
Diversifying of Two Pampus Species across the Indo–Pacific Barrier and the Origin of the Genus



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Laboratory Protocols
2.3. Single Nucleotide Polymorphism Calling
2.4. Population Structure
2.5. Phylogenetic Analysis
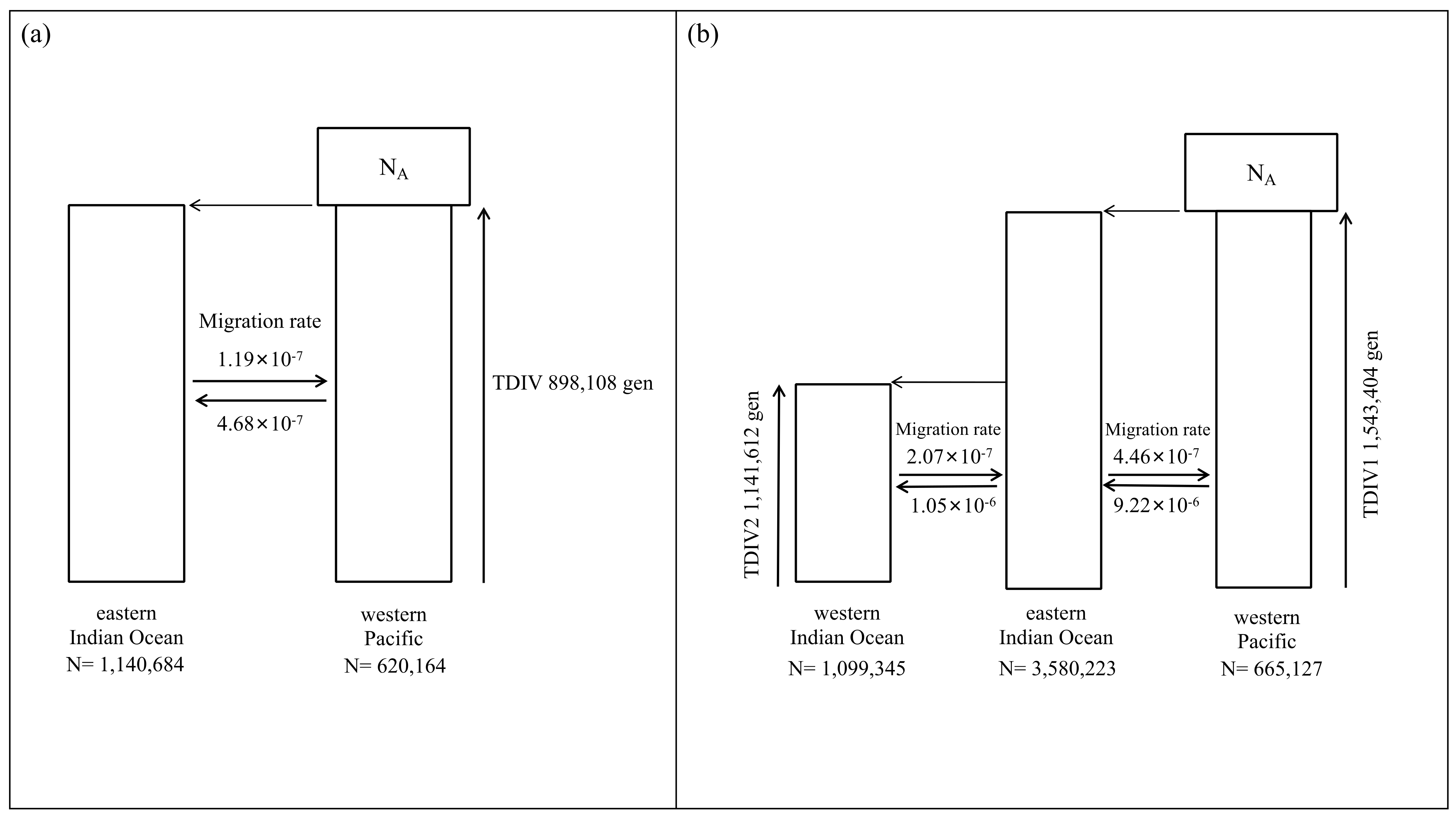
2.6. Divergence Time Dating and Demographic History
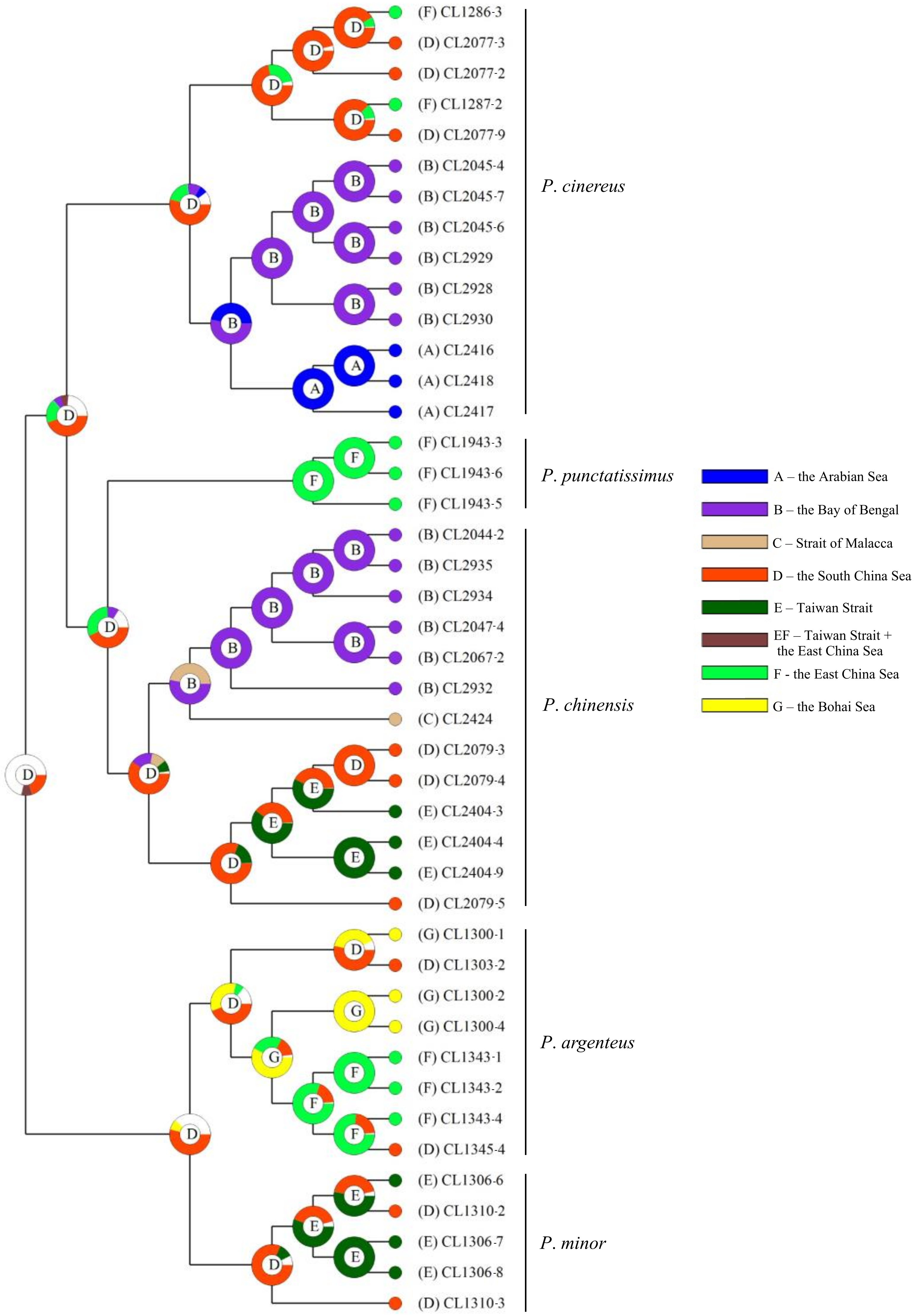
2.7. Infer Ancestral Area of Genus Pampus
3. Results
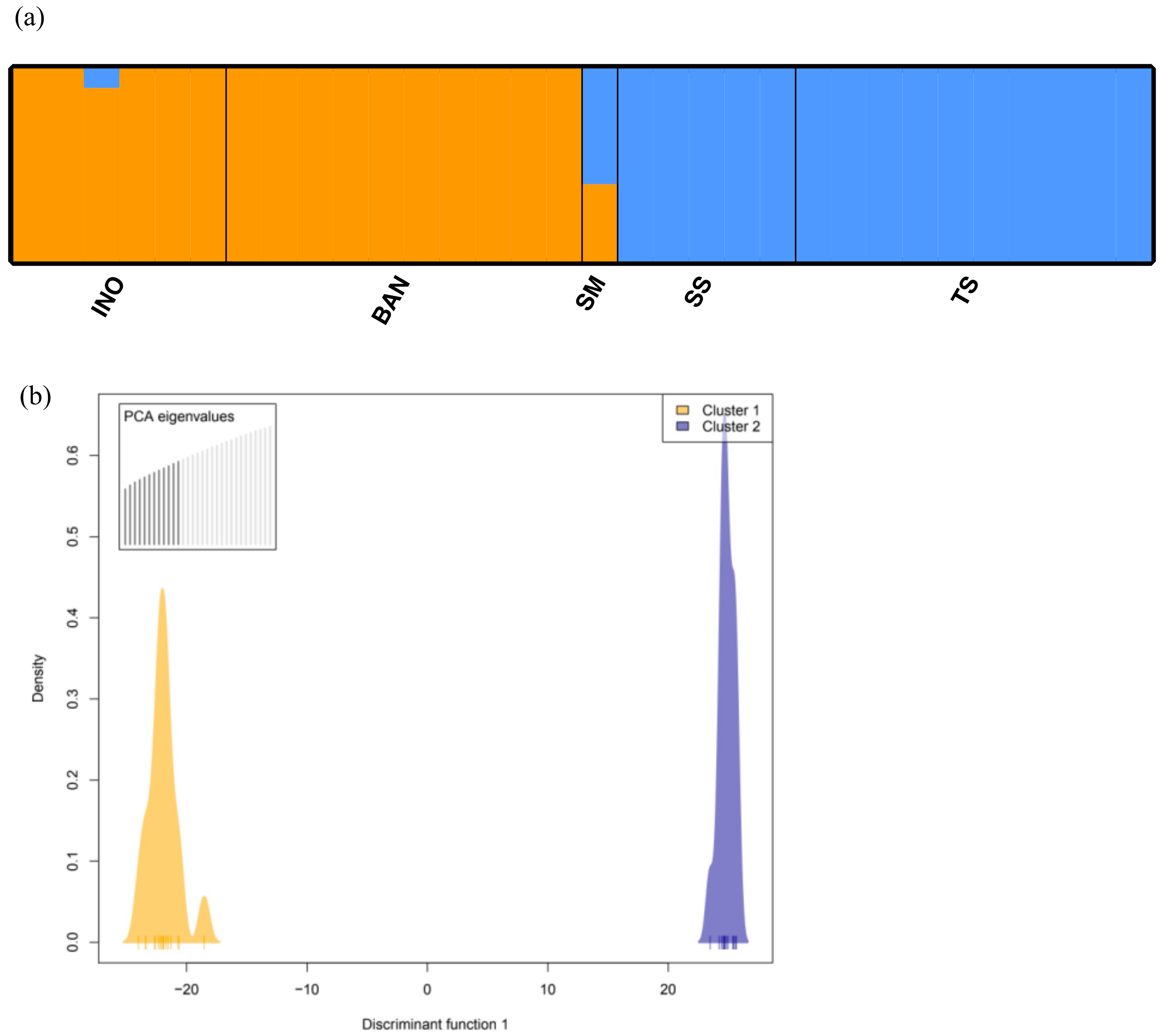
3.1. Population Structure
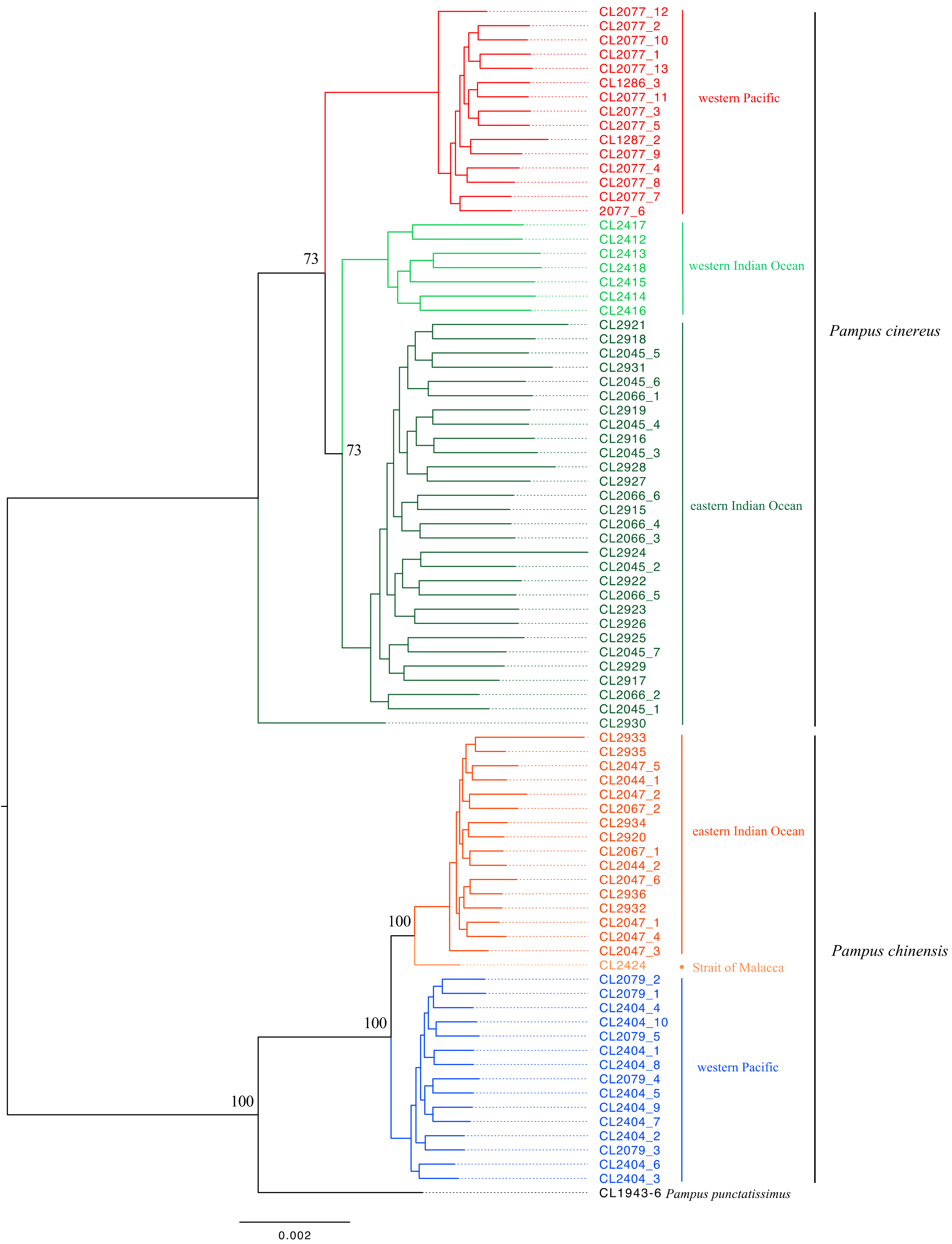
3.2. Phylogenetic Partitions
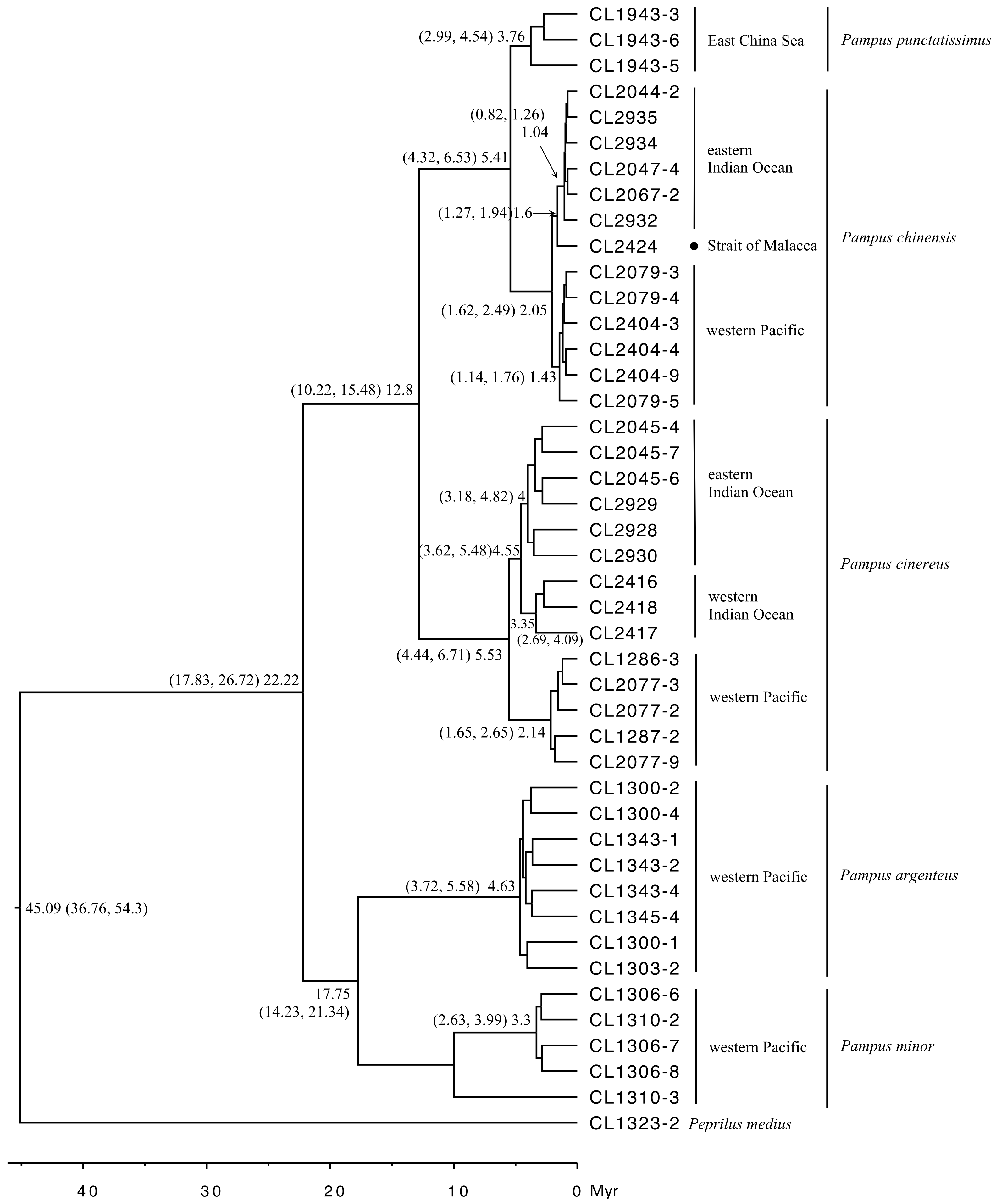
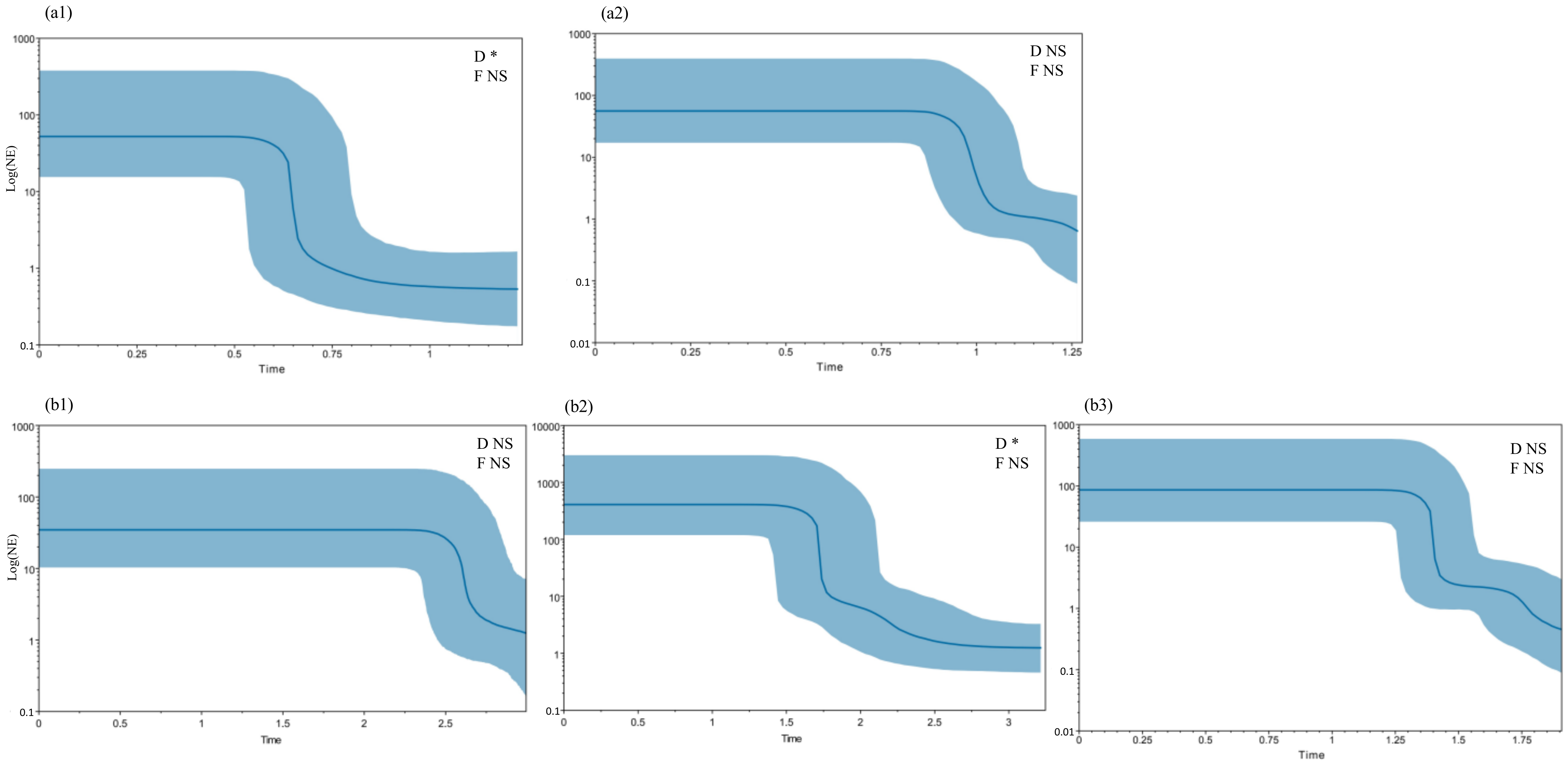
3.3. Divergence Time and Demographic History
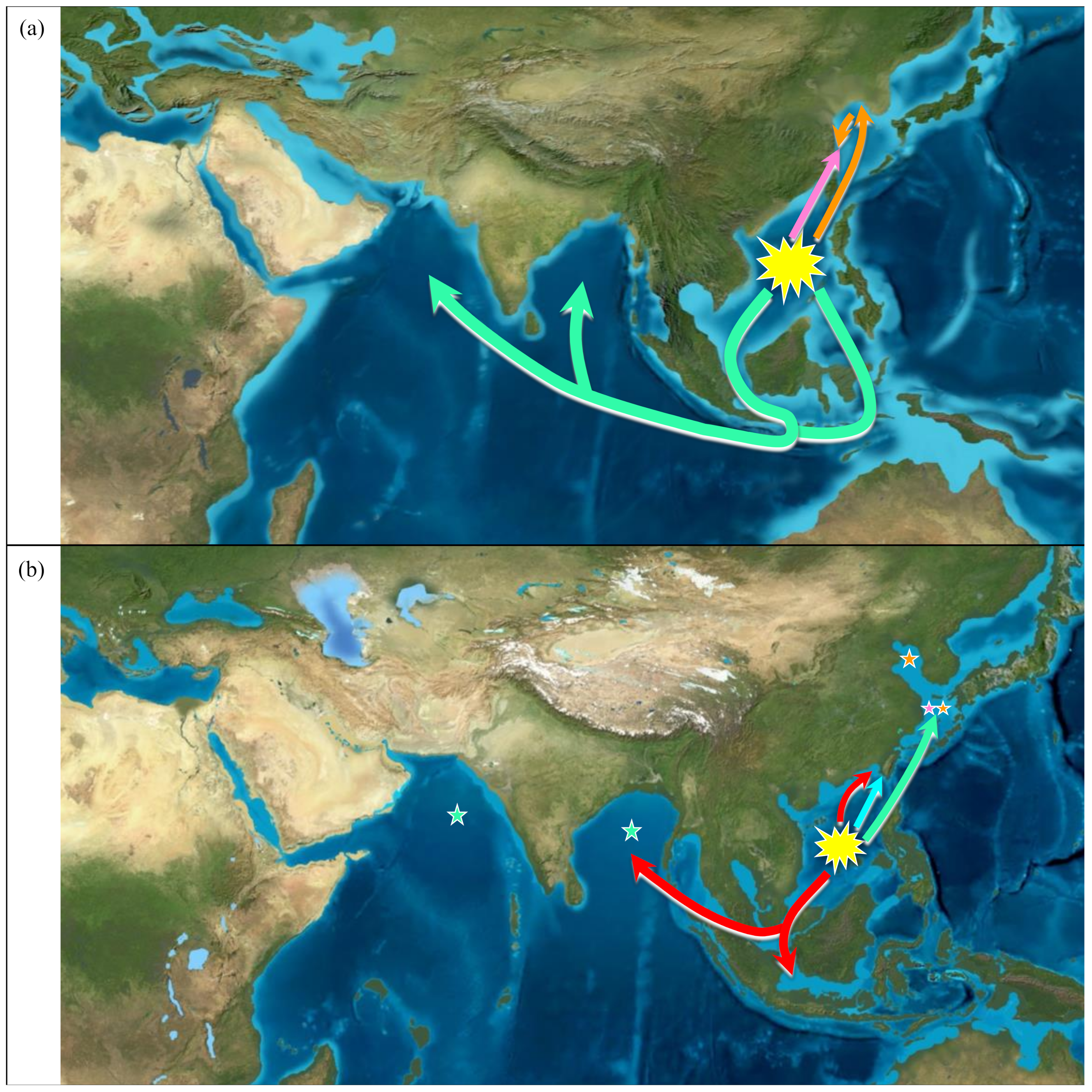
3.4. Ancestral Area
4. Discussion
4.1. Intraspecific Diversification of P. chinensis and P. cinereus
4.2. The Mechanism of Forming Different Intraspecific Lineages
4.3. The Origin and Dispersal Routes of Genus Pampus
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Briggs, J.C.; Bowen, B.W. A realignment of marine biogeographic provinces with particular reference to fish distributions. J. Biogeogr. 2012, 39, 12–30. [Google Scholar] [CrossRef]
- Bowen, B.W.; Gaither, M.R.; DiBattista, J.D.; Iacchei, M.; Andrews, K.R.; Grant, W.S.; Toonen, R.J.; Briggs, J.C. Comparative phylogeography of the ocean planet. Proc. Natl. Acad. Sci. USA 2016, 113, 7962–7969. [Google Scholar] [CrossRef] [Green Version]
- Waldrop, E.; Hobbs, J.P.A.; Randall, J.E.; DiBattista, J.D.; Rocha, L.A.; Kosaki, R.K.; Berumen, M.L.; Bowen, B.W. Phylogeography, population structure and evolution of coral-eating butterflyfishes (Family Chaetodontidae, genus Chaetodon, subgenus Corallochaetodon). J. Biogeogr. 2016, 43, 1116–1129. [Google Scholar] [CrossRef] [Green Version]
- Ahti, P.A.; Coleman, R.R.; DiBattista, J.D.; Berumen, M.L.; Rocha, L.A.; Bowen, B.W. Phylogeography of Indo-Pacific reef fishes: Sister wrasses Coris gaimard and C. cuvieri in the Red Sea, Indian Ocean and Pacific Ocean. J. Biogeogr. 2016, 43, 1103–1115. [Google Scholar] [CrossRef]
- Gaither, M.R.; Bowen, B.W.; Bordenave, T.R.; Rocha, L.A.; Newman, S.J.; Gomez, J.A.; van Herwerden, L.; Craig, M.T. Phylogeography of the reef fish Cephalopholis argus (Epinephelidae) indicates Pleistocene isolation across the Indo-Pacific Barrier with contemporary overlap in The Coral Triangle. BMC Evol. Biol. 2011, 11, 189. [Google Scholar] [CrossRef] [Green Version]
- Craig, M.T.; Eble, J.A.; Bowen, B.W.; Robertson, D.R. High genetic connectivity across the Indian and Pacific Oceans in the reef fish Myripristis berndti (Holocentridae). Mar. Ecol. Prog. Ser. 2007, 334, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Gaither, M.R.; Toonen, R.J.; Robertson, D.R.; Planes, S.; Bowen, B.W. Genetic evaluation of marine biogeographical barriers: Perspectives from two widespread Indo-Pacific snappers (Lutjanus kasmira and Lutjanus fulvus). J. Biogeogr. 2010, 37, 133–147. [Google Scholar] [CrossRef]
- Duncan, K.M.; Martin, A.P.; Bowen, B.W.; De Couet, H.G. Global phylogeography of the scalloped hammerhead shark (Sphyrna lewini). Mol. Ecol. 2006, 15, 2239–2251. [Google Scholar] [CrossRef]
- Reece, J.S.; Bowen, B.W.; Joshi, K.; Goz, V.; Larson, A. Phylogeography of two moray eels indicates high dispersal throughout the indo-pacific. J. Hered. 2010, 101, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Horne, J.B.; van Herwerden, L.; Choat, J.H.; Robertson, D.R. High population connectivity across the Indo-Pacific: Congruent lack of phylogeographic structure in three reef fish congeners. Mol. Phylogenetics Evol. 2008, 49, 629–638. [Google Scholar] [CrossRef]
- Ma, K.Y.; van Herwerden, L.; Newman, S.J.; Berumen, M.L.; Choat, J.H.; Chu, K.H.; de Mitcheson, Y.S. Contrasting population genetic structure in three aggregating groupers (Percoidei: Epinephelidae) in the Indo-West Pacific: The importance of reproductive mode. BMC Evol. Biol. 2018, 18, 180. [Google Scholar] [CrossRef]
- Taillebois, L.; Castelin, M.; Ovenden, J.R.; Bonillo, C.; Keith, P. Contrasting Genetic Structure among Populations of Two Amphidromous Fish Species (Sicydiinae) in the Central West Pacific. PLoS ONE 2013, 8, e75465. [Google Scholar] [CrossRef] [Green Version]
- Li, C.Y.; Olave, M.; Hou, Y.L.; Qin, G.; Schneider, R.F.; Gao, Z.X.; Tu, X.L.; Wang, X.; Qi, F.R.; Nater, A.; et al. Genome sequences reveal global dispersal routes and suggest convergent genetic adaptations in seahorse evolution. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Li, H.; Lin, H.; Ding, S.; Miyamoto, M.M. Phylogeography of the inshore fish, Bostrychus sinensis, along the Pacific coastline of China. Mol. Phylogenetics Evol. 2016, 96, 112–117. [Google Scholar] [CrossRef]
- Yin, G.; Pan, Y.; Anirban, S.; Mohammad, A.B.; Kim, J.-K.; Wu, H.; Li, C. Molecular systematics of Pampus (Perciformes: Stromateidae) based on thousands of nuclear loci using target-gene enrichment. Mol. Phylogenetics Evol. 2019, 140, 106595. [Google Scholar] [CrossRef]
- Almatar, S.M.; Lone, K.P.; Abu-Rezq, T.S.; Yousef, A.A. Spawning frequency, fecundity, egg weight and spawning type of silver pomfret, Pampus argenteus (Euphrasen) (Stromateidae), in Kuwait waters. J. Appl. Ichthyol. 2004, 20, 176–188. [Google Scholar] [CrossRef]
- Shi, Z.H.; Peng, S.M.; Sun, P.; Yin, F. Developmental potential and prospect of Pampus culture in China. Mod. Fish. Inf. 2009, 24, 3–4. [Google Scholar] [CrossRef]
- Liu, J.; Li, C.; Li, X. Phylogeny and biogeography of Chinese pomfret fishes (Pisces: Stromateidae). Studia Mar. Sin. 2002, 44, 235–239. [Google Scholar]
- Liu, J.; Li, C.S.; Ning, P. A redescription of grey pomfret Pampus cinereus (Bloch, 1795) with the designation of a neotype (Teleostei: Stromateidae). Chin. J. Oceanol. Limnol. 2013, 31, 140–145. [Google Scholar] [CrossRef]
- Divya, P.R.; Mohitha, C.; Rahul, G.K.; Shanis, C.P.R.; Basheer, V.S.; Gopalakrishnan, A. Molecular based phylogenetic species recognition in the genus Pampus (Perciformes: Stromateidae) reveals hidden diversity in the Indian Ocean. Mol. Phylogenetics Evol. 2017, 109, 240–245. [Google Scholar] [CrossRef]
- Divya, P.R.; Rahul, G.K.; Chelat, M.; Shanis, C.P.R.; Bineesh, K.K.; Basheer, V.S.; Achamveetil, G. Resurrection and Re-description of Pampus candidus (Cuvier), Silver Pomfret from the Northern Indian Ocean. Zool. Stud. 2019, 58, e7. [Google Scholar] [CrossRef]
- Li, Y.; Gao, T.X.; Zhou, Y.D.; Lin, L.S. Spatial genetic subdivision among populations of Pampus chinensis between China and Pakistan: Testing the barrier effect of the Malay Peninsula. Aquat. Living Resour. 2019, 32, 8. [Google Scholar] [CrossRef]
- Li, C.; Hofreiter, M.; Straube, N.; Corrigan, S.; Naylor, G.J. Capturing protein-coding genes across highly divergent species. Biotechniques 2013, 54, 321–326. [Google Scholar] [CrossRef]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 2010, 5448. [Google Scholar] [CrossRef]
- Fisher, S.; Barry, A.; Abreu, J.; Minie, B.; Nolan, J.; Delorey, T.M.; Young, G.; Fennell, T.J.; Allen, A.; Ambrogio, L.; et al. A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 2011, 12, R1. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Zhao, J.; Li, C. Species delimitation and phylogenetic reconstruction of the sinipercids (Perciformes: Sinipercidae) based on target enrichment of thousands of nuclear coding sequences. Mol. Phylogenetics Evol. 2017, 111, 44–55. [Google Scholar] [CrossRef]
- Marcel, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Yuan, H.; Atta, C.; Tornabene, L.; Li, C. Assexon: Assembling Exon Using Gene Capture Data. Evol Bioinform. 2019, 15, 1–13. [Google Scholar] [CrossRef]
- Jared, T.S.; Richard, D. Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 2012, 22, 14. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Jiang, J.; Jimenez, F.A.; Hoberg, E.P.; Cook, J.A.; Galbreath, K.E.; Li, C. Target gene enrichment in the cyclophyllidean cestodes, the most diverse group of tapeworms. Mol. Ecol. Resour 2016, 16, 1095–1106. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Proc, G.P.D. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assemly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. Structure Harvester: A website and program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and genotyping Loci de novo from short-read sequences. G3 (Bethesda) 2011, 1, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, T.; Tornabene, L.; Li, J.; Jiang, J.; Chakrabarty, P.; Sparks, J.S.; Naylor, G.J.P.; Li, C. Phylogenomic analysis on the exceptionally diverse fish clade Gobioidei (Actinopterygii: Gobiiformes) and data-filtering based on molecular clocklikeness. Mol. Phylogenetics Evol. 2018, 128, 192–202. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Mirarab, S.; Reaz, R.; Bayzid, M.S.; Zimmermann, T.; Swenson, M.S.; Warnow, T. ASTRAL: Genome-scale coalescent-based species tree estimation. Bioinformatics 2014, 30, i541–i548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018, 19, 153. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kuhnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Heled, J.; Drummond, A.J. Calibrated tree priors for relaxed phylogenetics and divergence time estimation. Syst. Biol. 2012, 61, 138–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miya, M.; Friedman, M.; Satoh, T.P.; Takeshima, H.; Sado, T.; Iwasaki, W.; Yamanoue, Y.; Nakatani, M.; Mabuchi, K.; Inoue, J.G.; et al. Evolutionary origin of the Scombridae (tunas and mackerels): Members of a paleogene adaptive radiation with 14 other pelagic fish families. PLoS ONE 2013, 8, e73535. [Google Scholar] [CrossRef] [PubMed]
- Rabosky, D.L.; Chang, J.; Title, P.O.; Cowman, P.F.; Sallan, L.; Friedman, M.; Kaschner, K.; Garilao, C.; Near, T.J.; Coll, M.; et al. An inverse latitudinal gradient in speciation rate for marine fishes. Nature 2018, 559, 392–395. [Google Scholar] [CrossRef]
- Rabosky, D.L.; Santini, F.; Eastman, J.; Smith, S.A.; Sidlauskas, B.; Chang, J.; Alfaro, M.E. Rates of speciation and morphological evolution are correlated across the largest vertebrate radiation. Nat. Commun. 2013, 4, 1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Lischer, H.E.L.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excofffier, L.; Marchi, N.; Marques, D.A.; Matthey-Doret, R.; Sousa, V.C. fastsimcoal2: Demographic inference under complex evolutionary scenarios. Bioinformatics 2021, 37, 4882–4885. [Google Scholar] [CrossRef]
- Excoffier, L.; Dupanloup, I.; Huerta-Sanchez, E.; Sousa, V.C.; Foll, M. Robust demographic inference from genomic and SNP data. PLoS Genet. 2013, 9, e1003905. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Blair, C.; He, X. RASP 4: Ancestral State Reconstruction Tool for Multiple Genes and Characters. Mol. Biol. Evol. 2020, 37, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Matzke, N.J. Model selection in historical biogeography reveals that founder-event speciation is a crucial process in Island Clades. Syst. Biol. 2014, 63, 951–970. [Google Scholar] [CrossRef] [PubMed]
- Ree, R.H.; Moore, B.R.; Webb, C.O.; Donoghue, M.J. A likelihood framework for inferring the evolution of geographic range on phylogenetic trees. Evolution 2005, 59, 2299–2311. [Google Scholar] [CrossRef] [PubMed]
- Ree, R.H.; Smith, S.A. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst. Biol. 2008, 57, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Hubert, N.; Meyer, C.P.; Bruggemann, H.J.; Guerin, F.; Komeno, R.J.; Espiau, B.; Causse, R.; Williams, J.T.; Planes, S. Cryptic diversity in Indo-Pacific coral-reef fishes revealed by DNA-barcoding provides new support to the centre-of-overlap hypothesis. PLoS ONE 2012, 7, e28987. [Google Scholar] [CrossRef]
- Wei, J.; Wu, R.; Xiao, Y.; Zhang, H.; Jawad, L.A.; Wang, Y.; Liu, J.; Al-Mukhtar, M.A. Validity of Pampus liuorum Liu & Li, 2013, Revealed by the DNA Barcoding of Pampus Fishes (Perciformes, Stromateidae). Diversity 2021, 13, 618. [Google Scholar] [CrossRef]
- Harold, K.V. Maps of Pleistocene sea levels in Southeast Asia: Shorelines, river systems and time durations. J. Biogeogr. 2000, 27, 1153–1167. [Google Scholar] [CrossRef] [Green Version]
- Greer, A.D. The hidden landscape: Evidence that sea-level change shaped the present population genomic patterns of marginal marine species. Mol. Ecol. 2021, 30, 1357–1360. [Google Scholar] [CrossRef]
- Rocha, L.A.; Craig, M.T.; Bowen, B.W. Phylogeography and the conservation of coral reef fishes. Coral Reefs 2007, 26, 501–502. [Google Scholar] [CrossRef]
- Shi, Z.H.; Wang, J.G.; Gao, L.J.; Lin, J.Z. Advances on the studies of reproductive biology and artificial breeding technology in silver pomfret Pampus argenteus. Mar. Fish. 2005, 27, 246–250. [Google Scholar] [CrossRef]
- Xue, H.J.; Chai, F.; Pettigrew, N.; Xu, D.Y.; Shi, M.; Xu, J.P. Kuroshio intrusion and the circulation in the South China Sea. J. Geophys. Res. Oceans 2004, 109, C02017(1–14). [Google Scholar] [CrossRef] [Green Version]
- Shankar, D.; Vinayachandran, P.N.; Unnikrishnan, A.S. The monsoon currents in the north Indian Ocean. Prog. Oceanogr. 2002, 52, 63–120. [Google Scholar] [CrossRef]
- Qiu, B.; Lukas, R. Seasonal and interannual variability of the North Equatorial Current, the Mindanao Current, and the Kuroshio along the Pacific western boundary. J. Geophys. Res. Oceans 1996, 101, 12315–12330. [Google Scholar] [CrossRef]
- Bellwood, D.R.; Meyer, C.P. Searching for heat in a marine biodiversity hotspot. J. Biogeogr. 2009, 36, 569–576. [Google Scholar] [CrossRef]
- Briggs, J.C. Diversity, endemism and evolution in the Coral Triangle. J. Biogeogr. 2009, 36, 2009–2010. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vouchers ID | Species | Sampling Site |
|---|---|---|
| CL2920, CL2932, CL2933, CL2934, CL2935, CL2936 | Pampus chinensis | Indian Ocean, India (INO) |
| CL2044-1, CL2044-2, CL2047-1, CL2047-2, CL2047-3, CL2047-4, CL2047-5, CL2047-6, CL2067-1, CL2067-2 | Pampus chinensis | Cox’s Bazar, Bangladesh (BAN) |
| CL2424 | Pampus chinensis | Strait of Malacca, Malaysia (SM) |
| CL2079-1, CL2079-2, CL2079-3, CL2079-4, CL2079-5 | Pampus chinensis | South China Sea, Beibu Gulf, Guangxi province, China (SS) |
| CL2404-1, CL2404-2, CL2404-3, CL2404-4, CL2404-5, CL2404-6, CL2404-7, CL2404-8, CL2404-9, CL2404-10 | Pampus chinensis | Taiwan Strait, Fujian province, China (TS) |
| CL2412, CL2413, CL2414, CL2415, CL2416, CL2417, CL2418 | Pampus cinereus | Arabian Sea, Iran (IRAN) |
| CL2915, CL2916, CL2917, CL2918, CL2919, CL2921, CL2922, CL2923, CL2924, CL2925, CL2926, CL2927, CL2928, CL2929, CL2930, CL2931 | Pampus cinereus | Indian Ocean, India (INO) |
| CL2045-1, CL2045-2, CL2045-3, CL2045-4, CL2045-5, CL2045-6, CL2045-7, CL2066-1, CL2066-2, CL2066-3, CL2066-4, CL2066-5, CL2066-6 | Pampus cinereus | Cox’s Bazar, Bangladesh (BAN) |
| CL2077-1, CL2077-2, CL2077-3, CL2077-4, CL2077-5, CL2077-6, CL2077-7, CL2077-8, CL2077-9, CL2077-10, CL2077-11, CL2077-12, CL2077-13 | Pampus cinereus | South China Sea, Beibu Gulf, Guangxi province, China (SS) |
| CL1286-3, CL1287-2 | Pampus cinereus | East China Sea, Shanghai, China (ES) |
| CL1310-2, CL1310-3 | Pampus minor | South China Sea, Beibu Gulf, Guangxi province, China (SS) |
| CL1306-6, CL1306-7, CL1306-8 | Pampus minor | Taiwan Strait, Fujian province, China (TS) |
| CL1303-2, CL1345-4 | Pampus argenteus | South China Sea, Beibu Gulf, Guangxi province, China (SS) |
| CL1343-1, CL1343-2, CL1343-4 | Pampus argenteus | East China Sea, Shanghai, China (ES) |
| CL1300-1, CL1300-2, CL1300-4 | Pampus argenteus | Bohai Sea, Liaoning province, China (BS) |
| CL1943-3, CL1943-5, CL1943-6 | Pampus punctatissimus | East China Sea, Shanghai, China (ES) |
| CL1323-2 | Peprilus medius | † |
| CL1346-6 | Psenopsis anomala | † |
| Pampus chinensis | |||||
|---|---|---|---|---|---|
| Population | INO | BAN | SM | SS | TS |
| Π | 0.123 | 0.102 | 0.141 | 0.113 | 0.120 |
| Pampus cinereus | |||||
| Population | IRAN | INO | BAN | SS | ES |
| Π | 0.103 | 0.094 | 0.088 | 0.049 | 0.049 |
| Pampus chinensis | ||||
|---|---|---|---|---|
| between Populations | BAN | SM | SS | TS |
| INO | 0.043 | 0.156 | 0.200 | 0.173 |
| BAN | 0.155 | 0.206 | 0.183 | |
| SM | 0.206 | 0.137 | ||
| SS | 0.041 | |||
| between groups | Pacific | |||
| eastern Indian Ocean | 0.131 | |||
| Pampus cinereus | ||||
| between populations | INO | BAN | SS | ES |
| IRAN | 0.068 | 0.080 | 0.183 | 0.186 |
| INO | 0.020 | 0.120 | 0.105 | |
| BAN | 0.140 | 0.133 | ||
| SS | 0.044 | |||
| between groups | western Indian Ocean | eastern Indian Ocean | ||
| Pacific | 0.180 | 0.094 | ||
| western Indian Ocean | 0.053 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, G.; Yin, G.; Sarker, A.; Li, C. Diversifying of Two Pampus Species across the Indo–Pacific Barrier and the Origin of the Genus. Diversity 2022, 14, 180. https://doi.org/10.3390/d14030180
Fan G, Yin G, Sarker A, Li C. Diversifying of Two Pampus Species across the Indo–Pacific Barrier and the Origin of the Genus. Diversity. 2022; 14(3):180. https://doi.org/10.3390/d14030180
Chicago/Turabian StyleFan, Gong, Guoxing Yin, Anirban Sarker, and Chenhong Li. 2022. "Diversifying of Two Pampus Species across the Indo–Pacific Barrier and the Origin of the Genus" Diversity 14, no. 3: 180. https://doi.org/10.3390/d14030180
APA StyleFan, G., Yin, G., Sarker, A., & Li, C. (2022). Diversifying of Two Pampus Species across the Indo–Pacific Barrier and the Origin of the Genus. Diversity, 14(3), 180. https://doi.org/10.3390/d14030180