Disentangling the Genetic Relationships of Three Closely Related Bandicoot Species across Southern and Western Australia

 , and
, and
Abstract
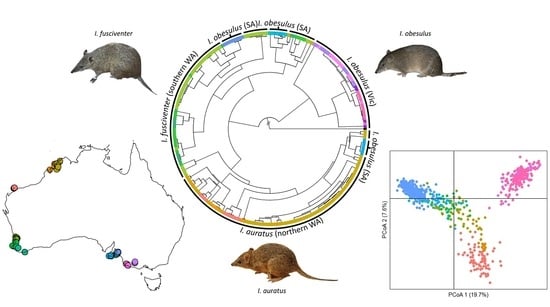
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection, Microsatellite Genotyping, and Mitochondrial DNA Sequencing
2.2. Data Analysis
2.2.1. Phylogenetic Analysis
2.2.2. Population Level Analysis
2.2.3. Genetic Variation Analysis
3. Results
3.1. Genetic Data Quality Assessment
3.2. Phylogeographic Structure across Southern and Western Australia
3.3. Population-Level Analysis
3.4. Population Genetic Diversity
4. Discussion
4.1. Distinction between I. fusciventer, I. auratus, and I. obesulus (SA)
4.2. Lack of Clarity on the Taxonomic Relationship of SA I. obesulus from Current Genetic Data
4.3. Genetic Diversity and Population Substructure
4.4. Recommendations for Conservation Management
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frankham, R. Challenges and opportunities of genetic approaches to biological conservation. Biol. Conserv. 2010, 143, 1919–1927. [Google Scholar] [CrossRef]
- Moritz, C. Strategies to protect biological diversity and the evolutionary processes that sustain it. Syst. Biol. 2002, 51, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R.; Ballou, J.D.; Eldridge, M.D.B.; Lacy, R.C.; Ralls, K.; Dudash, M.R.; Fenster, C.B. Predicting the probability of outbreeding depression. Conserv. Biol. 2011, 25, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, F.W.; Leary, R.F.; Spruell, P.; Wenburg, J.K. The problems with hybrids: Setting conservation guidelines. Trends Ecol. Evol. 2001, 16, 613–622. [Google Scholar] [CrossRef]
- Weeks, A.R.; Stoklosa, J.; Hoffmann, A.A. Conservation of genetic uniqueness of populations may increase extinction likelihood of endangered species: The case of Australian mammals. Front. Zoöl. 2016, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ryder, O.A. Species conservation and systematics: The dilemma of subspecies. Trends Ecol. Evol. 1986, 1, 9–10. [Google Scholar] [CrossRef]
- Moritz, C. Defining ‘evolutionarily significant units’ for conservation. Trends Ecol. Evol. 1994, 9, 373–375. [Google Scholar] [CrossRef]
- Fraser, D.J.; Bernatchez, L. Adaptive evolutionary conservation: Towards a unified concept for defining conservation units. Mol. Ecol. 2001, 10, 2741–2752. [Google Scholar] [CrossRef]
- Crandall, K.A.; Bininda-Emonds, O.R.; Mace, G.M.; Wayne, R.K. Considering evolutionary processes in conservation biology. Trends Ecol. Evol. 2000, 15, 290–295. [Google Scholar] [CrossRef]
- Gordon, G.; Hulbert, A.J. Peramelidae. In Fauna of Australia. Mammalia; Walton, D.W., Richardson, B.J., Eds.; Australian Government Publishing Service: Canberra, Australia, 1989; Volume 1b, pp. 603–624. [Google Scholar]
- Warburton, N.M.; Travouillon, K.J. The biology and palaeontology of the Peramelemorphia: A review of current knowledge and future research directions. Aust. J. Zoöl. 2016, 64, 151–181. [Google Scholar] [CrossRef]
- Cooper, S.; Ottewell, K.; Macdonald, A.J.; Adams, M.; Byrne, M.; Carthew, S.M.; Eldridge, M.D.B.; Li, Y.; Pope, L.C.; Saint, K.M.; et al. Phylogeography of southern brown and golden bandicoots: Implications for the taxonomy and distribution of endangered subspecies and species. Aust. J. Zoöl. 2018, 66, 379. [Google Scholar] [CrossRef]
- Ottewell, K.; Pitt, G.; Pellegrino, B.; Van Dongen, R.; Kinloch, J.; Willers, N.; Byrne, M. Remnant vegetation provides genetic connectivity for a critical weight range mammal in a rapidly urbanising landscape. Landsc. Urban Plan. 2019, 190, 103587. [Google Scholar] [CrossRef]
- Maclagan, S.J.; Coates, T.; Ritchie, E.G. Don’t judge habitat on its novelty: Assessing the value of novel habitats for an endangered mammal in a peri-urban landscape. Biol. Conserv. 2018, 223, 11–18. [Google Scholar] [CrossRef]
- Pope, L.C.; Storch, D.; Adams, M.; Moritz, C.; Gordon, G. A phylogeny for the genus Isoodon and a range extension for I. obesulus peninsulae based on mtDNA control region and morphology. Aust. J. Zoöl. 2001, 49, 411. [Google Scholar] [CrossRef]
- Wilson, D.E.; Reeder, D.M. Mammal Species of the World, 3rd ed.; A Taxonomic and Geographic Reference; Johns Hopkins University Press: Baltimore, MD, USA, 2005. [Google Scholar]
- Van Dyck, S.; Strahan, R. The Mammals of Australia, 3rd ed.; Reed New Holland: Sydney, Australia, 2008. [Google Scholar]
- Westerman, M.; Kear, B.; Aplin, K.; Meredith, R.; Emerling, C.; Springer, M. Phylogenetic relationships of living and recently extinct bandicoots based on nuclear and mitochondrial DNA sequences. Mol. Phylogenetics Evol. 2012, 62, 97–108. [Google Scholar] [CrossRef]
- Travouillon, K.J.; Phillips, M.J. Total evidence analysis of the phylogenetic relationships of bandicoots and bilbies (Marsupialia: Peramelemorphia): Reassessment of two species and description of a new species. Zootaxa 2018, 4378, 224–256. [Google Scholar] [CrossRef]
- Li, Y.; Lancaster, M.L.; Carthew, S.M.; Packer, J.G.; Cooper, S.J.B. Delineation of conservation units in an endangered marsupial, the southern brown bandicoot (Isoodon obesulus obesulus), in South Australia/western Victoria, Australia. Aust. J. Zoöl. 2014, 62, 345–359. [Google Scholar] [CrossRef]
- Zenger, K.R.; Eldridge, M.D.B.; Johnston, P.G. Phylogenetics, population structure and genetic diversity of the endangered southern brown bandicoot (Isoodon obesulus) in south-eastern Australia. Conserv. Genet. 2005, 6, 193–204. [Google Scholar] [CrossRef]
- Lyne, A.G.; Mort, P.A. A comparison of skull morphology in the marsupial bandicoot genus Isoodon: Its taxonomic implications and notes on a new species, Isoodon arnhemensis. Aust. Mammal. 1981, 4, 107–133. [Google Scholar]
- Menkhorst, P.; Knight, F. A Field Guide to the Mammals of Australia; Oxford University Press: Melbourne, Australia, 2011. [Google Scholar]
- Coates, T.; Nicholls, D.; Willig, R. The distribution of the southern brown bandicoot ’Isoodon obesulus’ in south central Victoria. Vic. Nat. 2008, 125, 128–139. [Google Scholar]
- Paull, D.J.; Mills, D.J.; Claridge, A.W. Fragmentation of the southern brown bandicoot Isoodon obesulus: Unraveling past climate change from vegetation clearing. Int. J. Ecol. 2013, 2013, 536524. [Google Scholar] [CrossRef]
- Burbidge, A.A.; Woinarski, J. Isoodon auratus. The IUCN Red List of Threatened Species 2016; Vol. e.T10863A115100163. Available online: http://dx.doi.org/10.2305/IUCN.UK.2016-3.RLTS.T10863A21966258.en (accessed on 23 July 2019).
- Burbidge, A.A.; Woinarski, J. Isoodon obesulus. The IUCN Red List of Threatened Species 2016; Vol. e.T40553A115173603. Available online: http://dx.doi.org/10.2305/IUCN.UK.2016-3.RLTS.T40553A21966368.en (accessed on 26 March 2019).
- Woinarski, J.C.Z.; Burbidge, A.A.; Harrison, P.; Kelly, J. The Action Plan for Australian Mammals 2012; CSIRO Publishing: Collingwood, Australia, 2014. [Google Scholar]
- Department of the Environment. Creating Safe Havens. Commonwealth of Australia. 2015. Available online: https://www.environment.gov.au/system/files/resources/f3e6ed38-6b27-46ca-a980-66ce3a1cdadc/files/factsheet-creating-safe-havens.pdf (accessed on 27 March 2019).
- Ottewell, K.; Dunlop, J.; Thomas, N.; Morris, K.; Coates, D.; Byrne, M. Evaluating success of translocations in maintaining genetic diversity in a threatened mammal. Biol. Conserv. 2014, 171, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, L.; Pope, L.C.; Taberlet, P.; Moritz, C. Versatile primers for the amplification of the mitochondrial DNA control region in marsupials. Mol. Ecol. 1997, 6, 1199–1201. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, F.; Oliver, J.; Marín, A.; Medina, J. The general stochastic model of nucleotide substitution. J. Theor. Biol. 1990, 142, 485–501. [Google Scholar] [CrossRef]
- Yang, Z. Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol. Evol. 1996, 11, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2018, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2011, 3, 217–223. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Dray, S.; Dufour, A.-B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Durand, E.; Forbes, F.; François, O. Bayesian clustering algorithms ascertaining spatial population structure: A new computer program and a comparison study. Mol. Ecol. Notes 2007, 7, 747–756. [Google Scholar] [CrossRef]
- François, O.; Ancelet, S.; Guillot, G. Bayesian clustering using hidden markov random fields in spatial population genetics. Genetics 2006, 174, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Jost, L. Gst and its relatives do not measure differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Slatkin, M.; Hudson, R.R. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 1991, 129, 555–562. [Google Scholar]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar] [CrossRef]
- Ray, N.; Currat, M.; Excoffier, L. Intra-deme molecular diversity in spatially expanding populations. Mol. Biol. Evol. 2003, 20, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L. Patterns of DNA sequence diversity and genetic structure after a range expansion: Lessons from the infinite-island model. Mol. Ecol. 2004, 13, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Pompanon, F.; Bonin, A.; Bellemain, E.; Taberlet, P. Genotyping errors: Causes, consequences and solutions. Nat. Rev. Genet. 2005, 6, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Goudet, J. Fstat, a Program to Estimate and Test Gene Diversities and Fixation Indices Version 2.9.3. 2001. Available online: http://www2.unil.ch/popgen/softwares/fstat.htm (accessed on 1 November 2018).
- Pohlert, T. The Pairwise Multiple Comparison of Mean Ranks Package (PMCMR); R package: Madison, WI, USA, 2014. [Google Scholar]
- Australia Wildlife Conservancy. No Data. Available online: http://www.australianwildlife.org/media/59016/golden-bandicoot-artesian-range-sanctuary-photo-ross-knowles-dsc_7435.jpg (accessed on 16 October 2020).
- Museums Victoria. Isoodon Obesulus Southern Brown Bandicoot in Museums Victoria Collection. 2017. Available online: https://collections.museumsvictoria.com.au/species/8401 (accessed on 9 December 2020).
- Frankham, R. Genetic rescue of small inbred populations: Meta-analysis reveals large and consistent benefits of gene flow. Mol. Ecol. 2015, 24, 2610–2618. [Google Scholar] [CrossRef]
- Cooper, M.L. Geographic variation in size and shape in the southern brown bandicoot, Isoodon obesulus (Peramelidae: Marsupialia), in Western Australia. Aust. J. Zool. 1998, 46, 145–152. [Google Scholar] [CrossRef]
- Cooper, M.L. Temporal variation in skull size and shape in the southern brown bandicoot, Isoodon obesulus (Peramelidae: Marsupialia) in Western Australia. Aust. J. Zoöl. 2000, 48, 47. [Google Scholar] [CrossRef]
- Dunlop, J.; Morris, K. Environmental determination of body size in mammals: Rethinking ‘island dwarfism’ in the golden bandicoot. Austral Ecol. 2018, 43, 817–827. [Google Scholar] [CrossRef]
- Hale, M. Inheritance of geographic variation in body size, and countergradient variation in growth rates, in the southern brown bandicoot Isoodon obesulus. Aust. Mammal. 2000, 22, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Avise, J.C. Phylogeography: Retrospect and prospect. J. Biogeogr. 2009, 36, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Kingman, J.F. Origins of the coalescent. 1974–1982. Genetics 2000, 156, 1461–1463. [Google Scholar]
- Pacioni, C.; Hunt, H.; Allentoft, M.E.; Vaughan, T.G.; Wayne, A.F.; Baynes, A.; Haouchar, D.; Dortch, J.; Bunce, M. Genetic diversity loss in a biodiversity hotspot: Ancient DNA quantifies genetic decline and former connectivity in a critically endangered marsupial. Mol. Ecol. 2015, 24, 5813–5828. [Google Scholar] [CrossRef] [PubMed]
- Czarnomska, S.D.; Niedziałkowska, M.; Borowik, T.; Jędrzejewska, B. Regional and local patterns of genetic variation and structure in yellow-necked mice—The roles of geographic distance, population abundance, and winter severity. Ecol. Evol. 2018, 8, 8171–8186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosser, S.; Abdelkrim, J.; Wing, J.; Robertson, B.C.; Gemmell, N.J. Strong isolation by distance argues for separate population management of endangered blue duck (Hymenolaimus malacorhynchos). Conserv. Genet. 2016, 18, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Rannala, B. Bayesian species delimitation using multilocus sequence data. Proc. Natl. Acad. Sci. USA 2010, 107, 9264–9269. [Google Scholar] [CrossRef] [Green Version]
- Knowles, L.L.; Carstens, B.C. Delimiting species without monophyletic gene trees. Syst. Biol. 2007, 56, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Bragg, J.G.; Potter, S.; Bi, K.; Catullo, R.A.; Donnellan, S.; Eldridge, M.D.B.; Joseph, L.; Keogh, J.S.; Oliver, P.M.; Rowe, K.C.; et al. Resources for phylogenomic analyses of Australian terrestrial vertebrates. Mol. Ecol. Resour. 2017, 17, 869–876. [Google Scholar] [CrossRef]
- Dolman, G.; Joseph, L. Evolutionary history of birds across southern Australia: Structure, history and taxonomic implications of mitochondrial DNA diversity in an ecologically diverse suite of species. Emu Austral Ornithol. 2015, 115, 35–48. [Google Scholar] [CrossRef]
- Li, Y.; Cooper, S.J.B.; Lancaster, M.L.; Packer, J.G.; Carthew, S.M. Comparative population genetic structure of the endangered southern brown bandicoot, Isoodon obesulus, in fragmented landscapes of Southern Australia. PLoS ONE 2016, 11, e0152850. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lancaster, M.L.; Cooper, S.J.B.; Taylor, A.C.; Carthew, S.M. Population structure and gene flow in the endangered southern brown bandicoot (Isoodon obesulus obesulus) across a fragmented landscape. Conserv. Genet. 2014, 16, 331–345. [Google Scholar] [CrossRef]
- Packer, J.G.; Delean, S.; Kueffer, C.; Prider, J.; Abley, K.; Facelli, J.M.; Carthew, S.M. Native faunal communities depend on habitat from non-native plants in novel but not in natural ecosystems. Biodivers. Conserv. 2016, 25, 503–523. [Google Scholar] [CrossRef]
- Lohr, C.; Algar, D. Managing feral cats through an adaptive framework in an arid landscape. Sci. Total. Environ. 2020, 720, 137631. [Google Scholar] [CrossRef] [PubMed]
- Moseby, K.E.; Read, J.; Paton, D.C.; Copley, P.; Hill, B.; Crisp, H. Predation determines the outcome of 10 reintroduction attempts in arid South Australia. Biol. Conserv. 2011, 144, 2863–2872. [Google Scholar] [CrossRef]
- Gardiner, R.; Bain, G.; Hamer, R.; Jones, M.E.; Johnson, C.N. Habitat amount and quality, not patch size, determine persistence of a woodland-dependent mammal in an agricultural landscape. Landsc. Ecol. 2018, 33, 1837–1849. [Google Scholar] [CrossRef]
- Palmer, C. All Good—Wankura have 2 New Island Homes; NT Department of Natural Resources, Environment, the Arts and Sport: Darwin, Australia, 2009.
- Department of Land Resource Management. Nt Action Plan. Golden Bandicoot Isoodon auratus. 2015. Available online: https://nt.gov.au/__data/assets/pdf_file/0011/269147/nt-action-plan-golden-bandicoot.pdf (accessed on 26 March 2019).
- Brown, G.W.; Main, M.L. National Recovery Plan for the Southern Brown Bandicoot Isoodon obesulus obesulus; Victorian Government Department of Sustainability and Environment (DSE): Melbourne, Australia, 2010.
- Haby, N.; Long, K. Recovery Plan for the Southern Brown Bandicoot in the Mount Lofty Ranges, South Australia, 2004–2009; Department of Environment and Heritage: Adelaide, South Australia, Australia, 2005.
- Department of Environment and Conservation. Gorgon Gas Development—Threatened and Priority Species Translocation and Reintroduction Program—Annual Report 2010/11; Department of Environment and Conservation: Perth, Australia, 2011.
- Department of Environment and Conservation. Barrow Group Nature Reserves Draft Management Plan 2011; Department of Environment and Conservation: Perth, Australia, 2011.
- Department of Environment and Conservation (NSW). Southern Brown Bandicoot (Isoodon obesulus) Recovery Plan; NSW DEC: Hurstville, New South Wales, Australia, 2006.
 indicates sampling locations. Each pie chart represents haplotype frequencies within each sampling site, and size of pie chart represents numbers of haplotypes. Colour codes indicate the sites where haplotypes were mostly found. Pattern codes represent individual haplotypes found within each site.
indicates sampling locations. Each pie chart represents haplotype frequencies within each sampling site, and size of pie chart represents numbers of haplotypes. Colour codes indicate the sites where haplotypes were mostly found. Pattern codes represent individual haplotypes found within each site.
indicates sampling locations. Each pie chart represents haplotype frequencies within each sampling site, and size of pie chart represents numbers of haplotypes. Colour codes indicate the sites where haplotypes were mostly found. Pattern codes represent individual haplotypes found within each site.
indicates sampling locations. Each pie chart represents haplotype frequencies within each sampling site, and size of pie chart represents numbers of haplotypes. Colour codes indicate the sites where haplotypes were mostly found. Pattern codes represent individual haplotypes found within each site.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Source of Variation | Variance Components | Percentage | p-Value |
|---|---|---|---|---|
| BWI, KIMI, KIMM | Among population | 2.1 | 48.3 | <0.001 |
| Within population | 2.3 | 51.7 | ||
| Perth metro, SW WA Coast, | Among population | 1.7 | 27.9 | <0.001 |
| SW WA inland | Within population | 4.3 | 72.1 | |
| MLR, FP, KI | Among population | 5.6 | 73.3 | <0.001 |
| Within population | 2.0 | 26.7 | ||
| LG, The Grampians, Mount Burr | Among population | 1.0 | 51.6 | <0.001 |
| Within population | 1.0 | 48.4 |
| WA | SA | VIC | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| State | Sampling Site | Barrow Island | Kimberley Islands | Kimberley Mainland | Perth Metro | SW WA Coast | SW WA Inland | Kangaroo Island | Fleurieu Peninsula | Mount Lofty Ranges | The Grampians | Lower Glenelg | Mount Burr |
| WA | Barrow Island | ─ | 0.436 | 0.478 | 0.701 | 0.682 | 0.902 | 0.817 | 0.817 | 0.868 | 0.862 | 0.783 | |
| Kimberley islands | 0.391 | ─ | 0.331 | 0.791 | 0.821 | 0.888 | 0.895 | 0.877 | 0.874 | 0.876 | 0.764 | ||
| Kimberley mainland | 0.495 | 0.462 | ─ | 0.656 | 0.623 | 0.788 | 0.776 | 0.761 | 0.785 | 0.846 | 0.705 | ||
| Perth metro | 0.471 | 0.435 | 0.579 | ─ | 0.111 | 0.611 | 0.550 | 0.423 | 0.853 | 0.880 | 0.792 | ||
| SW WA Coast | 0.582 | 0.533 | 0.685 | 0.318 | ─ | 0.602 | 0.450 | 0.359 | 0.783 | 0.894 | 0.778 | ||
| SW WA inland | 0.437 | 0.389 | 0.564 | 0.262 | 0.231 | ─ | |||||||
| SA | Kangaroo Island | 0.815 | 0.711 | 0.781 | 0.502 | 0.646 | 0.579 | ─ | 0.503 | 0.535 | 0.894 | 0.970 | 0.899 |
| Fleurieu Peninsula | 0.558 | 0.465 | 0.66 | 0.424 | 0.565 | 0.375 | 0.922 | ─ | 0.183 | 0.870 | 0.971 | 0.895 | |
| Mount Lofty Ranges | 0.400 | 0.284 | 0.445 | 0.415 | 0.520 | 0.383 | 0.754 | 0.404 | ─ | 0.913 | 0.960 | 0.896 | |
| VIC | Grampians | 0.869 | 0.824 | 0.843 | 0.739 | 0.804 | 0.756 | 0.971 | 0.933 | 0.828 | ─ | 0.303 | 0.302 |
| Lower Glenelg | 0.877 | 0.835 | 0.843 | 0.751 | 0.824 | 0.765 | 0.981 | 0.946 | 0.846 | 0.765 | ─ | 0.177 | |
| Mount Burr | 0.778 | 0.746 | 0.803 | 0.703 | 0.744 | 0.687 | 0.899 | 0.830 | 0.747 | 0.555 | 0.085 | ─ | |
| State | Region | Location | N D-loop | Nh | uh | h | ∏ | N | H | Ar | A | FIS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WA | 159 | 61 | 61 | 0.916 ± 0.017 | 0.023 ± 0.011 | 172 | 0.835 ± 0.030 | 18.1 ± 2.2 | 19.0 ± 2.3 | ||||
| North WA | 87 | 36 | 36 | 0.761 ± 0.051 | 0.013 ± 0.007 | 109 | 0.744 ± 0.064 A,C | 13.1 ± 1.7 A | 15.0 ± 2.0 A | ||||
| Barrow Island | 11 | 7 | 7 | 0.873 ± 0.089 | 0.010 ± 0.006 | 77 | 0.642 ± 0.084 | 3.2 ± 0.3 | 8.1 ± 1.3 | 0.056 | |||
| Kimberley Island | 11 | 8 | 8 | 0.927 ± 0.067 | 0.015 ± 0.009 | 16 | 0.649 ± 0.051 | 3.1 ± 0.2 | 7.0 ± 0.9 | 0.277 | |||
| Kimberley mainland | 65 | 21 | 21 | 0.585 ± 0.075 | 0.008 ± 0.005 | 16 | 0.861 ± 0.030 | 4.4 ± 0.3 | 10.8 ± 1.5 | 0.075 | |||
| South WA | 71 | 25 | 25 | 0.939 ± 0.014 | 0.021 ± 0.011 | 63 | 0.786 ± 0.044 A | 12.0 ± 1.7 A | 12.1 ± 1.8 A | ||||
| Perth metro | 40 | 12 | 10 | 0.853 ± 0.039 | 0.018 ± 0.009 | 39 | 0.759 ± 0.051 | 3.8 ± 0.3 | 9.6 ± 1.0 | 0.175 | |||
| South West Coast | 13 | 5 | 3 | 0.782 ± 0.079 | 0.016 ± 0.009 | 7 | 0.767 ± 0.071 | 3.8 ± 0.4 | 6.0 ± 0.8 | 0.231 | |||
| South West inland | 19 | 10 | 10 | 0.906 ± 0.041 | 0.017 ± 0.009 | 17 | 0.787 ± 0.030 | 3.8 ± 0.2 | 7.6 ± 0.9 | 0.128 | |||
| SA | 21 | 9 | 9 | 0.857 ± 0.056 | 0.023 ± 0.012 | 373 | 0.623 ± 0.050 B | 5.7 ± 0.7 B | 6.9 ± 0.8 B | ||||
| Mount Lofty Ranges | 9 | 4 | 4 | 0.778 ± 0.110 | 0.015 ± 0.009 | 311 | 0.597 ± 0.052 | 2.7 ± 0.2 | 5.7 ± 0.8 | 0.251 | |||
| Fleurieu Peninsula | 4 | 3 | 3 | 0.833 ± 0.222 | 0.008 ± 0.006 | 45 | 0.567 ± 0.056 | 2.6 ± 0.2 | 4.4 ± 0.5 | 0.458 | |||
| Kangaroo Island | 8 | 2 | 2 | 0.250 ± 0.180 | 0.001 ± 0.001 | 17 | 0.422 ± 0.079 | 2.1 ± 0.3 | 3.1 ± 0.5 | 0.230 | |||
| VIC | 38 | 7 | 7 | 0.634 ± 0.060 | 0.008 ± 0.005 | 186 | 0.627 ± 0.059 B,C | 5.8 ± 0.5 B | 6.7 ± 0.6 B | ||||
| Grampians | 11 | 2 | 1 | 0.182 ± 0.144 | 0.002 ± 0.002 | 27 | 0.486 ± 0.078 | 2.3 ± 0.3 | 3.1 ± 0.4 | 0.243 | |||
| Lower Glenelg | 12 | 2 | 0 | 0.167 ± 0.134 | 0.001 ± 0.001 | 12 | 0.444 ± 0.098 | 2.0 ± 0.2 | 2.4 ± 0.4 | 0.353 | |||
| Mount Burr | 11 | 2 | 1 | 0.327 ± 0.153 | 0.009 ± 0.005 | 147 | 0.612 ± 0.057 | 2.9 ± 0.2 | 6.0 ± 0.6 | 0.290 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thavornkanlapachai, R.; Levy, E.; Li, Y.; Cooper, S.J.B.; Byrne, M.; Ottewell, K. Disentangling the Genetic Relationships of Three Closely Related Bandicoot Species across Southern and Western Australia. Diversity 2021, 13, 2. https://doi.org/10.3390/d13010002
Thavornkanlapachai R, Levy E, Li Y, Cooper SJB, Byrne M, Ottewell K. Disentangling the Genetic Relationships of Three Closely Related Bandicoot Species across Southern and Western Australia. Diversity. 2021; 13(1):2. https://doi.org/10.3390/d13010002
Chicago/Turabian StyleThavornkanlapachai, Rujiporn, Esther Levy, You Li, Steven J. B. Cooper, Margaret Byrne, and Kym Ottewell. 2021. "Disentangling the Genetic Relationships of Three Closely Related Bandicoot Species across Southern and Western Australia" Diversity 13, no. 1: 2. https://doi.org/10.3390/d13010002
APA StyleThavornkanlapachai, R., Levy, E., Li, Y., Cooper, S. J. B., Byrne, M., & Ottewell, K. (2021). Disentangling the Genetic Relationships of Three Closely Related Bandicoot Species across Southern and Western Australia. Diversity, 13(1), 2. https://doi.org/10.3390/d13010002