

Microbiome Profile of the Mediterranean Mussel (Mytilus galloprovincialis) from Northern Aegean Sea (Greece) Culture Areas, Based on a 16S rRNA Next Generation Sequencing Approach



Abstract
1. Introduction
2. Materials and Methods
2.1. Site Information and Sampling
2.2. Sample Preparation
2.3. DNA Isolation
2.4. 16S rRNA Gene Library Preparation
2.5. Library Purification and Amplicon Sequencing
2.6. Bioinformatic and Statistical Analysis
3. Results
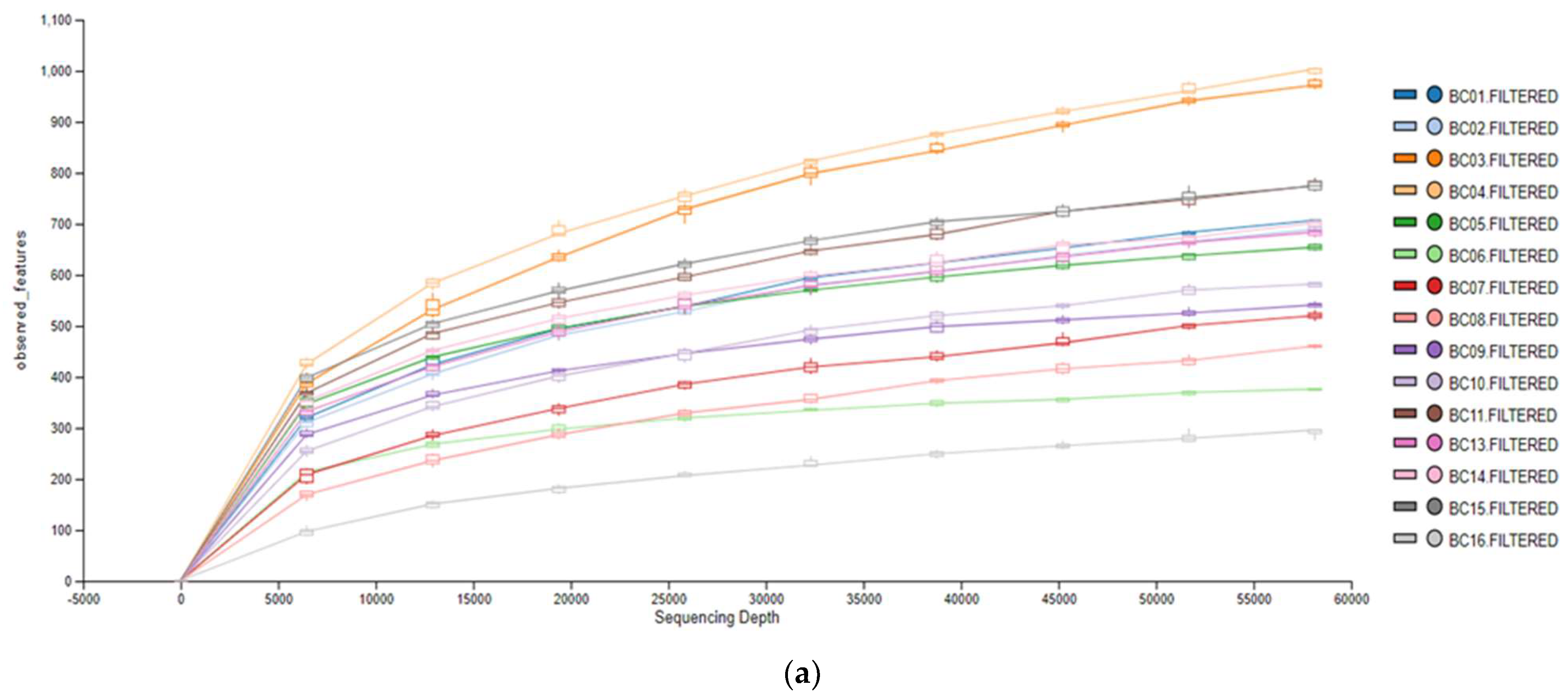
3.1. Microbiological Analysis
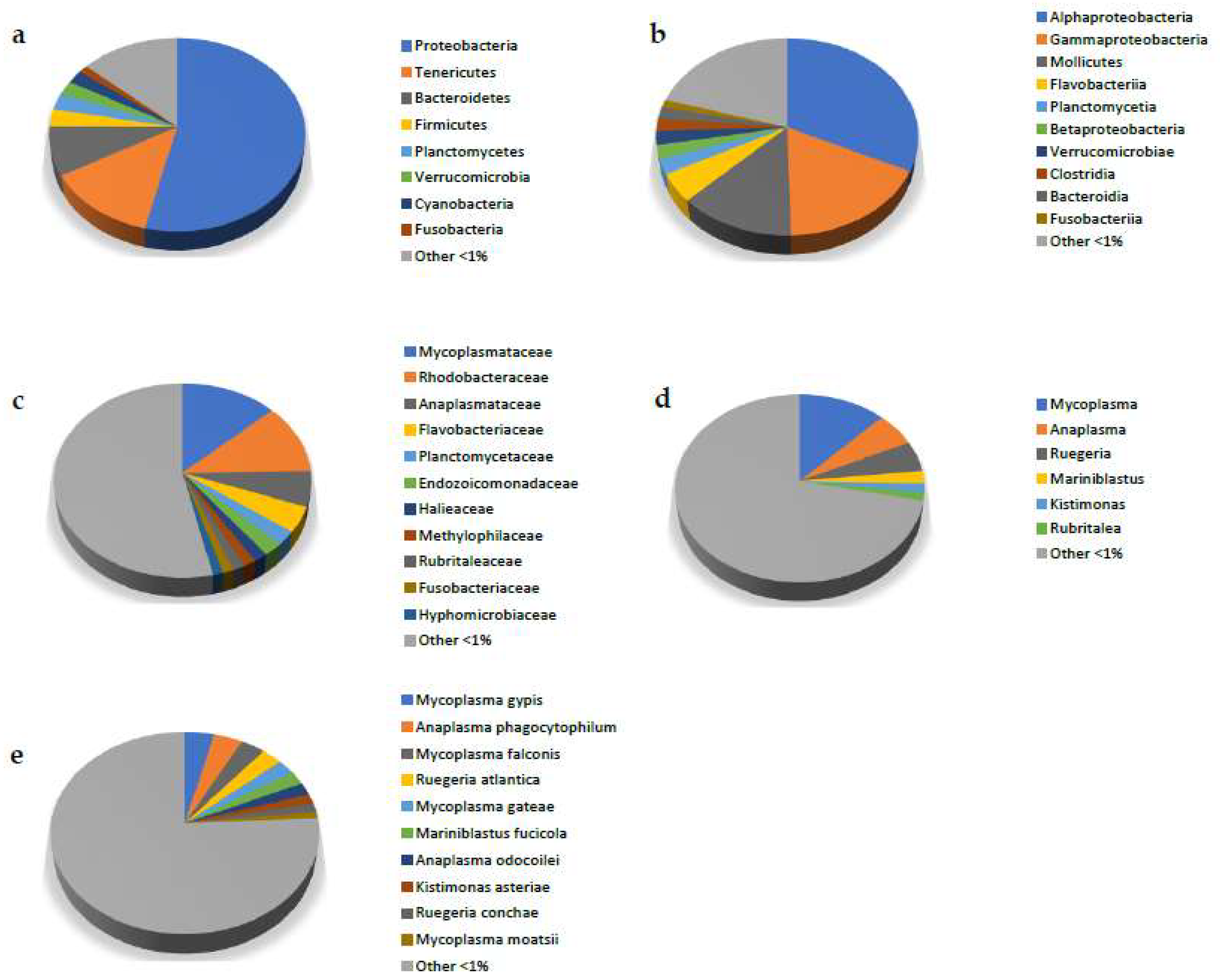
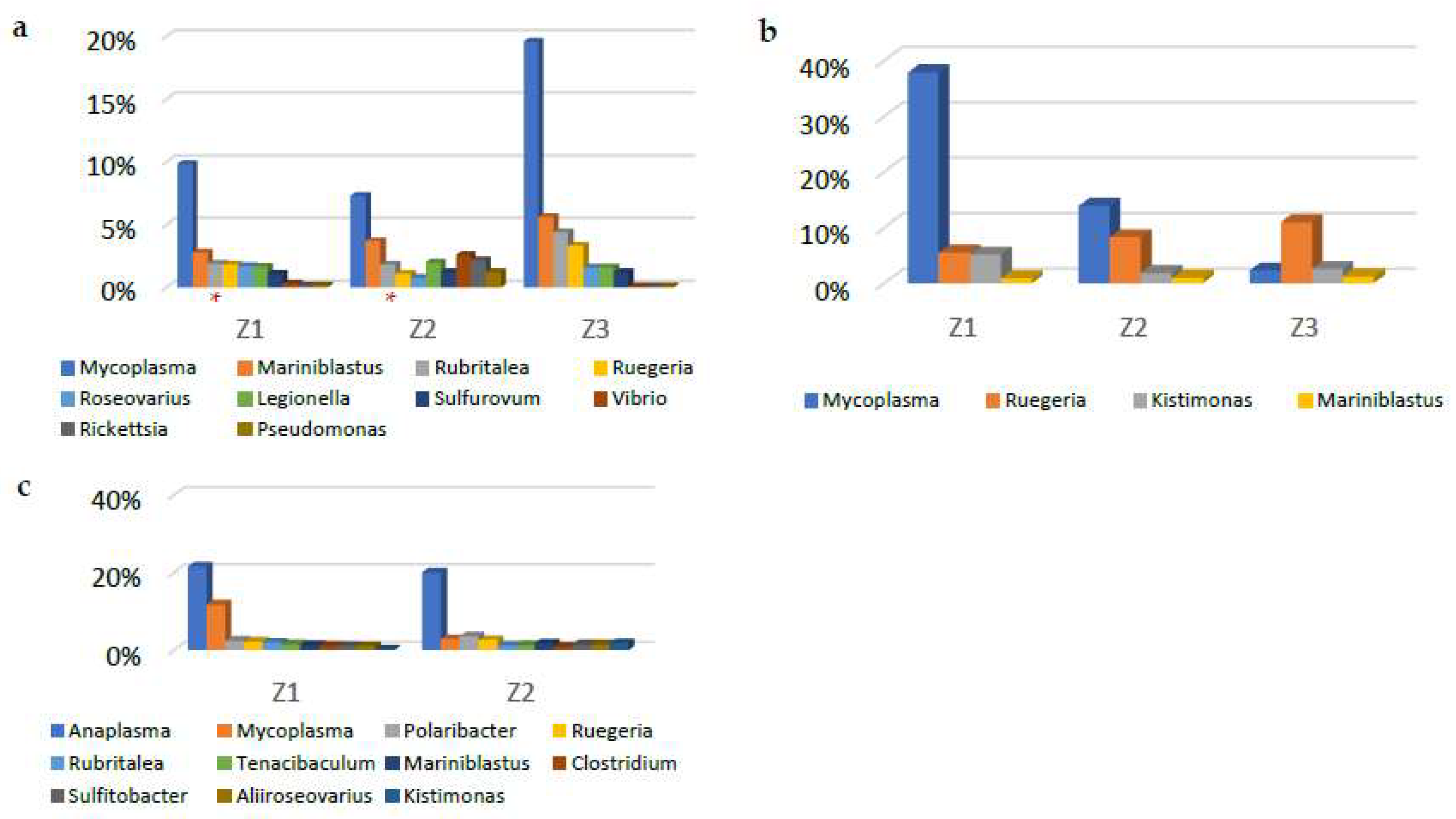
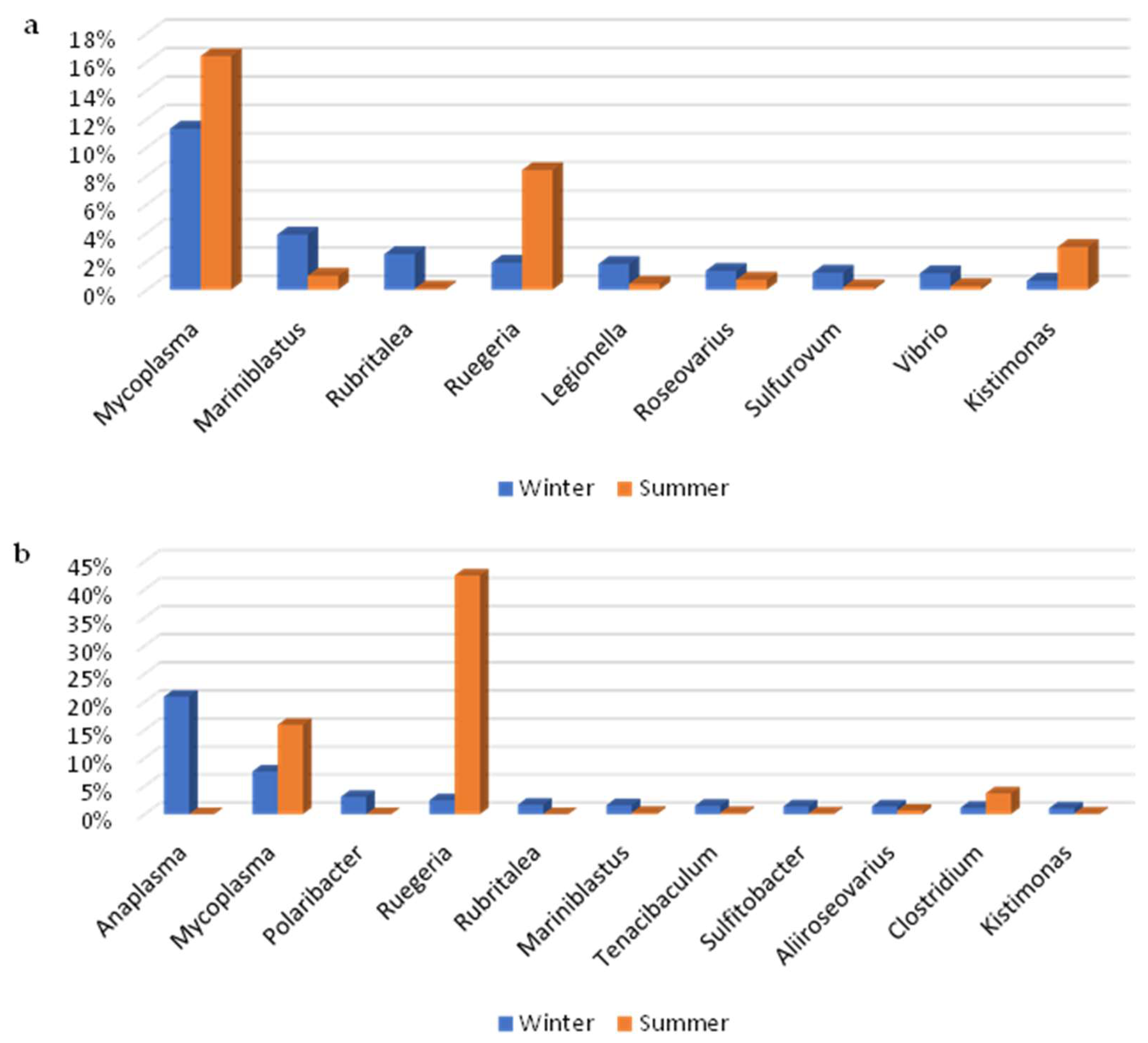
3.2. Metagenomic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Buer, A.L.; Maar, M.; Nepf, M.; Ritzenhofen, L.; Dahlke, S.; Friedland, R.; Krost, P.; Peine, F.; Schernewski, G. Potential and feasibility of Mytilus spp. farming along a salinity gradient. Front. Mar. Sci. 2020, 7, 371. [Google Scholar] [CrossRef]
- Bozcal, E.; Dagdeviren, M. Bacterial metagenome analysis of Mytilus galloprovincialis collected from Istanbul and Izmir coastal stations of Turkey. Environ. Monit. Assess. 2020, 192, 186. [Google Scholar] [CrossRef] [PubMed]
- Musella, M.; Wathsala, R.; Tavella, T.; Rampelli, S.; Barone, M.; Palladino, G.; Biagi, E.; Brigidi, P.; Turroni, S.; Franzellitti, S.; et al. Tissue-scale microbiota of the Mediterranean mussel (Mytilus galloprovincialis) and its relationship with the environment. Sci. Total Environ. 2020, 717, 137209. [Google Scholar] [CrossRef]
- Figueras, A.; Moreira, R.; Sendra, M.; Novoa, B. Genomics and immunity of the Mediterranean mussel Mytilus galloprovincialis in a changing environment. Fish Shellfish Immunol. 2019, 90, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Dumbauld, B.R.; Ruesink, J.L.; Rumsrill, S.S. The ecological role of bivalve shellfish aquaculture in the estuarine environment: A review with application to oyster and clam culture in West Coast (USA) estuaries. Aquaculture 2009, 290, 196–223. [Google Scholar] [CrossRef]
- Li, L.L.; Amara, R.; Souissi, S.; Dehaut, A.; Duflos, G.; Monchy, S. Impacts of microplastics exposure on mussel (Mytilus edulis) gut microbiota. Sci. Total Environ. 2020, 745, 141018. [Google Scholar] [CrossRef]
- Pierce, M.L.; Ward, J.E. Microbial ecology of the bivalvia, with an emphasis on the family ostreidae. J. Shellfish Res. 2018, 37, 793–806. [Google Scholar] [CrossRef]
- Paillard, C.; Gueguen, Y.; Wegner, K.M.; Bass, D.; Pallavicini, A.; Vezzulli, L.; Arzul, I. Recent advances in bivalve-microbiota interactions for disease prevention in aquaculture. Curr. Opin. Biotechnol. 2022, 73, 225–232. [Google Scholar] [CrossRef]
- Wathsala, R.H.G.R.; Musella, M.; Valbonesi, P.; Candela, M.; Franzellitti, S. Variability of metabolic, protective, antioxidant, and lysosomal gene transcriptional profiles and microbiota composition of Mytilus galloprovincialis farmed in the North Adriatic Sea (Italy). Mar. Pollut. Bull. 2021, 172, 112847. [Google Scholar] [CrossRef]
- Voultsiadou, E.; Ampatzopoulos, T.; Antonopoulou, E.; Gkanias, K.; Gkelis, S.; Staikou, A.; Triantafyllidis, A. Bivalve Culture. Aquaculture—Organisms, Production Systems, Perspectives; Kallipos, Open Academic Editions: Thessaloniki, Greece, 2015; pp. 209–239. Available online: http://hdl.handle.net/11419/5083 (accessed on 7 November 2022).
- Karageorgis, A.P.; Skourtos, M.S.; Kapsimalis, V.; Kontogianni, A.D.; Skoulikidis, N.T.; Pagou, K.; Nikolaidis, N.P.; Drakopoulou, P.; Zanou, B.; Karamanos, H.; et al. An integrated approach to watershed management within the DPSIR framework: Axios River catchment and Thermaikos Gulf. Reg. Environ. Change 2005, 5, 138–160. [Google Scholar] [CrossRef]
- Androulidakis, Y.; Kolovoyiannis, V.; Makris, C.; Krestenitis, Y.; Baltikas, V.; Stefanidou, N.; Chatziantoniou, A.; Topouzelis, K.; Moustaka-Gouni, M. Effects of ocean circulation on the eutrophication of a Mediterranean gulf with river inlets: The Northern Thermaikos Gulf. Cont. Shelf Res. 2021, 221, 104416. [Google Scholar] [CrossRef]
- Figdor, D.; Gulabivala, K. Survival against the odds: Microbiology of root canals associated with post-treatment disease. Endod. Top. 2008, 18, 62–77. [Google Scholar] [CrossRef]
- Fricke, W.F.; Cebula, T.A.; Ravel, J. Chapter 28—Genomics. In Microbial Forensics, 2nd ed.; Budowle, B., Schutzer, S.E., Breeze, R.G., Keim, P.S., Morse, S.A., Eds.; Academic Press: San Diego, CA, USA, 2011; pp. 479–492. [Google Scholar] [CrossRef]
- Gensberger, E.; Gössl, E.; Antonielli, L.; Sessitsch, A.; Kostić, T. Effect of different heterotrophic plate count methods on the estimation of the composition of the culturable microbial community. PeerJ 2015, 3, e862. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, I.; Vadstein, O. Evaluation of plate count methods for determination of maximum specific growth rate in mixed microbial communities, and its possible application for diversity assessment. J. Appl. Microbiol. 2000, 88, 442–448. [Google Scholar] [CrossRef]
- Schloss, P.D.; Handelsman, J. Metagenomics for studying unculturable microorganisms: Cutting the Gordian knot. Genome Biol. 2005, 6, 229. [Google Scholar] [CrossRef]
- Auguste, M.; Lasa, A.; Balbi, T.; Pallavicini, A.; Vezzulli, L.; Canesi, L. Impact of nanoplastics on hemolymph immune parameters and microbiota composition in Mytilus galloprovincialis. Mar. Environ. Res. 2020, 159, 105017. [Google Scholar] [CrossRef]
- Balbi, T.; Vezzulli, L.; Lasa, A.; Pallavicini, A.; Canesi, L. Insight into the microbial communities associated with first larval stages of Mytilus galloprovincialis: Possible interference by estrogenic compounds. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2020, 237, 108833. [Google Scholar] [CrossRef]
- Vezzulli, L.; Stagnaro, L.; Grande, C.; Tassistro, G.; Canesi, L.; Pruzzo, C. Comparative 16SrDNA gene-based microbiota profiles of the Pacific oyster (Crassostrea gigas) and the Mediterranean mussel (Mytilus galloprovincialis) from a shellfish farm (Ligurian Sea, Italy). Microb. Ecol. 2018, 75, 495–504. [Google Scholar] [CrossRef]
- Winand, R.; Bogaerts, B.; Hoffman, S.; Lefevre, L.; Delvoye, M.; Van Braekel, J.; Fu, Q.; Roosens, N.H.; De Keersmaecker, S.C.; Vanneste, K. Targeting the 16S rRNA gene for bacterial identification in complex mixed samples: Comparative evaluation of second (Illumina) and third (Oxford Nanopore Technologies) generation sequencing technologies. Int. J. Mol. Sci. 2020, 21, 298. [Google Scholar] [CrossRef]
- Maestri, S. Development of Novel Bioinformatic Pipelines for MinION-Based DNA Barcoding. Ph.D. Thesis, Università degli Studi di Verona, Verona, Italy, 2021. Available online: https://iris.univr.it/retrieve/handle/11562/1042782/205364/ (accessed on 19 November 2022).
- Maestri, S.; Cosentino, E.; Paterno, M.; Freitag, H.; Garces, J.M.; Marcolungo, L.; Alfano, M.; Njunjić, I.; Schilthuizen, M.; Slik, F.; et al. A rapid and accurate MinION-based workflow for tracking species biodiversity in the field. Genes 2019, 10, 468. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Techn. J. 1948, 27, 379–423, 623–656. [Google Scholar] [CrossRef]
- Kalaitzidou, M.P.; Alvanou, M.V.; Papageorgiou, K.V.; Lattos, A.; Sofia, M.; Kritas, S.K.; Petridou, E.; Giantsis, I.A. Pollution indicators and HAB-associated halophilic bacteria alongside harmful cyanobacteria in the largest mussel cultivation area in Greece. Int. J. Environ. Res. Public Health 2022, 19, 5285. [Google Scholar] [CrossRef] [PubMed]
- Polymenakou, P.N.; Bertilsson, S.; Tselepides, A.; Stephanou, E.G. Bacterial community composition in different sediments from the eastern mediterranean sea: A Comparison of four 16S ribosomal DNA clone libraries. Microbial Ecol. 2005, 50, 447–462. [Google Scholar] [CrossRef]
- King, G.M.; Judd, C.; Kuske, C.R.; Smith, C. Analysis of stomach and gut microbiomes of the eastern oyster (Crassostrea virginica) from Coastal Louisiana, USA. PLoS ONE 2012, 7, e51475. [Google Scholar] [CrossRef]
- Lokmer, A.; Kuenzel, S.; Baines, J.F.; Wegner, K.M. The role of tissue-specific microbiota in initial establishment success of Pacific oysters. Environ. Microbiol. 2016, 18, 970–987. [Google Scholar] [CrossRef]
- Arfken, A.; Song, B.; Bowman, J.S.; Piehler, M. Denitrification potential of the eastern oyster microbiome using a 16S rRNA gene based metabolic inference approach. PLoS ONE 2017, 12, e0185071. [Google Scholar] [CrossRef] [PubMed]
- Lattos, A.; Giantsis, I.A.; Karagiannis, D.; Theodorou, J.A.; Michaelidis, B. Gut symbiotic microbial communities in the IUCN critically endangered pinna nobilis suffering from mass mortalities, revealed by 16S rRNA amplicon NGS. Pathogens 2020, 9, 1002. [Google Scholar] [CrossRef]
- Cano, I.; Ryder, D.; Webb, S.C.; Jones, B.J.; Brosnahan, C.L.; Carrasco, N.; Bodinier, B.; Furones, D.; Pretto, T.; Carella, F.; et al. Cosmopolitan distribution of endozoicomonas-like organisms and other intracellular microcolonies of bacteria causing infection in marine mollusks. Front. Microbiol. 2020, 11, 577481. [Google Scholar] [CrossRef]
- Arahal, D.R.; Lucena, T.; Rodrigo-Torres, L.; Pujalte, M.J. Ruegeria denitrificans sp. nov. a marine bacterium in the family Rhodobacteraceae with the potential ability for cyanophycin synthesis. Int. J. Syst. Evol. Microbiol. 2018, 68, 2515–2522. [Google Scholar] [CrossRef]
- Santisi, S.; Genovese, M.; Bonsignore, M.; Fiumara, E.; Maricchiolo, G.; Mancuso, M.; Genovese, L.; Giuliano, L.; Cappello, S. Study of bacterial communities in mussel Mytilus galloprovincialis by a combination of 16S crDNA and 16S rDNA sequencing. Int. J. Microbiol. Appl. 2015, 2, 18. [Google Scholar]
- Lage, O.M.; Albuquerque, L.; Lobo-da Cunha, A.; da Costa, M.S. Mariniblastus fucicola gen. nov. sp. nov. a novel planctomycete associated with macroalgae. Int. J. Syst. Evol. Microbiol. 2017, 67, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Faria, M.; Bordin, N.; Kizina, J.; Harder, J.; Devos, D.; Lage, O.M. Planctomycetes attached to algal surfaces: Insight into their genomes. Genomics 2018, 110, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Mannas, H.; Mimouni, R.; Chaouqy, N.; Hamadi, F.; Martinez-Urtaza, J. Occurrence of vibrio and Salmonella species in mussels (Mytilus galloprovincialis) collected along the Moroccan Atlantic coast. SpringerPlus 2014, 3, 265. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Xu, J.K.; Chen, Y.W.; Ding, W.Y.; Shao, A.Q.; Liang, X.; Zhu, Y.T.; Yang, J.L. Characterization of gut microbiome in the mussel Mytilus galloprovincialis in response to thermal stress. Front. Physiol. 2019, 10, 1086. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Number of Mussels | Zone | Region | Month | Collection Depth (m) | Water Surface Temperature (°C) | Salinity (psu) |
|---|---|---|---|---|---|---|---|
| M1 | 10 | Z1M | Makrigialos | December | 3–4 | 15.7 | 37–38 |
| M2 | 12 | Z2M | |||||
| M3 | 12 | Z1M | Makrigialos | January | 3–4 | 14.0 | 37–38 |
| M4 | 12 | Z2M | |||||
| M5 | 12 | Z1M | Makrigialos | February | 3–4 | 11.7 | 37–38 |
| M6 | 12 | Z2M | |||||
| M7 | 11 | Z1C | Chalastra | February | 4–8 | 11.0 | 37–38 |
| M8 | 12 | Z2C | |||||
| M9 | 12 | Z3C | |||||
| M10 | 12 | Z1C | Chalastra | February | 4–8 | 12.1 | 37–38 |
| Μ11 | 12 | Z2C | |||||
| Μ13 | 12 | Z1C | Chalastra | July | 36–37 | ||
| Μ14 | 11 | Z2C | 4–8 | 26.0 | |||
| Μ15 | 12 | Z3C | |||||
| Μ16 | 12 | Z2M | Makrigialos | August | 3–4 | 27.5 | 36–37 |
| Sample | M1 | M2 | M3 | M4 | M5 | M6 | M7 | M8 |
| Barcode | BC01 | BC02 | BC03 | BC04 | BC07 | BC08 | BC05 | BC06 |
| Sample | M9 | M10 | M11 | M13 | M14 | M15 | M16 | |
| Barcode | BC09 | BC10 | BC11 | BC13 | BC14 | BC15 | BC16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoinas, K.; Konstantou, V.; Bompou, E.; Floros, G.; Chatziplis, D.; Imsiridou, A.; Loukovitis, D. Microbiome Profile of the Mediterranean Mussel (Mytilus galloprovincialis) from Northern Aegean Sea (Greece) Culture Areas, Based on a 16S rRNA Next Generation Sequencing Approach. Diversity 2023, 15, 463. https://doi.org/10.3390/d15030463
Schoinas K, Konstantou V, Bompou E, Floros G, Chatziplis D, Imsiridou A, Loukovitis D. Microbiome Profile of the Mediterranean Mussel (Mytilus galloprovincialis) from Northern Aegean Sea (Greece) Culture Areas, Based on a 16S rRNA Next Generation Sequencing Approach. Diversity. 2023; 15(3):463. https://doi.org/10.3390/d15030463
Chicago/Turabian StyleSchoinas, Konstantinos, Vasiliki Konstantou, Emmanouela Bompou, George Floros, Dimitrios Chatziplis, Anastasia Imsiridou, and Dimitrios Loukovitis. 2023. "Microbiome Profile of the Mediterranean Mussel (Mytilus galloprovincialis) from Northern Aegean Sea (Greece) Culture Areas, Based on a 16S rRNA Next Generation Sequencing Approach" Diversity 15, no. 3: 463. https://doi.org/10.3390/d15030463
APA StyleSchoinas, K., Konstantou, V., Bompou, E., Floros, G., Chatziplis, D., Imsiridou, A., & Loukovitis, D. (2023). Microbiome Profile of the Mediterranean Mussel (Mytilus galloprovincialis) from Northern Aegean Sea (Greece) Culture Areas, Based on a 16S rRNA Next Generation Sequencing Approach. Diversity, 15(3), 463. https://doi.org/10.3390/d15030463