
Vital Role of PINK1/Parkin-Mediated Mitophagy of Pulmonary Epithelial Cells in Severe Pneumonia Induced by IAV and Secondary Staphylococcus aureus Infection

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
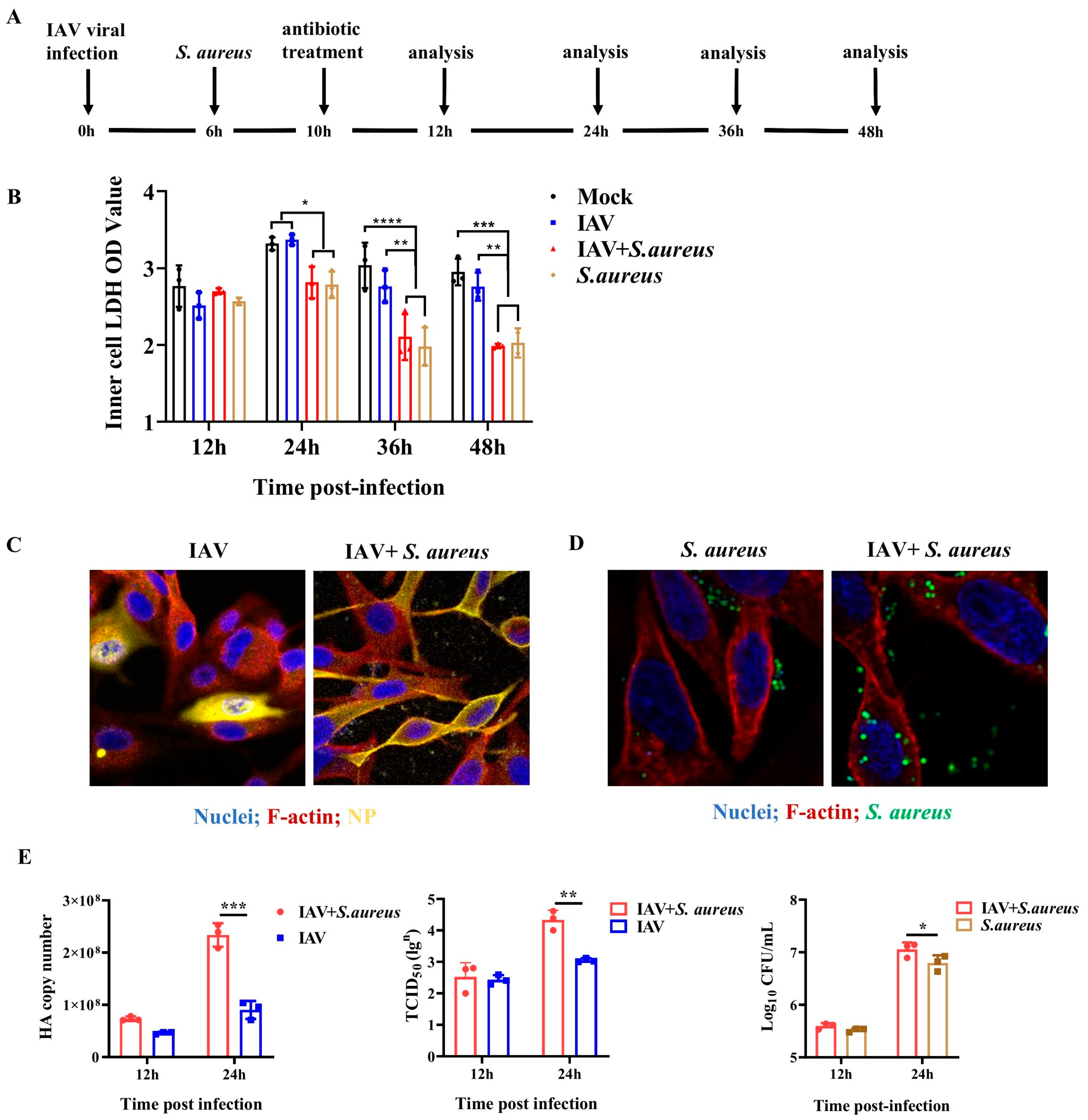
2.1. IAV and Secondary S. aureus Infection Reduce the Viability of Pulmonary Epithelial Cells and Promote the Intracellular Proliferation of Viruses and Bacteria
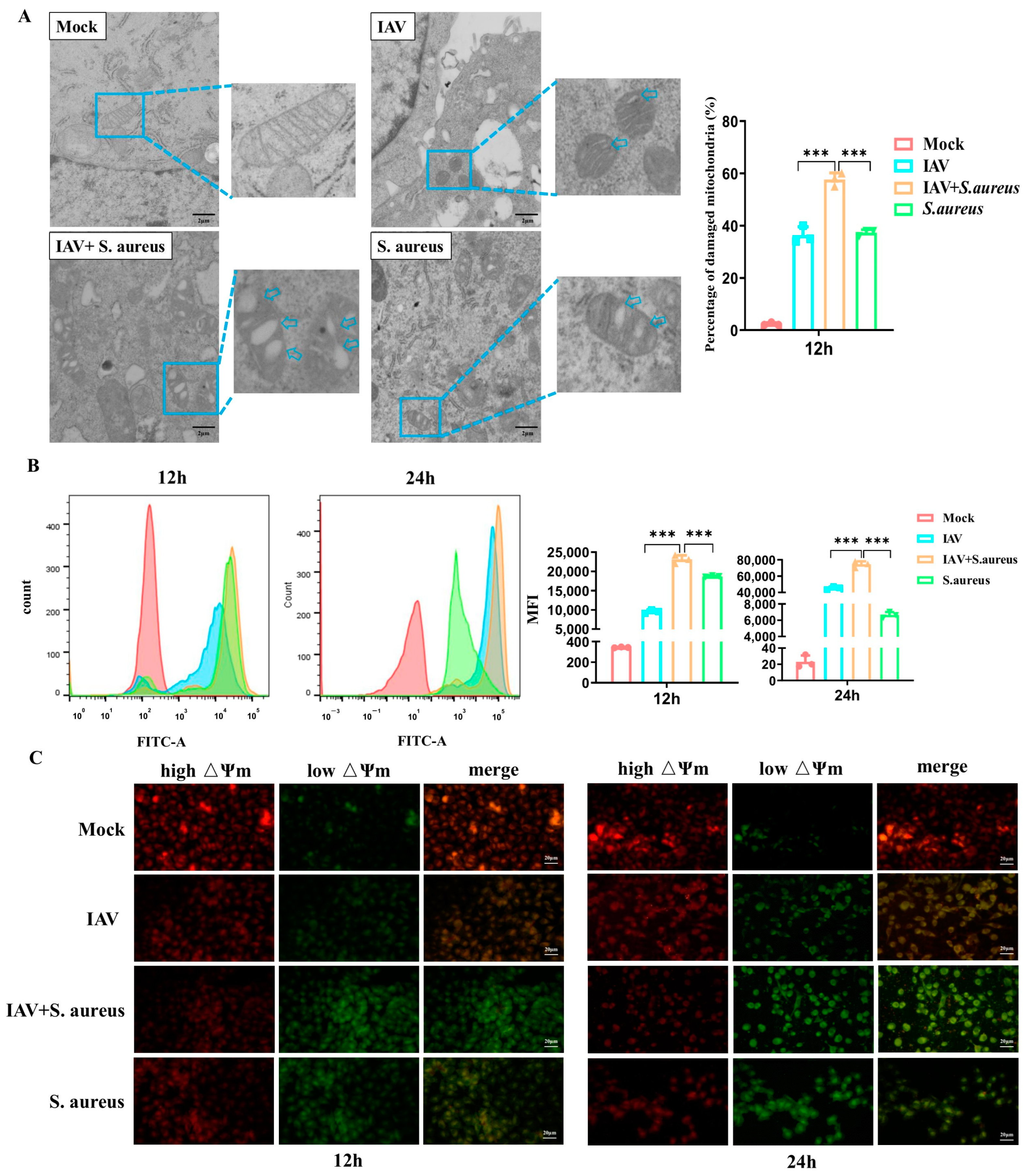
2.2. IAV and Secondary S. aureus Infection Cause the Mitochondria Damage in Pulmonary Epithelial Cells
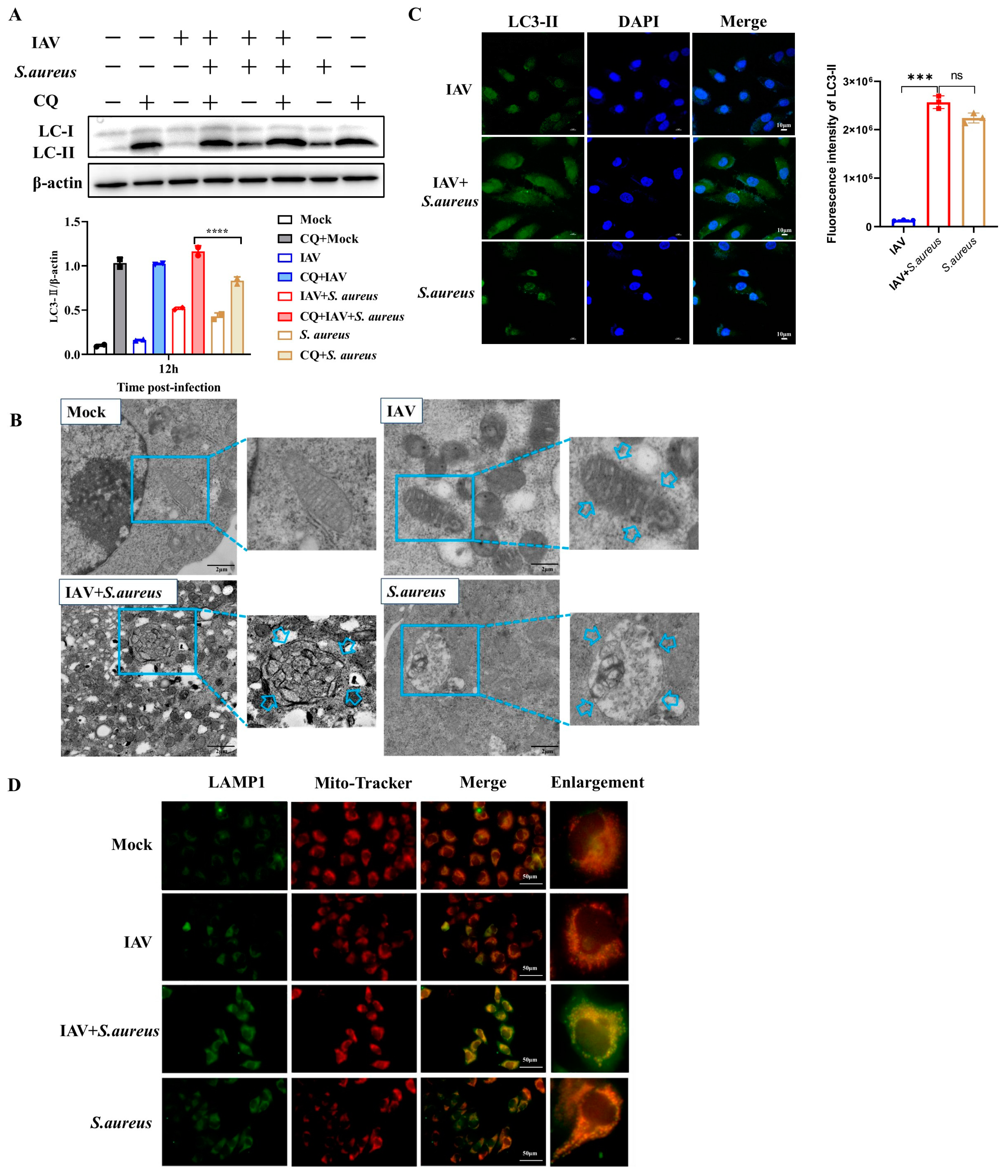
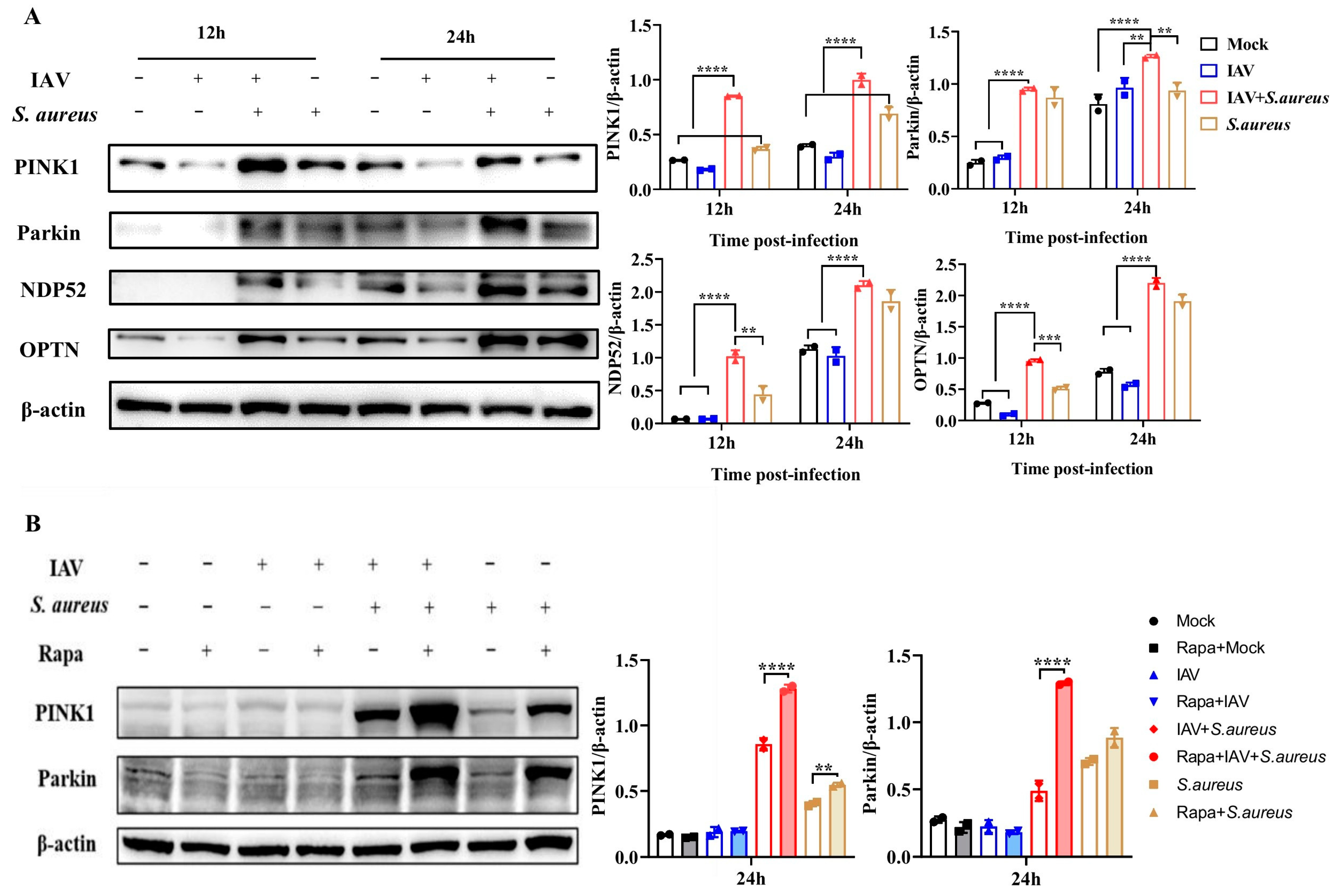
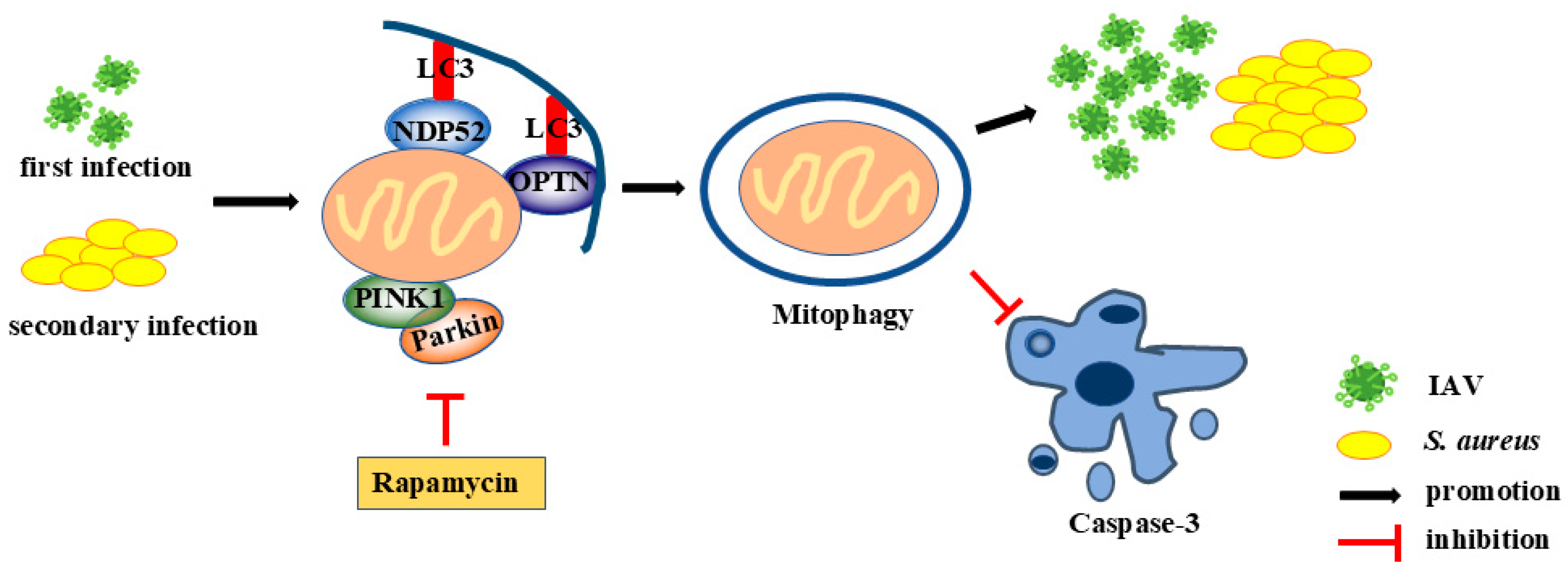
2.3. IAV and Secondary S. aureus Infection Induce the PINK1/Parkin-Mediated Mitophagy in Pulmonary Epithelial Cells
2.4. Mitophagy Facilitates Pathogen Proliferation in Pulmonary Epithelial Cells with IAV and Secondary S. aureus Infection
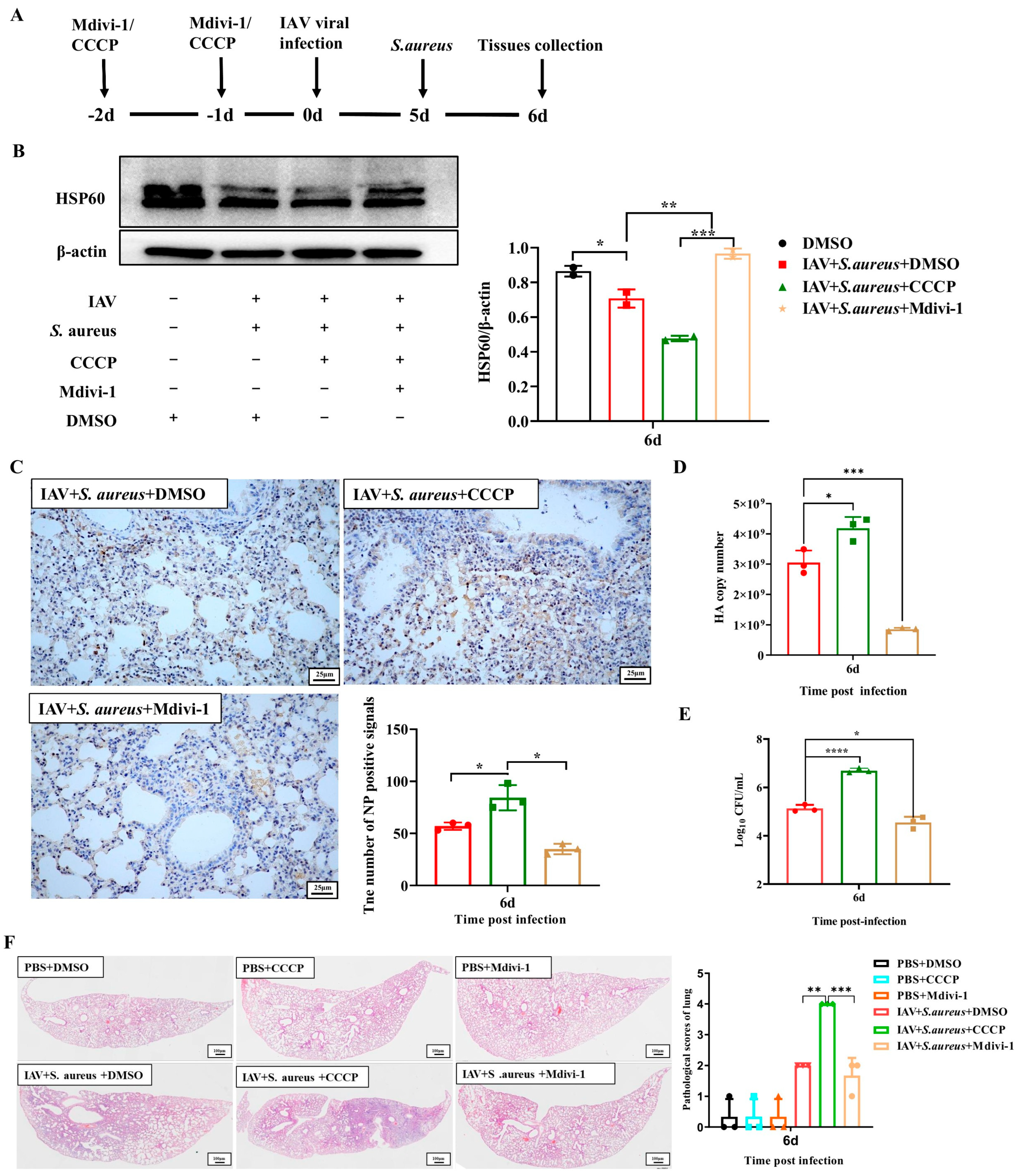
2.5. Mitophagy Exacerbates the Lung Injury in Mice with IAV and Secondary S. aureus Infection
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Virus, Bacterial Strains, and Cell Lines
4.3. Infection of Cells and Transmission Electron Microscopy (TEM) In Vitro
4.4. Lactic Dehydrogenase (LDH) Release Assay
4.5. Real-Time Quantitative PCR (RT-qPCR)
4.6. Virus Titration
4.7. Immunofluorescent Staining
4.8. Measurement of ROS Production
4.9. Mitochondrial Membrane Potential (MMP) Measurement
4.10. Rapamycin (Rapa), CCCP, and Mdivi-1 Treatment Model In Vitro
4.11. Western Blot Analysis
4.12. Intracellular Bacteria Colony Counting
4.13. Terminal-Deoxynucleoitidyl Transferase Mediated Nick End Labeling (TUNEL)
4.14. Infection of Mice In Vivo
4.15. CCCP and Mdivi-1 Treatment Model In Vivo
4.16. Histology and Immunochemistry
4.17. Bacterial Load Detection of Lungs
4.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lakdawala, S.S.; Lamirande, E.W.; Suguitan, A.L., Jr.; Wang, W.; Santos, C.P.; Vogel, L.; Matsuoka, Y.; Lindsley, W.G.; Jin, H.; Subbarao, K. Eurasian-origin gene segments contribute to the transmissibility, aerosol release, and morphology of the 2009 pandemic H1N1 influenza virus. PLoS Pathog. 2011, 7, e1002443. [Google Scholar] [CrossRef]
- McAuley, J.L.; Hornung, F.; Boyd, K.L.; Smith, A.M.; McKeon, R.; Bennink, J.; Yewdell, J.W.; McCullers, J.A. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2007, 2, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Estenssoro, E.; Rios, F.G.; Apezteguia, C.; Reina, R.; Neira, J.; Ceraso, D.H.; Orlandi, C.; Valentini, R.; Tiribelli, N.; Brizuela, M.; et al. Pandemic 2009 influenza A in Argentina: A study of 337 patients on mechanical ventilation. Am. J. Respir. Crit. Care Med. 2010, 182, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Farias, J.A.; Fernandez, A.; Monteverde, E.; Vidala, N.; Arias, P.; Montes, M.J.; Rodriguez, G.; Allasia, M.; Ratto, M.E.; Jaen, R.; et al. Critically ill infants and children with influenza A (H1N1) in pediatric intensive care units in Argentina. Intensive Care Med. 2010, 36, 1015–1022. [Google Scholar] [CrossRef]
- Howden, B.P.; Giulieri, S.G.; Wong Fok Lung, T.; Baines, S.L.; Sharkey, L.K.; Lee, J.Y.H.; Hachani, A.; Monk, I.R.; Stinear, T.P. Staphylococcus aureus host interactions and adaptation. Nat. Rev. Microbiol. 2023, 21, 380–395. [Google Scholar] [CrossRef]
- Al Amir Dache, Z.; Thierry, A.R. Mitochondria-derived cell-to-cell communication. Cell Rep. 2023, 42, 112728. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Teodorof-Diedrich, C.; Spector, S.A. Human Immunodeficiency Virus Type 1 gp120 and Tat Induce Mitochondrial Fragmentation and Incomplete Mitophagy in Human Neurons. J. Virol. 2018, 92, e00993-18. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, Y.; Yin, L.; Liang, W.; Zhang, J.; Ma, C.; Zhang, Y.; Liu, B.; Wang, J.; Zhao, W.; et al. The nonstructural protein 1 of respiratory syncytial virus hijacks host mitophagy as a novel mitophagy receptor to evade the type I IFN response in HEp-2 cells. mBio 2023, 14, e0148023. [Google Scholar] [CrossRef]
- Lee, J.; Ou, J.J. HCV-induced autophagy and innate immunity. Front. Immunol. 2024, 15, 1305157. [Google Scholar] [CrossRef]
- Xu, D.; Hu, G.; Luo, J.; Cheng, J.; Wu, D.; Cheng, L.; Huang, X.; Fu, S.; Liu, J. Staphylococcus aureus induces mitophagy to promote its survival within bovine mammary epithelial cells. Vet. Microbiol. 2023, 280, 109697. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Yi, S.; Zheng, B.; Zhu, Y.; Cai, Y.; Sun, H.; Zhou, J. Melatonin ameliorates excessive PINK1/Parkin-mediated mitophagy by enhancing SIRT1 expression in granulosa cells of PCOS. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E91–E101. [Google Scholar] [CrossRef]
- Borsa, M.; Obba, S.; Richter, F.C.; Zhang, H.; Riffelmacher, T.; Carrelha, J.; Alsaleh, G.; Jacobsen, S.E.W.; Simon, A.K. Autophagy preserves hematopoietic stem cells by restraining MTORC1-mediated cellular anabolism. Autophagy 2023, 20, 45–57. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef]
- Fan, S.; Wu, K.; Zhao, M.; Yuan, J.; Ma, S.; Zhu, E.; Chen, Y.; Ding, H.; Yi, L.; Chen, J. LDHB inhibition induces mitophagy and facilitates the progression of CSFV infection. Autophagy 2021, 17, 2305–2324. [Google Scholar] [CrossRef]
- Tang, Y.; Su, R.; Gu, Q.; Hu, Y.; Yang, H. PI3K/AKT-mediated autophagy inhibition facilitates mast cell activation to enhance severe inflammatory lung injury in influenza A virus- and secondary Staphylococcus aureus-infected mice. Antiviral Res. 2023, 209, 105502. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef]
- Wei, Y.; Lan, B.; Zheng, T.; Yang, L.; Zhang, X.; Cheng, L.; Tuerhongjiang, G.; Yuan, Z.; Wu, Y. GSDME-mediated pyroptosis promotes the progression and associated inflammation of atherosclerosis. Nat. Commun. 2023, 14, 929. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Lin, X.; Ren, C.; Zhao, J.; Wang, F.; Gao, X.; Xiao, R.; Zhao, L.; Chen, H.; et al. Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy 2019, 15, 1163–1181. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Choi, S.M.; McHugh, K.J.; Mandalapu, S.; Enelow, R.I.; Kolls, J.K.; Alcorn, J.F. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. J. Immunol. 2013, 191, 5153–5159. [Google Scholar] [CrossRef]
- Kudva, A.; Scheller, E.V.; Robinson, K.M.; Crowe, C.R.; Choi, S.M.; Slight, S.R.; Khader, S.A.; Dubin, P.J.; Enelow, R.I.; Kolls, J.K.; et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 2011, 186, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Small, C.L.; Shaler, C.R.; McCormick, S.; Jeyanathan, M.; Damjanovic, D.; Brown, E.G.; Arck, P.; Jordana, M.; Kaushic, C.; Ashkar, A.A.; et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J. Immunol. 2010, 184, 2048–2056. [Google Scholar] [CrossRef]
- Tashiro, M.; Ciborowski, P.; Klenk, H.D.; Pulverer, G.; Rott, R. Role of Staphylococcus protease in the development of influenza pneumonia. Nature 1987, 325, 536–537. [Google Scholar] [CrossRef]
- Koshiba, T.; Yasukawa, K.; Yanagi, Y.; Kawabata, S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci. Signal 2011, 4, ra7. [Google Scholar] [CrossRef]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2021, 17, 496–511. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.L.; Liu, F.L.; Sun, J.; Li, X.; He, X.Y.; Zheng, H.Y.; Zhou, Y.H.; Yan, Q.; Chen, L.; Yu, G.Y.; et al. SARS-CoV-2-triggered mast cell rapid degranulation induces alveolar epithelial inflammation and lung injury. Signal Transduct. Target. Ther. 2021, 6, 428. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Xu, C.; Zhang, W.; Cao, Y.; Ye, J.; Li, B.; Jia, S.; Weng, L.; Liu, Y.; Liu, L.; et al. FUNDC1-mediated mitophagy and HIF1alpha activation drives pulmonary hypertension during hypoxia. Cell Death Dis. 2022, 13, 634. [Google Scholar] [CrossRef]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, Y.; Qiu, X.; Wang, G.; Hu, Z.; Chen, S.; Wu, Z.; Yuan, N.; Gao, H.; Wang, J.; et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 2019, 20, 433–446. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Huo, C.; Xiao, K.; Zhang, S.; Tang, Y.; Wang, M.; Qi, P.; Xiao, J.; Tian, H.; Hu, Y. H5N1 Influenza a Virus Replicates Productively in Pancreatic Cells and Induces Apoptosis and Pro-Inflammatory Cytokine Response. Front. Cell Infect. Microbiol. 2018, 8, 386. [Google Scholar] [CrossRef]
- Huo, C.; Xiao, J.; Xiao, K.; Zou, S.; Wang, M.; Qi, P.; Liu, T.; Hu, Y. Pre-Treatment with Zirconia Nanoparticles Reduces Inflammation Induced by the Pathogenic H5N1 Influenza Virus. Int. J. Nanomed. 2020, 15, 661–674. [Google Scholar] [CrossRef]
- Meng, D.; Huo, C.; Wang, M.; Xiao, J.; Liu, B.; Wei, T.; Dong, H.; Zhang, G.; Hu, Y.; Sun, L. Influenza A Viruses Replicate Productively in Mouse Mastocytoma Cells (P815) and Trigger Pro-inflammatory Cytokine and Chemokine Production through TLR3 Signaling Pathway. Front. Microbiol. 2016, 7, 2130. [Google Scholar] [CrossRef] [PubMed]
- Gopal, R.; Lee, B.; McHugh, K.J.; Rich, H.E.; Ramanan, K.; Mandalapu, S.; Clay, M.E.; Seger, P.J.; Enelow, R.I.; Manni, M.L.; et al. STAT2 Signaling Regulates Macrophage Phenotype During Influenza and Bacterial Super-Infection. Front. Immunol. 2018, 9, 2151. [Google Scholar] [CrossRef] [PubMed]
- Huo, C.; Tang, Y.; Li, X.; Han, D.; Gu, Q.; Su, R.; Liu, Y.; Reiter, R.J.; Liu, G.; Hu, Y.; et al. Melatonin alleviates lung injury in H1N1-infected mice by mast cell inactivation and cytokine storm suppression. PLoS Pathog. 2023, 19, e1011406. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, C.; Li, Y.; Tang, Y.; Su, R.; Xu, J.; Dong, H.; Hu, Y.; Yang, H. Vital Role of PINK1/Parkin-Mediated Mitophagy of Pulmonary Epithelial Cells in Severe Pneumonia Induced by IAV and Secondary Staphylococcus aureus Infection. Int. J. Mol. Sci. 2025, 26, 4162. https://doi.org/10.3390/ijms26094162
Huo C, Li Y, Tang Y, Su R, Xu J, Dong H, Hu Y, Yang H. Vital Role of PINK1/Parkin-Mediated Mitophagy of Pulmonary Epithelial Cells in Severe Pneumonia Induced by IAV and Secondary Staphylococcus aureus Infection. International Journal of Molecular Sciences. 2025; 26(9):4162. https://doi.org/10.3390/ijms26094162
Chicago/Turabian StyleHuo, Caiyun, Yuli Li, Yuling Tang, Ruijing Su, Jiawei Xu, Hong Dong, Yanxin Hu, and Hanchun Yang. 2025. "Vital Role of PINK1/Parkin-Mediated Mitophagy of Pulmonary Epithelial Cells in Severe Pneumonia Induced by IAV and Secondary Staphylococcus aureus Infection" International Journal of Molecular Sciences 26, no. 9: 4162. https://doi.org/10.3390/ijms26094162
APA StyleHuo, C., Li, Y., Tang, Y., Su, R., Xu, J., Dong, H., Hu, Y., & Yang, H. (2025). Vital Role of PINK1/Parkin-Mediated Mitophagy of Pulmonary Epithelial Cells in Severe Pneumonia Induced by IAV and Secondary Staphylococcus aureus Infection. International Journal of Molecular Sciences, 26(9), 4162. https://doi.org/10.3390/ijms26094162