

Application of Multiomics in Perinatology: A Metabolomics Integration-Focused Review


Abstract
1. Introduction
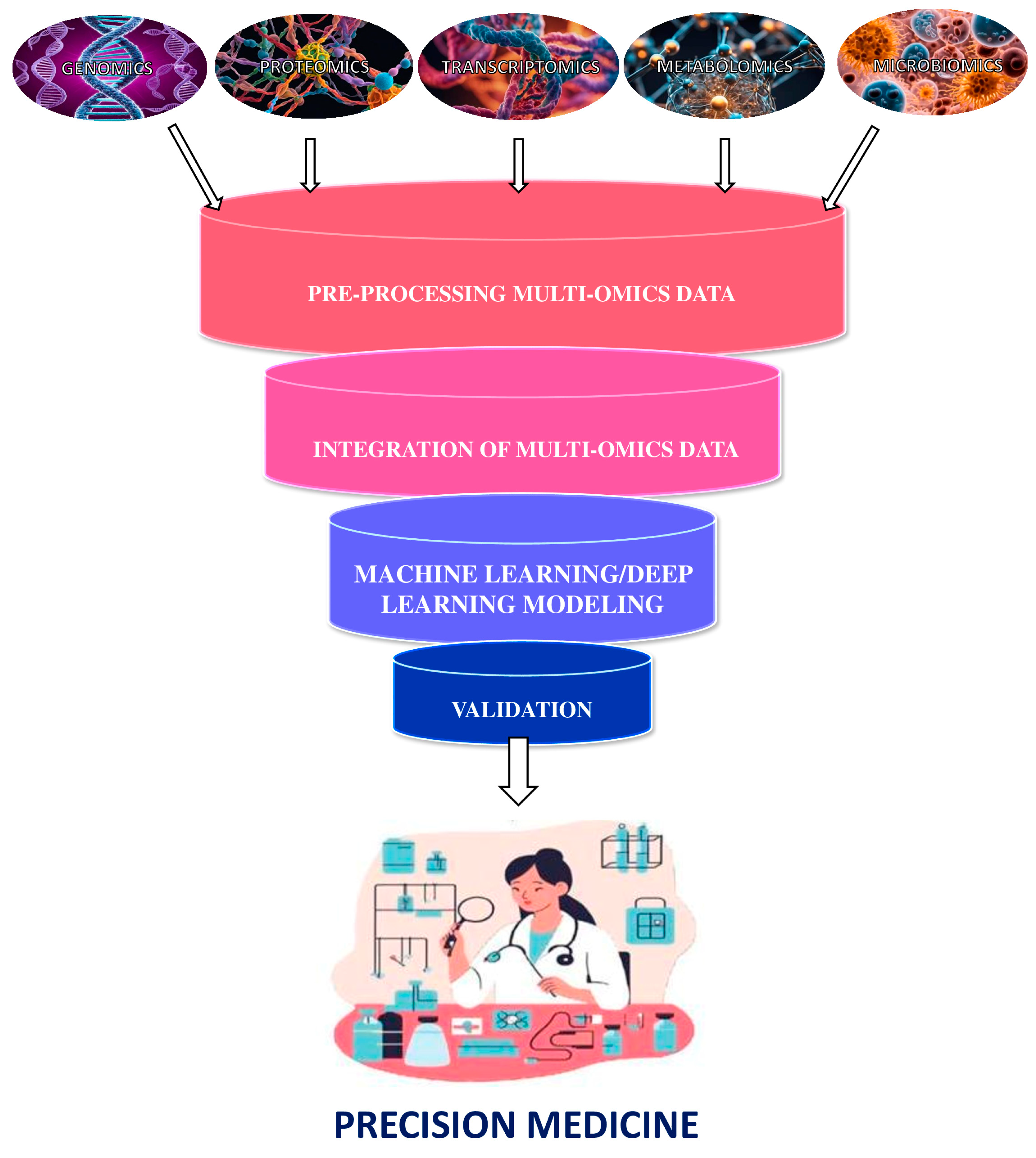
2. Multiomics Integration: From Spearman Correlation Analysis to Artificial Intelligence
3. Multiomics Studies in Perinatology
3.1. Pregnancy
3.2. Newborns
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnson, K.B.; Wei, W.Q.; Weeraratne, D.; Frisse, M.E.; Misulis, K.; Rhee, K.; Zhao, J.; Snowdon, J.L. Precision Medicine, AI, and the Future of Personalized Health Care. Clin. Transl. Sci. 2021, 14, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Caudle, K.E.; Klein, T.E.; Hoffman, J.M.; Muller, D.J.; Whirl-Carrillo, M.; Gong, L.; McDonagh, E.M.; Sangkuhl, K.; Thorn, C.F.; Schwab, M.; et al. Incorporation of pharmacogenomics into routine clinical practice: The Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr. Drug Metab. 2014, 15, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Hartmaier, R.J.; Albacker, L.A.; Chmielecki, J.; Bailey, M.; He, J.; Goldberg, M.E.; Ramkissoon, S.; Suh, J.; Elvin, J.A.; Chiacchia, S.; et al. High-Throughput Genomic Profiling of Adult Solid Tumors Reveals Novel Insights into Cancer Pathogenesis. Cancer Res. 2017, 77, 2464–2475. [Google Scholar] [CrossRef]
- Wörheide, M.A.; Krumsiek, J.; Kastenmüller, G.; Arnold, M. Multi-omics integration in biomedical research—A metabolomics-centric review. Anal. Chim. Acta 2021, 1141, 144–162. [Google Scholar] [CrossRef]
- Romero, R.; Espinoza, J.; Gotsch, F.; Kusanovic, J.P.; Friel, L.A.; Erez, O.; Mazaki-Tovi, S.; Than, N.G.; Hassan, S.; Tromp, G. The use of high-dimensional biology (genomics, transcriptomics, proteomics, and metabolomics) to understand the preterm parturition syndrome. BJOG 2006, 113 (Suppl. 3), 118–135. [Google Scholar] [CrossRef]
- Csala, A.; Zwinderman, A.H.; Hof, M.H. Multiset sparse partial least squares path modeling for high dimensional omics data analysis. BMC Bioinform. 2020, 21, 9. [Google Scholar] [CrossRef]
- Bersanelli, M.; Mosca, E.; Remondini, D.; Giampieri, E.; Sala, C.; Castellani, G.; Milanesi, L. Methods for the integration of multi-omics data: Mathematical aspects. BMC Bioinform. 2016, 17 (Suppl. 2), 15. [Google Scholar] [CrossRef] [PubMed]
- Paczkowska, M.; Barenboim, J.; Sintupisut, N.; Fox, N.S.; Zhu, H.; Abd-Rabbo, D.; Mee, M.W.; Boutros, P.C.; PCAWG Drivers and Functional Interpretation Working Group; Reimand, J.; et al. Integrative pathway enrichment analysis of multivariate omics data. Nat. Commun. 2020, 11, 735. [Google Scholar] [CrossRef]
- Bosco, A.; Piu, C.; Picciau, M.E.; Pintus, R.; Fanos, V.; Dessì, A. Metabolomics in NEC: An Updated Review. Metabolites 2023, 14, 14. [Google Scholar] [CrossRef]
- Hotea, I.; Sirbu, C.; Plotuna, A.M.; Tîrziu, E.; Badea, C.; Berbecea, A.; Dragomirescu, M.; Radulov, I. Integrating (nutri-)metabolomics into the One Health Tendency—The Key for Personalized Medicine Advancement. Metabolites 2023, 13, 800. [Google Scholar] [CrossRef]
- Thompson, P.T.; Moseley, H.N.B. MESSES: Software for Transforming Messy Research Datasets into Clean Submissions to Metabolomics Workbench for Public Sharing. Metabolites 2023, 13, 842. [Google Scholar] [CrossRef] [PubMed]
- Rocca-Serra, P.; Salek, R.M.; Arita, M.; Correa, E.; Dayalan, S.; Gonzalez-Beltran, A.; Ebbels, T.; Goodacre, R.; Hastings, J.; Haug, K.; et al. Data standards can boost metabolomics research, and if there is a will, there is a way. Metabolomics 2016, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Bardanzellu, F.; Fanos, V. How could metabolomics change pediatric health? Ital. J. Pediatr. 2020, 46, 37. [Google Scholar] [CrossRef]
- Dessì, A.; Tognazzi, C.; Bosco, A.; Pintus, R.; Fanos, V. Metabolomic profiles and microbiota of GDM offspring: The key for future perspective? Front. Pediatr. 2022, 10, 941800. [Google Scholar] [CrossRef]
- Wadhwa, P.D.; Buss, C.; Entringer, S.; Swanson, J.M. Developmental origins of health and disease: Brief history of the approach and current focus on epigenetic mechanisms. Semin. Reprod. Med. 2009, 27, 358–368. [Google Scholar] [CrossRef]
- Kang, M.; Ko, E.; Mersha, T.B. A roadmap for multi-omics data integration using deep learning. Brief Bioinform. 2022, 23, bbab454. [Google Scholar] [CrossRef]
- Pammi, M.; Aghaeepour, N.; Neu, J. Multiomics, artificial intelligence, and precision medicine in perinatology. Pediatr. Res. 2023, 93, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef]
- Poirion, O.B.; Jing, Z.; Chaudhary, K.; Huang, S.; Garmire, L.X. DeepProg: An ensemble of deep-learning and machine-learning models for prognosis prediction using multi-omics data. Genome Med. 2021, 13, 112. [Google Scholar] [CrossRef]
- Wei, L.; Niraula, D.; Gates, E.D.H.; Fu, J.; Luo, Y.; Nyflot, M.J.; Bowen, S.R.; El Naqa, I.M.; Cui, S. Artificial intelligence (AI) and machine learning (ML) in precision oncology: A review on enhancing discoverability through multiomics integration. Br. J. Radiol. 2023, 96, 20230211. [Google Scholar] [CrossRef]
- Theodosiou, A.A.; Read, R.C. Artificial intelligence, machine learning and deep learning: Potential resources for the infection clinician. J. Infect. 2023, 87, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Srivastava, D.; Sahu, M.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Artificial intelligence to deep learning: Machine intelligence approach for drug discovery. Mol. Divers. 2021, 25, 1315–1360. [Google Scholar] [CrossRef]
- MacEachern, S.J.; Forkert, N.D. Machine learning for precision medicine. Genome 2021, 64, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Wanichthanarak, K.; Fahrmann, J.F.; Grapov, D. Genomic, Proteomic, and Metabolomic Data Integration Strategies. Biomark. Insights 2015, 10 (Suppl. 4), 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Sovio, U.; Aye, I.L.; Gaccioli, F.; Dopierala, J.; Johnson, M.D.; Wood, A.M.; Cook, E.; Jenkins, B.J.; Koulman, A.; et al. Placental polyamine metabolism differs by fetal sex, fetal growth restriction, and preeclampsia. JCI Insight 2018, 3, e120723. [Google Scholar] [CrossRef]
- Barak, O.; Lovelace, T.; Piekos, S.; Chu, T.; Cao, Z.; Sadovsky, E.; Mouillet, J.F.; Ouyang, Y.; Parks, W.T.; Hood, L.; et al. Integrated unbiased multiomics defines disease-independent placental clusters in common obstetrical syndromes. BMC Med. 2023, 21, 349. [Google Scholar] [CrossRef]
- Marić, I.; Contrepois, K.; Moufarrej, M.N.; Stelzer, I.A.; Feyaerts, D.; Han, X.; Tang, A.; Stanley, N.; Wong, R.J.; Traber, G.M.; et al. Early prediction and longitudinal modeling of preeclampsia from multiomics. Patterns 2022, 3, 100655. [Google Scholar] [CrossRef]
- Kelly, R.S.; Croteau-Chonka, D.C.; Dahlin, A.; Mirzakhani, H.; Wu, A.C.; Wan, E.S.; McGeachie, M.J.; Qiu, W.; Sordillo, J.E.; Al-Garawi, A.; et al. Integration of metabolomic and transcriptomic networks in pregnant women reveals biological pathways and predictive signatures associated with preeclampsia. Metabolomics 2017, 13, 7. [Google Scholar] [CrossRef]
- Odenkirk, M.T.; Stratton, K.G.; Gritsenko, M.A.; Bramer, L.M.; Webb-Robertson, B.M.; Bloodsworth, K.J.; Weitz, K.K.; Lipton, A.K.; Monroe, M.E.; Ash, J.R.; et al. Unveiling molecular signatures of preeclampsia and gestational diabetes mellitus with multi-omics and innovative cheminformatics visualization tools. Mol. Omics 2020, 16, 521–532. [Google Scholar] [CrossRef]
- Easton, Z.J.W.; Sarr, O.; Zhao, L.; Buzatto, A.Z.; Luo, X.; Zhao, S.; Li, L.; Regnault, T.R.H. An integrated multi-omics approach highlights elevated non-esterified fatty acids impact BeWo trophoblast metabolism and lipid processing. Metabolites 2023, 13, 883. [Google Scholar] [CrossRef]
- Dong, L.; Han, L.; Duan, T.; Lin, S.; Li, J.; Liu, X. Integrated microbiome-metabolome analysis reveals novel associations between fecal microbiota and hyperglycemia-related changes of plasma metabolome in gestational diabetes mellitus. RSC Adv. 2020, 10, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhai, X.; Ding, L.; Yu, M.; Zhang, Q.; Liu, J.; Song, Y.; Ma, L.; Xiao, X. Landscapes of maternal and neonatal gut microbiome and plasma metabolome signatures and their interaction in gestational diabetes mellitus. J. Nutr. Biochem. 2024, 134, 109716. [Google Scholar] [CrossRef]
- Fu, Y.; Gou, W.; Wu, P.; Lai, Y.; Liang, X.; Zhang, K.; Shuai, M.; Tang, J.; Miao, Z.; Chen, J.; et al. Landscape of the gut mycobiome dynamics during pregnancy and its relationship with host metabolism and pregnancy health. Gut 2024, 73, 1302–1312. [Google Scholar] [CrossRef]
- Tao, Z.; Chen, Y.; He, F.; Tang, J.; Zhan, L.; Hu, H.; Ding, Z.; Ruan, S.; Chen, Y.; Chen, B.; et al. Alterations in the gut microbiome and metabolisms in pregnancies with fetal growth restriction. Microbiol. Spectr. 2023, 11, e0007623. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Li, D.; Peng, J.; Yang, W.; Zhang, X.; Li, H. Potential association of gut microbial metabolism and circulating mRNA based on multiomics sequencing analysis in fetal growth restriction. Mediat. Inflamm. 2024, 2024, 9986187. [Google Scholar] [CrossRef]
- Jehan, F.; Sazawal, S.; Baqui, A.H.; Nisar, M.I.; Dhingra, U.; Khanam, R.; Ilyas, M.; Dutta, A.; Mitra, D.K.; Mehmood, U.; et al. Multiomics characterization of preterm birth in low- and middle-income countries. JAMA Netw. Open 2020, 3, e2029655. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, C.A.; Khan, W.; Khanam, R.; Das, S.; Khalid, J.; Pervin, J.; Kasaro, M.P.; Contrepois, K.; Chang, A.L.; Phongpreecha, T.; et al. Multiomic signals associated with maternal epidemiological factors contributing to preterm birth in low- and middle-income countries. Sci. Adv. 2023, 9, eade7692. [Google Scholar] [CrossRef]
- Wu, M.; Li, F.; Zhou, T.; Zhao, J.; Jiang, L.; Zhang, H.; Wang, W.; Cheng, X.; Wu, X.; Xiong, M. Dysregulation of mucosal-associated invariant T cells correlates with altered placental microenvironment in preterm birth. Mol. Hum. Reprod. 2024, 30, gaae006. [Google Scholar] [CrossRef]
- Ghaemi, M.S.; DiGiulio, D.B.; Contrepois, K.; Callahan, B.; Ngo, T.T.M.; Lee-McMullen, B.; Lehallier, B.; Robaczewska, A.; Mcilwain, D.; Rosenberg-Hasson, Y.; et al. Multiomics modeling of the immunome, transcriptome, microbiome, proteome and metabolome adaptations during human pregnancy. Bioinformatics 2019, 35, 95–103. [Google Scholar] [CrossRef]
- Stelzer, I.A.; Ghaemi, M.S.; Han, X.; Ando, K.; Hédou, J.J.; Feyaerts, D.; Peterson, L.S.; Rumer, K.K.; Tsai, E.S.; Ganio, E.A.; et al. Integrated trajectories of the maternal metabolome, proteome, and immunome predict labor onset. Sci. Transl. Med. 2021, 13, eabd9898. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Sonek, J.; McKenna, D.; Cool, D.; Aydas, B.; Turkoglu, O.; Bjorndahl, T.; Mandal, R.; Wishart, D.; Friedman, P.; et al. Artificial intelligence and amniotic fluid multiomics: Prediction of perinatal outcome in asymptomatic women with short cervix. Ultrasound Obs. Gynecol. 2019, 54, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Orchanian, S.B.; Gauglitz, J.M.; Wandro, S.; Weldon, K.C.; Doty, M.; Stillwell, K.; Hansen, S.; Jiang, L.; Vargas, F.; Rhee, K.E.; et al. Multiomic analyses of nascent preterm infant microbiomes differentiation suggest opportunities for targeted intervention. Adv. Biol. 2022, 6, e2101313. [Google Scholar] [CrossRef] [PubMed]
- Young, G.; Berrington, J.E.; Cummings, S.; Dorling, J.; Ewer, A.K.; Frau, A.; Lett, L.; Probert, C.; Juszczak, E.; Kirby, J.; et al. Mechanisms affecting the gut of preterm infants in enteral feeding trials: A nested cohort within a randomised controlled trial of lactoferrin. Arch. Dis. Child Fetal Neonatal Ed. 2023, 108, 272–279. [Google Scholar] [CrossRef]
- Morrow, A.L.; Lagomarcino, A.J.; Schibler, K.R.; Taft, D.H.; Yu, Z.; Wang, B. Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants. Microbiome 2013, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.J.; Embleton, N.D.; Marrs, E.C.; Smith, D.P.; Nelson, A.; Abdulkadir, B.; Skeath, T.; Petrosino, J.F.; Perry, J.D.; Berrington, J.E.; et al. Temporal bacterial and metabolic development of the preterm gut reveals specific signatures in health and disease. Microbiome 2016, 4, 67. [Google Scholar] [CrossRef]
- Brehin, C.; Dubois, D.; Dicky, O.; Breinig, S.; Oswald, E.; Serino, M. Evolution of gut microbiome and metabolome in suspected necrotizing enterocolitis: A case-control study. J. Clin. Med. 2020, 9, 2278. [Google Scholar] [CrossRef]
- Du, T.T.; Liu, X.C.; He, Y.; Gao, X.; Liu, Z.Z.; Wang, Z.L.; Li, L.Q. Changes of gut microbiota and tricarboxylic acid metabolites may be helpful in early diagnosis of necrotizing enterocolitis: A pilot study. Front. Microbiol. 2023, 14, 1119981. [Google Scholar] [CrossRef]
- Stewart, C.J.; Embleton, N.D.; Marrs, E.C.L.; Smith, D.P.; Fofanova, T.; Nelson, A.; Skeath, T.; Perry, J.D.; Petrosino, J.F.; Berrington, J.E.; et al. Longitudinal development of the gut microbiome and metabolome in preterm neonates with late onset sepsis and healthy controls. Microbiome 2017, 5, 75. [Google Scholar] [CrossRef]
- Wandro, S.; Osborne, S.; Enriquez, C.; Bixby, C.; Arrieta, A.; Whiteson, K. The microbiome and metabolome of preterm infant stool are personalized and not driven by health outcomes, including necrotizing enterocolitis and late-onset sepsis. mSphere 2018, 3, e00104-18. [Google Scholar] [CrossRef]
- Stewart, C.J.; Nelson, A.; Treumann, A.; Skeath, T.; Cummings, S.P.; Embleton, N.D.; Berrington, J.E. Metabolomic and proteomic analysis of serum from preterm infants with necrotising entercolitis and late-onset sepsis. Pediatr. Res. 2016, 79, 425–431. [Google Scholar] [CrossRef]
- Bi, Y.; Yu, W.; Bian, W.; Jin, M.; He, Y.; Wang, J.; Miao, X.; Guo, T.; Ma, X.; Gong, P.; et al. Metabolic and microbial dysregulation in preterm infants with neonatal respiratory distress syndrome: An early developmental perspective. J. Proteome Res. 2024, 23, 3460–3468. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xiang, M.; Cai, X.; Wu, B.; Chen, C.; Cai, N.; Ao, D. Multi-omics analyses of gut microbiota via 16S rRNA gene sequencing, LC-MS/MS and diffusion tension imaging reveal aberrant microbiota-gut-brain axis in very low or extremely low birth weight infants with white matter injury. BMC Microbiol. 2023, 23, 387. [Google Scholar] [CrossRef] [PubMed]
- Chatziioannou, A.C.; Wolters, J.C.; Sarafidis, K.; Thomaidou, A.; Agakidis, C.; Govorukhina, N.; Kuivenhoven, J.A.; Bischoff, R.; Theodoridis, G. Targeted LC-MS/MS for the evaluation of proteomics biomarkers in the blood of neonates with necrotizing enterocolitis and late-onset sepsis. Anal. Bioanal. Chem. 2018, 410, 7163–7175. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Author and Year | Participants | Samples | Study Design | Omics | Main Results | Multiomics Correlation Analysys |
|---|---|---|---|---|---|---|
| Gong et al. (2018) [25] | 4212 nulliparous women, 134 PE, 162 IUGR e 259 controls | Placental samples, serum | Prospective cohort study | Placental transcriptome, placental methylome Mother’s serum metabolome | Different placental polyamine metabolism by fetal sex | No correlation analysis (multiomic approach to identify polyamine synthesis pathway implicated in the pathophysiology of placentally related com- plications of human pregnancy) |
| Barack et al. (2023) [26] | 75 severe PE 40 IUGR 33 IUGR + HDP 72 sPTD 2 control groups: −113 term −16 preterm without PE, IUGR or sPTD | Placental samples | Retrospective cohort study | Transcriptomic, proteomic, metabolomics (biobank) | Omics-based placental dysfunctions clustering showed a better correlation with histopathology than the predefined disease clusters and could improve diagnostic accuracy | Unbiased AI-based SNF |
| Marić et al. (2022) [27] | 33 women in the discovery cohort (17 PE, 16 normotensive) 16 women in the validation cohort (12 PE, 4 normotensive) | Plasma, vaginal swab, urine | Longitudinal, prospective cohort study | Six omics datasets cell-free RNA (cfRNA)/transcriptome, proteome, me- tabolome, lipidome and microbiome | Prediction models from urine metabolome and from proteome had the best accuracy | ML (Elastic net) |
| Kelly et al. [28] | 47 pregnant women from VDAART cohort who developed PE and 62 healthy controls | Plasma | Prospective cohort study | Metabolomics and transcriptomic | Predictive PE serum metabolites from the first trimester of pregnancy with moderate to good discriminatory ability in the third trimester Integration with transcriptomic data provided a deeper understanding of the biology underlying PE | Correlation clustering analysis (WGCNA) |
| Odenkirk et al. (2020) [29] | 191 women (98 controls, 45 GDM, 48 PE) | Plasma (Peribank) | Case control study | Proteomics and lipidomics | Direct relationship between proteins and lipids, suggesting a strong interdependence in the origin and progression of GDM and PE | Multi-omic assay/comparison |
| Easton et al. (2023) [30] | - | BeWo cell line model | In vitro model + Multiomics approach | Transcriptomic (mRNA microarray), metabolomic, and lipidomic | ↑ certain dietary fats in maternal circulation involved in facilitating the aberrant placental function in obese and GDM pregnancies Saturated and unsaturated FA species differentially impact placental function | Pathways enrichement analysis software (MetaboAnalyst v5) |
| Dong et al. (2020) [31] | 40 pregnant women (20 GDM and 20 non-diabetic control) | Plasma, faecal samples | Case control study | Metabolomics and microbiomics | Alterations in faecal microbiota associated with hyperglicemia-related changes of the plasma metabolome of GDM women suggesting novel therapies against gut microbiota to alleviate GDM | Correlation analysis (co-inertia, procrustes and redundancy analysis) |
| Liu et al. (2024) [32] | 89 pregnant women and their neonates (44 with GDM, 45 controls) | Maternal faecal and blood samples, umbilical cord blood and neonatal meconium | Prospective cohort study | Metabolomics and microbiomics | Distinct microbial and metabolic signatures in mothers with GDM and their infants Metabolites related to glucose, lipid and energy metabolism were affected differently in GDM, underlying metabolic transmission across generations | Correlation analysis (Spearman, R packages corrplot, ggalluvial, and ggplot) |
| Fu et al. (2024) [33] | 750, 748 and 709 pregnant women had ITS2 sequencing data, 16S sequencing data and serum metabolome data available, respectively, across all trimesters. | Stool samples and blood samples | Cohort study | Metabolomics, microbiomics and mycobiomics | Significant interactions between the intestinal mycobiome, biological function, serum metabolites and the course of pregnancy Correlation between the presence of Mucor and the onset of GDM /fetal macrosomia Pre-pregnancy overweight influences both altered intestinal mycobiome composition and metabolic remodelling during pregnancy | Correlation analysis (regression, network and Spearman analysis) |
| Tao et al. (2023) [34] | 70 pregnant women (35 IUGR, 35 healthy controls) | 70 faecal samples, 50 serum samples (31 from controls group, 19 from IUGR group) | Cohort study | Metabolomics and microbiomics | Intestinal dysbiosis and metabolic disorders in IUGR patients, which contribute to the pathogenesis of the disease Both microbial species and altered metabolites in IUGR are closely related to fetal measures and maternal clinical variables | Correlation analysis (Spearman) |
| Tang et al. (2024) [35] | 20 pregnant women (11 healthy pregnant women, 9 IUGR) | Umbilical cord blood, maternal serum, feces, placental tissue samples | Cohort study | Metabolomics, transcrptomics and microbiomics | Metabolomic, microbiomic and transcriptomic alterations mediate the interaction between the gut and the circulatory system in IUGR. | Correlation analysis (Spearman) |
| Jehan et al. (2020) [36] | 81 pregnant women (39 PTD, 42 term pregnancies) from 5 different regions | Urine, plasma | Prospective cohort study | Metabolomics, transcriptomics and proteomics | An inflammatory component (from transcriptomics and proteomics data) and specific alterations in the urinary metabolome were the main PTD associated features. Predictive accuracy for PTD increases due to a multiomics approach | Higher-level ML |
| Espinosa et al. (2023) [37] | 231 pregnant women: (113 PTD, 118 with a term delivery) | Blood samples | Cohort study | Metabolomics, lipidomics and proteomics | ML models showed robust performance for the prediction of PTD, time-to-delivery, maternal age, gravidity and BMI | ML |
| Wu et al. (2024) [38] | 35 pregnant women (13 normal pregnancy but indicated for caesarian section because of a scarred uterus, 22 pathological pregnancy) | Peripheral blood, umbilical cord blood and placenta samples | Observational and comparative study | Metabolomics, lipidomics and transciptomics | Aberrant immune activation and an abnormal increase of lipids and lipid-like metabolites in the placental microenvironment in PTD The proportion and activation of MAIT cells were positively correlated with the abnormal increase in lipids and lipid-like metabolites | Correlation analysis |
| Ghaemi et al., 2019 [39] | 17 pregnant women, delivering at term | Plasma and serum samples, whole blood samples and vaginal swabs, stool, saliva and tooth/gum samples | Prospective cohort study | Metabolomics, immunomics transcriptomic, microbiomics and proteomics | A singlle model created from 7 high-throughput longitudinal biological assays of the same patient cohort showed both significantly greater predictive power of gestational age and new interactions between different biological modalities | ML (stacked generalization algorithm) |
| Stelzer et al. (2021) [40] | 63 healthy pregnant women with spontaneous labour (of which 5 preterm) | Serial blood samples collected during the last 100 days of pregnancy | Longitudinal observational study | Metabolomics, proteomics and immunomics | Co-ordinated alterations in the metabolome, proteome and maternal immunome marked a molecular transition from pregnancy maintenance to pre-birth biology 2 to 4 weeks before delivery | ML (stacked generalization algorithm) |
| Bahado-Singh et al. (2019) [41] | 26 pregnant women asymptomatic with short CL (<15 mm) of these, 11 patients delivered ≥ 34 weeks, while 15 delivered < 34 weeks | Amniotic fluid | Retrospective observational study | Metabolomics and proteomics | Good to excellent prediction of perinatal outcome in asymptomatic pregnant women with short CL in the second trimester with combined omics, demographic and clinical data, machine learning, particularly deep learning | DL, RF, SVM, generalized linear model, PAM, LDA |
| Clinical Condition | Possible Biomarkers or Main Involved Pathways | Multiple Testing Correction Method | Authors |
|---|---|---|---|
| PE-IUGR | DiAcSpm is the first circulating maternal metabolites demonstrating opposite associations with PE and IUGR (higher maternal serum levels of DiAcSpm associated with ↑ PE risk but ↓ IUGR risk) | No FDR correction but unadjusted p-value < 0.01 for metabolites FDR-adjusted p-value < 0.05 for other analysis (Benjamini–Hochberg procedure) | Gong et al. [25] |
| Significantly altered metabolites included mainly lipids and amino acids: ↑ sphingolipid species associated with IUGR and HDP involvement in interleukin signaling, interferon and glucose metabolism | FDR-adjusted p-value < 0.05 (Benjamini–Hochberg procedure) for metabolic pathway enrichment analysis and canonical pathways | Barak et al. [26] | |
| PE | Lipid and amino acid metabolism changes: metabolism of glycerophospholipids, fatty acids and arachidonic acid metabolic imbalances related to fatty acid synthesis pathways regulation of immune function and circulatory system | No correction for multiple testing Some metabolites and biological pathways were confirmed through replication between first- and third-trimester samples | Kelly et al. [28] |
| Tryptophan metabolism alteration involving kynurenine pathways Fatty acid and arachidonic acid metabolism changes together with steroid hormone metabolism alteration | FDR-adjusted p-value < 0.05 (Benjamini–Hochberg procedure) | Marić et al. [27] | |
| IUGR | Possibile mediators in the interaction between the gut and the circulatory system in IUGR: altered metabolites (including methionine and alanine) changes in some microbial species (Tyzzerella) alteration of circulating mRNAs (TRIM34, SMOX, FAM83A, NAPG) | No correction for multiple testing Unadjusted p-value < 0.05 in Spearman’s correlation analysis | Tang et al. [35] |
| Significant association between 25 major bacterial species and 13 serum metabolites (allantoin, pinitol, nicotinic acid, malic acid, phosphatidic lysoacid, maltose, 9-hexa-decene, glycol-diacid-2-phosphate) together with 4 of faecal origin (dodecanoic glycol, dodecanoic glycol IV and pyrraline) Both faecal (physangulin E, Ginkgolide C and pyrraline) and serum metabolites (allantoin and malic acid) altered in IUGR were also found to be associated with specific clinical phenotypes (multiple fetal measures and maternal biochemical markers) | FDR adjusted p-value thresholds: <0.05, <0.01, <0.001 for microbiota analysis and <0.001 for differences in serum metabolites | Tao et al. [34] | |
| GDM | Correlation between the faecal microbiota and the plasma metabolome in GDM related to five plasma metabolites (↑ lactic acid, proline, galactitol and ↓ glycerolmethylmalonic acid) and 98 members of the faecal microbiota (mainly members of the Firmicutes) | FDR-adjusted p-value < 0.05 | Dong et al. [31] |
| Alteration in bile acid metabolism, certain lipid metabolites related to the metabolic pathway of unsaturated fatty acids and lipid biosynthesis in both mothers and diabetic offspring: ↓ glycerophospholipids (e.g., PC(18:0/20:3(5Z,8Z,11Z)) in both mothers and infants with GDM ↓ polyunsaturated fatty acids (e.g., PE (18:4/22:6)) in diabetic mothers but ↑ GDM infants | FDR-adjusted p-value < 0.05 (Benjamini–Hochberg procedure) | Liu et al. [32] | |
| PE-GDM | PE group: ↑ coagulation protein and complement cascade protein ↓ phosphatidylinositol GDM group: ↑ phosphatidylethanolamine plasmanogel species ↓ phosphatidylethanolamine-binding protein 1 (PEBP1) | ANOVA with Dunnett correction G-test with Bonferroni correction FDR-adjusted p-value < 0.05 | Odenkirk et al. [29] |
| PTD | Specific alterations in the PTD urinary metabolome: glutamine and glutamate metabolism valine, leucine and isoleucine biosynthesis pathways From proteomic and transcriptomic data: ↑ inflammatory pathways | Fisher’s tests for enrichment of metabolic pathways: unadjusted p-value < 4.4 × 10−⁹ for glutamine and glutamate metabolism, unadjusted p-value < 7.3 × 10−6 for valine, leucine and isoleucine biosynthesis Bonferroni-adjusted p-values of < 0.05 (transcrittomic analysis) FDR-adjusted p-value < 1.0 × 10−6 (proteomic analysis but some tests report incorrect p values as more specific or limited comparisons) | Jehan et al. [36] |
| Specific fetal proteins (e.g., ALPP, AFP, and PGF) and immune proteins (e.g., PD-L1, CCL28, and LIFR) showed correlation with time to delivery | Bonferroni-adjusted p-values of <0.05 and |Rho| > 0.38 | Espinosa et al. [37] | |
| Aberrant immune activation and abnormal increase of lipids and lipid-like metabolites in the placental microenvironment Heatmap correlation analysis showed that placental MAIT cell activation and expression of pro-inflammatory tissue molecules were positively correlated with the levels of most elevated metabolites (sphingolipids, lysolipids, acylcarnitines) | No correction for multiple testing Unadjusted p-value < 0.05 for differences in metabolites | Wu et al. [38] | |
| Time of Delivery | Plasma proteomics has the highest predictive power among the individual datasets Correlation between pregnanolone sulfate and immune system and between the oral microbiome and the TCRγδ⁺ cells | Bonferroni correction with significance threshold adjusted according to the number of comparisons made in the dataset | Ghaemi et al. [39] |
| ↑ in steroid hormone metabolites and interleukin-1 type 4 receptor before labour, coinciding with the shift from immune activation to regulation of inflammatory responses | FDR-adjusted p-value < 0.05 | Stelzer et al. [40] |
| Author and Year | Participants | Samples | Study Design | Omics | Results | Multiomics Correlation Analysys |
|---|---|---|---|---|---|---|
| Orchanian et al. (2022) [42] | 75 preterm infants: 29 LP-C, 28 LP-V, and 18 VLBW-C | gut, oral, and skin samples | Cohort study | Metabolomics and microbiomics | Skin microbiome is robust to early perturbations, whereas direct exposure of infants to antibiotics, rather than presumed maternal transmission, delays microbiome development and prevents early differentiation according to body site, regardless of mode of delivery | Mantel test |
| Young et al. (2023) [43] | 479 infants born <32 weeks (gut microbiome 201 infants, metabolites 83 infants for stools and 90 for urine, VOCs 117 infants) | Fecal and urine samples | Nested cohort study (within a randomised control trial) | Metabolomics and microbiomics | Minimal impacts of lactoferrin but much larger impacts of hospital site and postnatal age on the gut microbiome and metabolome of preterm infants | No correlation analysis |
| Morrow et al. (2013) [44] | 11 NEC vs. 21 controls | Fecal and urine samples | Prospective observational study | Metabolomics and microbiomics | Urinary metabolites variations are closely related to the specific dysbiosis preceding disease development | Correlation analysis (Spearman analysis, Kruskal–Wallis test) |
| Stewart et al. (2016) [45] | 7 NEC vs. 28 controls (of which 6 NEC and 10 controls also performed metabolomic analysis) | Stool samples | Cohort study | Metabolomics and microbiomics | Absence of a NEC uniform microbial signature but metabolomic profiling showed 5 discriminant metabolites in NEC samples | Correlation analysis |
| Brehin et al. (2020) [46] | 11 NEC stage I vs. 21 controls | Fecal samples | Prospective cohort study | Metabolomics and microbiomics | Changes in the gut microbiota and microbiome in NEC-1 appear more evident from day 20 onwards, compared to healthy controls, identifying this precise time window as a therapeutic/diagnostic target for NEC | No correlation analysis |
| Du et al. (2023) [47] | 16 NEC vs. 16 non NEC controls | Fecal samples | Prospective cohort study | Metabolomics and microbiomics | Interdependence between metabolome and faecal microbiota changes Potential value for NEC early diagnosis for ↓ unclassified microbiota species as well as ↑ TCA metabolites | Correlation analysis (Spearman) |
| Stewart et al. (2017) [48] | 7 LOS and 28 matched healthy (no LOS or NEC) controls | Fecal samples | Observational longitudinal cohort study | Metabolomics and microbiomics | Multi-omic analysis showed Bifidobacterium was positively correlated with metabolites significantly ↑ in control infants | Empirical correlation analysis software (MixOmics, through sPLS-DA) |
| Wandro et al. (2018) [49] | 32 VLBW (3 NEC 8 LOS and 21 controls) | Fecal samples | Retrospective observational cohort study | Metabolomics and microbiomics | Bacterial abundances varied over 4 orders of magnitude and were ↓ in infants that developed NEC or LOS No metabolites or microbiome signature associated with NEC or LOS | Mantel test and Pearson correlation |
| Stewart et al. (2015) [50] | 19 patients (6 NEC, 4 LOS vs. 9 controls) | Serum samples | Prospective cohort study | Metabolomics and proteomics | No single protein or metabolite was detected in all NEC or LOS cases, which was absent from controls Notably, the only child who died during the study had a unique proteomic and metabolomic profile | No correlation analysis |
| Bi et al. (2024) [51] | 13 preterm infants with NRDS and 12 preterm without NRDS | Blood and faecal samples | Longitudinal cohort study | Metabolomics and microbiomics | Presence of specific metabolic and microbiota alterations associated with the NRDS Inverse relationship between tryptophan concentrations and the abundance of the anaerobic genus Blautia (from integrated multiomics analysis) | Empirical correlation analysis software (MixOmic version 6.0.0.) |
| Liu et al. (2023) [52] | 23 preterm with and 48 patients without WMI (WMI group included 12 cases of mild WMI, 7 cases of moderate WMI, and 4 cases of severe WMI) | Meconium (within 24 h after birth) and stool samples | Prospective cohort study | Metabolomics and microbiomics + MRI and DTI | Significant difference in the expression of 139 metabolic markers and severe gut dysbiosis in the WMI group Certain groups characteristic of WMI-related dysbiosis strongly associated with alterations in the faecal metabolome (from multiomics correlation analysis), with possible causal links to the damage found in the white matter of the brain | Correlation analysis (Spearman) |
| Neonatal Disorders | Possible Biomarkers or Main Involved Pathways | Multiple Testing Correction Method | Authors |
|---|---|---|---|
| Prematurity | Metabolomic analyses showed a common developmental trend of all gut metabolomes of preterm infants towards the profiles of term infants significant ↑ in primary bile acid metabolism occurs only in late-preterm infants not treated with antibiotics and born vaginally | No correction for multiple testing | Orchanian et al. [42] |
| NEC | only high urinary alanine:histidine ratios provide a good prediction of overall NEC, thus representing a potential biomarker alanine positively associated with NEC cases preceded by Firmicutes dysbiosis histidine inversely associated with NEC cases preceded by Proteobacteria dysbiosis | FDR-adjusted p-value < 0.05 (Benjamini–Hochberg procedure) | Morrow et al. [44] |
| 5 discriminant metabolites in NEC samples involved into C-21 steroid hormones, linoleate, prostaglandines, leucotrienes pathways with increased intensity before diagnosis and decrease afterwards | FDR-adjusted p-value applied but threshold not specified | Stewart et al. [45] | |
| The faecal metabolome of NEC-1 was more divergent from the second month of life the first changes were evidenced with a significant ↓ in serine levels in the second 10 days of life (11–20 days) ending with ↓ ethanol and leucine concentrations in the second month of life | Two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli Significance threshold: p < 0.05 | Brehin et al. [46] | |
| ↓ in unclassified Staphylococcus, Lactobacillaceae and Bifidobacterium animalis subsp. lactis at the species level together with ↑ in the content of some TCA metabolites (succinate, L-malic acid and oxaloacetate) may represent possible early biomarkers of the disease interdependence between metabolome and faecal microbiota changes especially for two metabolites, L-malic acid and oxaloacetate | No correction for multiple testing | Du et al. [47] | |
| NEC + LOS | No metabolites or microbiome signature associated with NEC or LOS | No correction for multiple testing | Wandro et al. [49] |
| No discriminating metabolomic and proteomic biomarkers for NEC or LOS | No correction for multiple testing | Stewart et al. [50] | |
| LOS | Changes in the faecal metabolome were significant at diagnosis and 7 days later, but not in the 7 days prior to diagnosis. 7 faecal metabolites were significantly altered between infants with LOS and control infants at diagnosis, after correction for confounding factors raffinose, sucrose and acetic acid were closely related to the gut microbiota, whereas 18-hydroxycortisol, L-glutamate and 18-oxocortisol could be related to metabolic stress or inflammatory responses. | FDR-adjusted p-value applied but threshold not specified (Benjamini–Hochberg procedure) | Stewart et al. [48] |
| NRDS | In NRDS group: marked impairment of tryptophan metabolism alterations in cortisol metabolism (↑ levels of intermediates, such as pregnenolone and 17α-hydroxyprogesterone) accumulation of medium and long-chain fatty acids (probable association with mitochondrial dysfunction) high relative abundance of Haemophilus, Fusicatenibacter and Vibrio | FDR-adjusted p-value < 0.05 | Bi et al. [51] |
| WMI | Presence of characteristic groups in WMI-related dysbiosis (Bacteroidetes, Staphylococcus and Acinetobacter) strongly associated with alterations in the faecal metabolome (↓ colic and allocholine acid, ↑ cinobufagin, adenosine-3-monophosphate and N-acetylneuramic acid) mainly related to arginine and bile acid biosynthesis pathways | No correction for multiple testing | Liu et al. [52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosco, A.; Arru, F.; Abis, A.; Fanos, V.; Dessì, A. Application of Multiomics in Perinatology: A Metabolomics Integration-Focused Review. Int. J. Mol. Sci. 2025, 26, 4164. https://doi.org/10.3390/ijms26094164
Bosco A, Arru F, Abis A, Fanos V, Dessì A. Application of Multiomics in Perinatology: A Metabolomics Integration-Focused Review. International Journal of Molecular Sciences. 2025; 26(9):4164. https://doi.org/10.3390/ijms26094164
Chicago/Turabian StyleBosco, Alice, Francesca Arru, Alessandra Abis, Vassilios Fanos, and Angelica Dessì. 2025. "Application of Multiomics in Perinatology: A Metabolomics Integration-Focused Review" International Journal of Molecular Sciences 26, no. 9: 4164. https://doi.org/10.3390/ijms26094164
APA StyleBosco, A., Arru, F., Abis, A., Fanos, V., & Dessì, A. (2025). Application of Multiomics in Perinatology: A Metabolomics Integration-Focused Review. International Journal of Molecular Sciences, 26(9), 4164. https://doi.org/10.3390/ijms26094164