ENSG00000077080 | 4.645848 | 1.06 × 10−96 |
| XKR7 | ENSG00000260903 | 4.393692 | 3.97 × 10−85 |
| MAPK8IP2 | ENSG00000008735 | 2.266975 | 1.26 × 10−72 |
| CPLX1 | ENSG00000168993 | 3.580086 | 2.59 × 10−71 |
| NFE2L2 | ENSG00000116044 | −1.23975 | 2.19 × 10−59 |
| UNC79 | ENSG00000133958 | 2.025565 | 1.71 × 10−57 |
| MAPK8IP1 | ENSG00000121653 | 1.378141 | 3.56 × 10−55 |
| PAX5 | ENSG00000196092 | 3.248805 | 3.04 × 10−52 |
| CHGA | ENSG00000100604 | 3.290491 | 2.07 × 10−49 |
| AC005696.4 | ENSG00000277200 | 3.286964 | 1.39 × 10−48 |
| FAM57B | ENSG00000149926 | 2.656496 | 3.01 × 10−48 |
| INIP | ENSG00000148153 | −1.25455 | 9.29 × 10−47 |
| DPY19L1 | ENSG00000173852 | −1.36383 | 2.2 × 10−44 |
| siRNA-MMP3 plus cisplatin vs. NC-siRNA plus cis-platin | |||
| CDC25A | ENSG00000164045 | −1.0636 | 4.63 × 10−44 |
| ANKRD1 | ENSG00000148677 | −1.0090 | 3.96 × 10−39 |
| ACTR2 | ENSG00000138071 | −1.1531 | 3.37 × 10−37 |
| GFPT1 | ENSG00000198380 | −1.0276 | 1.58 × 10−34 |
| KCNC3 | ENSG00000131398 | 1.1819 | 9.43 × 10−33 |
| CARNMT1 | ENSG00000156017 | −1.0228 | 6.55 × 10−32 |
| PLEKHB2 | ENSG00000115762 | −1.0282 | 2.73 × 10−31 |
| PODXL2 | ENSG00000114631 | 1.0052 | 1.41 × 10−27 |
| LIMA1 | ENSG00000050405 | −1.0028 | 5.53 × 10−27 |
| REEP5 | ENSG00000129625 | −1.0534 | 3.17 × 10−26 |
| UEVLD | ENSG00000151116 | −1.0062 | 8.64 × 10−25 |
| SBK1 | ENSG00000188322 | 1.1215 | 9.09 × 10−22 |
| POLR3G | ENSG00000113356 | −1.0585 | 7.21 × 10−21 |
| ADAMTSL4 | ENSG00000143382 | 1.0051 | 7.96 × 10−19 |
| MOSPD3 | ENSG00000106330 | 1.0839 | 3.99 × 10−18 |
| TRIP11 | ENSG00000100815 | −1.0167 | 3.2 × 10−17 |
| TIMM10 | ENSG00000134809 | −1.1039 | 1.03 × 10−16 |
| PDCD4 | ENSG00000150593 | −1.0309 | 1.07 × 10−16 |
| F11R | ENSG00000158769 | −1.0167 | 1.46 × 10−16 |
| CNNM1 | ENSG00000119946 | 1.0436 | 1.82 × 10−15 |
| Gene Symbol | Description | MW [kDa] | calc. pI | ENSEMBL Gene ID |
|---|---|---|---|---|
| unique intracellular proteins in cisplatin-treated cells | ||||
| ALB | Albumin | 69.3 | 6.28 | ENSG00000163631 |
| C18orf63 | Uncharacterized protein C18orf63 | 77.2 | 9.8 | ENSG00000206043 |
| CCDC22 | Coiled-coil domain-containing protein 22 | 70.7 | 6.74 | ENSG00000101997 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | 36 | 8.46 | ENSG00000111640 |
| H2BC21 | Histone H2B type 2-E | 13.9 | 10.32 | ENSG00000184678 |
| HSPA9 | Stress-70 protein, mitochondrial | 73.6 | 6.16 | ENSG00000113013 |
| KCTD3 | BTB/POZ domain-containing protein KCTD3 | 88.9 | 7.03 | ENSG00000136636 |
| MYL12B | Myosin regulatory light chain 12B | 19.8 | 4.84 | ENSG00000118680 |
| RPL27A | Large ribosomal subunit protein uL15 | 16.6 | 11 | ENSG00000166441 |
| RPL36AL | Ribosomal protein eL42-like | 12.5 | 10.65 | ENSG00000165502 |
| RPS15 | Small ribosomal subunit protein uS19 | 17 | 10.39 | ENSG00000115268 |
| RPS23 | Small ribosomal subunit protein uS12 | 15.8 | 10.49 | ENSG00000186468 |
| RPS29 | Small ribosomal subunit protein uS14 | 6.7 | 10.13 | ENSG00000213741 |
| RPS4Y2 | Small ribosomal subunit protein eS4, Y isoform 2 | 29.3 | 10.08 | ENSG00000280969 |
| RPS6 | Small ribosomal subunit protein eS6 | 28.7 | 10.84 | ENSG00000137154 |
| SRXN1 | Sulfiredoxin-1 | 14.3 | 8.19 | ENSG00000271303 |
| Common proteins in cisplatin-treated and cisplatin-untreated cells | ||||
| ACTB | Actin, cytoplasmic 1 | 41.7 | 5.48 | ENSG00000075624 |
| ATXN2 | Ataxin-2 | 140.2 | 9.57 | ENSG00000204842 |
| BANF1 | Barrier-to-autointegration factor | 10.1 | 6.09 | ENSG00000175334 |
| CLTC | Clathrin heavy chain 1 | 191.5 | 5.69 | ENSG00000141367 |
| GRN | Progranulin | 63.5 | 6.83 | ENSG00000030582 |
| H2AC20 | Histone H2A type 2-C | 14 | 10.9 | ENSG00000184260 |
| H4C1; H4C11; H4C12; H4C13; H4C14; H4C15; H4C16; H4C2; H4C3; H4C4; H4C5; H4C6; H4C8; H4C9 | Histone H4 | 11.4 | 11.36 | ENSG00000158406; ENSG00000197061; ENSG00000197238; ENSG00000197837; ENSG00000270276; ENSG00000270882; ENSG00000273542; ENSG00000274618; ENSG00000275126; ENSG00000276180; ENSG00000276966; ENSG00000277157; ENSG00000278637; ENSG00000278705 |
| IGHG1 | Isoform 1 of Immunoglobulin heavy constant gamma 1 | 36.1 | 8.19 | ENSG00000211896 |
| MYH10 | Myosin-10 | 228.9 | 5.54 | ENSG00000133026 |
| MYH9 | Myosin-9 | 226.4 | 5.6 | ENSG00000100345 |
| MYL6 | Myosin light polypeptide 6 | 16.9 | 4.65 | ENSG00000092841 |
| RPL13 | Large ribosomal subunit protein eL13 | 24.2 | 11.65 | ENSG00000167526 |
| RPL15 | Large ribosomal subunit protein eL15 | 24.1 | 11.62 | ENSG00000174748 |
| RPL35 | Large ribosomal subunit protein uL29 | 14.5 | 11.05 | ENSG00000136942 |
| RPL39 | Large ribosomal subunit protein eL39 | 6.4 | 12.56 | ENSG00000198918 |
| RPS11 | Small ribosomal subunit protein uS17 | 18.4 | 10.3 | ENSG00000142534 |
| RPS13 | Small ribosomal subunit protein uS15 | 17.2 | 10.54 | ENSG00000110700 |
| RPS14 | Small ribosomal subunit protein uS11 | 16.3 | 10.05 | ENSG00000164587 |
| RPS18 | Small ribosomal subunit protein uS13 | 17.7 | 10.99 | ENSG00000096150; ENSG00000223367; ENSG00000226225; ENSG00000231500; ENSG00000235650 |
| RPS24 | Small ribosomal subunit protein eS24 | 15.4 | 10.78 | ENSG00000138326 |
| RPS25 | Small ribosomal subunit protein eS25 | 13.7 | 10.11 | ENSG00000118181; ENSG00000280831 |
| RPS3 | Small ribosomal subunit protein uS3 | 26.7 | 9.66 | ENSG00000149273 |
| unique proteins in cisplatin untreated cells | ||||
| FLG | Filaggrin | 434.9 | 9.25 | ENSG00000143631 |
| H2AX | Histone H2AX | 15.1 | 10.74 | ENSG00000188486 |
| H3-3A; H3-3B | Histone H3.3 | 15.3 | 11.27 | ENSG00000132475; ENSG00000163041 |
| HMGA2 | High mobility group protein HMGI-C | 11.8 | 10.62 | ENSG00000149948 |
| WDFY1 | WD repeat and FYVE domain-containing protein 1 | 46.3 | 7.33 | ENSG00000085449 |
| Gene Symbol | Description | MW [kDa] | calc. pI | ENSEMBL Gene ID |
|---|---|---|---|---|
| unique intracellular proteins in cisplatin-treated cells | ||||
| ACTB | Actin, cytoplasmic 1 | 41.7 | 5.48 | ENSG00000075624 |
| CCDC22 | Coiled-coil domain-containing protein 22 | 70.7 | 6.74 | ENSG00000101997 |
| HNRNPAB | Heterogeneous nuclear ribonucleoprotein A/B | 36.2 | 8.21 | ENSG00000197451 |
| ALPK2 | Alpha-protein kinase 2 | 236.9 | 5.24 | ENSG00000198796 |
| RPS25 | Small ribosomal subunit protein eS25 | 13.7 | 10.11 | ENSG00000118181; ENSG00000280831 |
| KIAA0232 | Uncharacterized protein KIAA0232 | 154.7 | 4.78 | ENSG00000170871 |
| IGHG4 | Immunoglobulin heavy constant gamma 4 | 43.8 | 6.24 | ENSG00000211892; ENSG00000277016 |
| H4C1; H4C11; H4C12; H4C13; H4C14; H4C15; H4C16; H4C2; H4C3; H4C4; H4C5; H4C6; H4C8; H4C9 | Histone H4 | 11.4 | 11.36 | ENSG00000158406; ENSG00000197061; ENSG00000197238; ENSG00000197837; ENSG00000270276; ENSG00000270882; ENSG00000273542; ENSG00000274618; ENSG00000275126; ENSG00000276180; ENSG00000276966; ENSG00000277157; ENSG00000278637; ENSG00000278705 |
| POTEE | POTE ankyrin domain family member E | 121.3 | 6.2 | ENSG00000188219 |
| RPL26 | Large ribosomal subunit protein uL24 | 17.2 | 10.55 | ENSG00000161970 |
| DST | Dystonin | 860.1 | 5.25 | ENSG00000151914 |
| NPAP1 | Nuclear pore-associated protein 1 | 120.9 | 8.69 | ENSG00000185823 |
| ATP10B | Phospholipid-transporting ATPase VB | 165.3 | 6.89 | ENSG00000118322 |
| Common proteins in cisplatin-treated and cisplatin-untreated cells | ||||
| IGHG1 | Isoform 1 of Immunoglobulin heavy constant gamma 1 | 36.1 | 8.19 | ENSG00000211896 |
| ATXN2 | Ataxin-2 | 140.2 | 9.57 | ENSG00000204842 |
| IGLC1 | Immunoglobulin lambda-1 light chain | 22.8 | 6.76 | ENSG00000211675 |
| LAMA5 | Laminin subunit alpha-5 | 399.5 | 7.02 | ENSG00000130702 |
| ALB | Albumin | 69.3 | 6.28 | ENSG00000163631 |
| FLG | Filaggrin | 434.9 | 9.25 | ENSG00000143631 |
| C1R | Complement C1r subcomponent | 80.1 | 6.21 | ENSG00000159403 |
| HSPA7 | Putative heat shock 70 kDa protein 7 | 40.2 | 7.87 | ENSG00000225217 |
| unique proteins in cisplatin untreated cells | ||||
| AFF1 | AF4/FMR2 family member 1 | 131.3 | 9.2 | ENSG00000172493 |
| C18orf63 | Uncharacterized protein C18orf63 | 77.2 | 9.8 | ENSG00000206043 |
| CCDC25 | Coiled-coil domain-containing protein 25 | 24.5 | 6.8 | ENSG00000147419 |
| DEFA1; DEFA1B | Neutrophil defensin 1 | 10.2 | 6.99 | ENSG00000206047; ENSG00000240247; ENSG00000284983; ENSG00000285176 |
| DSC1 | Desmocollin-1 | 99.9 | 5.43 | ENSG00000134765 |
| FAM228B | Protein FAM228B | 38 | 8.73 | ENSG00000219626 |
| GRN | Progranulin | 63.5 | 6.83 | ENSG00000030582 |
| HBE1 | Hemoglobin subunit epsilon | 16.2 | 8.63 | ENSG00000213931 |
| IGLV2-11 | Immunoglobulin lambda variable 2–11 | 12.6 | 7.24 | ENSG00000211668 |
| LILRB3 | Leukocyte immunoglobulin-like receptor subfamily B member 3 | 69.3 | 6.93 | ENSG00000204577; ENSG00000274587; ENSG00000275019 |
| MGAT5B | Alpha-1,6-mannosylglycoprotein 6-beta-N-acetylglucosaminyltransferase B | 89.5 | 8.35 | ENSG00000167889 |
| MORC4 | MORC family CW-type zinc finger protein 4 | 106.3 | 7.46 | ENSG00000133131 |
| PABPN1L | Isoform 2 of Embryonic polyadenylate-binding protein 2 | 27.4 | 4.72 | |
| POTEF | POTE ankyrin domain family member F | 121.4 | 6.2 | ENSG00000196604 |
| RECQL4 | ATP-dependent DNA helicase Q4 | 133 | 8.09 | ENSG00000160957 |
| RP1L1 | Retinitis pigmentosa 1-like 1 protein | 252.1 | 4.45 | ENSG00000183638 |
| SMARCD2 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily D member 2 | 58.9 | 9.64 | ENSG00000108604 |
| SMIM35 | Small integral membrane protein 35 | 9.4 | 5.87 | ENSG00000255274 |
| SORBS1 | Sorbin and SH3 domain-containing protein 1 | 142.4 | 6.84 | ENSG00000095637 |
| SPTAN1 | Spectrin alpha chain, non-erythrocytic 1 | 284.4 | 5.35 | ENSG00000197694 |
| TRIM41 | E3 ubiquitin-protein ligase TRIM41 | 71.6 | 5.06 | ENSG00000146063 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera-Serrano, M.; Flores-Colón, M.; Valiyeva, F.; Meléndez, L.M.; Vivas-Mejía, P.E. Upregulation of MMP3 Promotes Cisplatin Resistance in Ovarian Cancer. Int. J. Mol. Sci. 2025, 26, 4012. https://doi.org/10.3390/ijms26094012
Rivera-Serrano M, Flores-Colón M, Valiyeva F, Meléndez LM, Vivas-Mejía PE. Upregulation of MMP3 Promotes Cisplatin Resistance in Ovarian Cancer. International Journal of Molecular Sciences. 2025; 26(9):4012. https://doi.org/10.3390/ijms26094012
Chicago/Turabian StyleRivera-Serrano, Mariela, Marienid Flores-Colón, Fatima Valiyeva, Loyda M. Meléndez, and Pablo E. Vivas-Mejía. 2025. "Upregulation of MMP3 Promotes Cisplatin Resistance in Ovarian Cancer" International Journal of Molecular Sciences 26, no. 9: 4012. https://doi.org/10.3390/ijms26094012
APA StyleRivera-Serrano, M., Flores-Colón, M., Valiyeva, F., Meléndez, L. M., & Vivas-Mejía, P. E. (2025). Upregulation of MMP3 Promotes Cisplatin Resistance in Ovarian Cancer. International Journal of Molecular Sciences, 26(9), 4012. https://doi.org/10.3390/ijms26094012