
First Genomic Survey of Pleurocryptella fimbriata Provides Preliminary Insights into Genome Characteristics and Evolution of a Deep-Sea Parasitic Isopod


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, Library Construction, and Sequencing
2.3. K-Mer Analysis and Genome Assembly
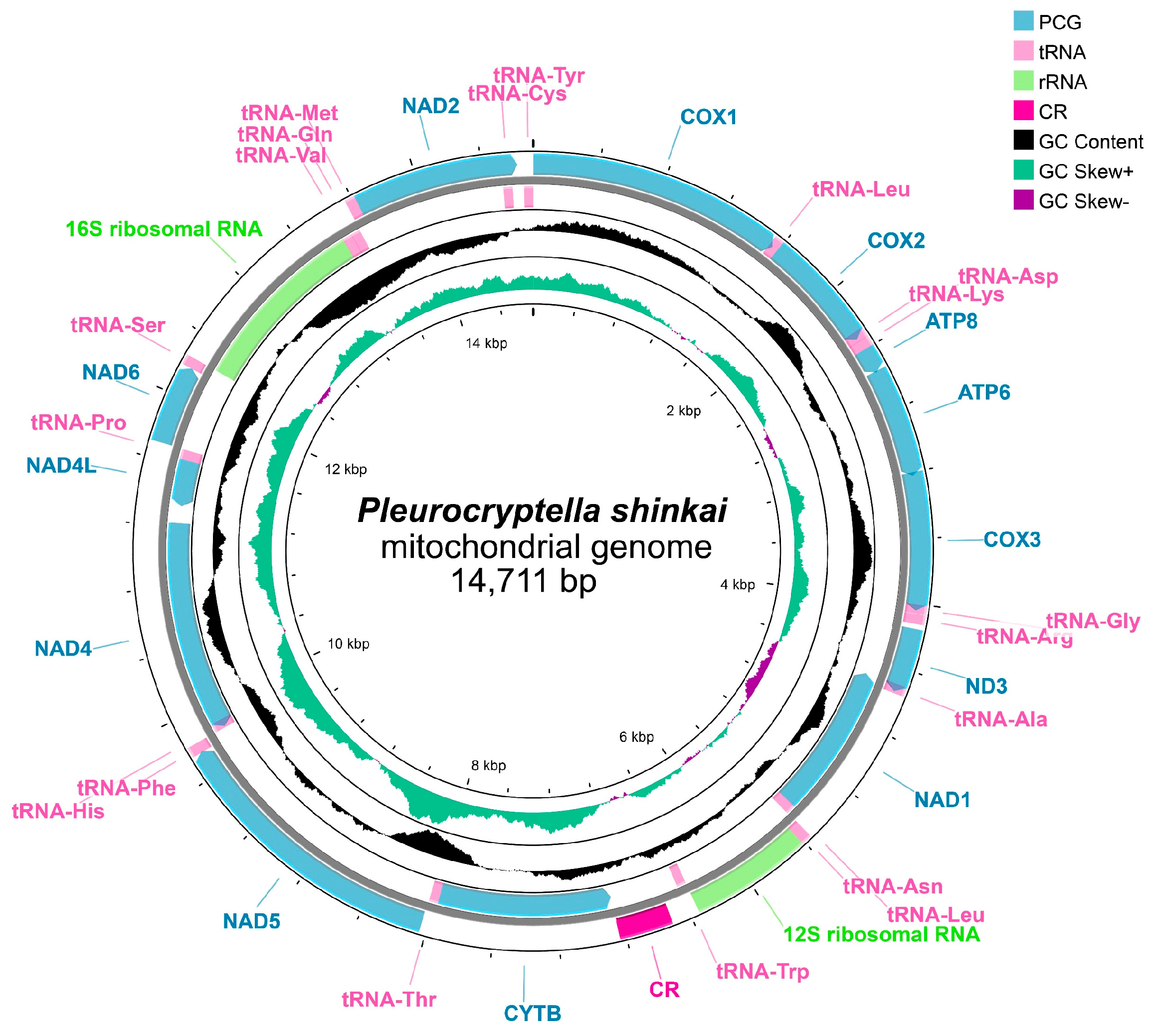
2.4. Mitogenome Assembly and SNP Screening
2.5. Phylogenetic Analysis
2.6. Positive Selection Analysis
3. Results
3.1. Characteristics of P. fimbriata and S. crosnieri Specimens
3.2. Sequencing and Quality Evaluation
3.3. K-Mer-Analysis-Based Genome Characteristics
3.4. Genome Assembly Statistics
3.5. Mitogenome Features and Candidate SNPs
3.6. Phylogenetic and Selective Pressure Analyses of Mitochondrial PCGs
4. Discussion
4.1. Characteristics of the Parasitic P. fimbriata Genome
4.2. Phylogeny-Based Insights into the Origin and Evolution of Bopyrids
4.3. Adaptations of the Mitochondrial Genome to Deep-Sea Environments in P. fimbriata
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, J.D.; Boyko, C.B. The global diversity of parasitic isopods associated with crustacean hosts (Isopoda: Bopyroidea and Cryptoniscoidea). PLoS ONE 2012, 7, e35350. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xing, Y.; Chen, X.; Zhong, S.; Pengsakul, T.; Qiao, Y. Integration of transcriptomic and metabolomic analyses reveal the molecular responses of the mud crab Scylla paramamosain to infection by an undescribed endoparasite Portunion sp. Fish. Shellfish. Immunol. 2023, 140, 108978. [Google Scholar] [CrossRef]
- Niza, N.N.M.; Yusoff, N.A.H.; Tosin, O.V.; Norhan, N.A.-S.; Husin, N.M.; Ikhwanuddin, M.; Abdullah, F.; Ishak, A.N.; Hassan, M. Prevalence, molecular characterization and the effects of boyprid isopod Probopyrus buitendijki infestation on the Macrobrachium rosenbergii from Nyatoh River, Terengganu, Malaysia. Reg. Stud. Mar. Sci. 2024, 69, 103311. [Google Scholar] [CrossRef]
- Miranda, I.; Mantelatto, F. Temporal dynamic of the relationship between the parasitic isopod Aporobopyrus curtatus (Crustacea: Isopoda: Bopyridae) and the anomuran crab Petrolisthes armatus (Crustacea: Decapoda: Porcellanidae) in southern Brazil. Lat. Am. J. Aquat. Res. 2010, 38, 210–217. [Google Scholar] [CrossRef]
- de Barros, M.S.F.; da Silva Neto, L.S.; Calado, T.C.d.S. First record of parasitism by Probopyrus pandalicola (Isopoda, Bopyridae) on the freshwater prawn Macrobrachium acanthurus (Decapoda, Palaemonidae) and ecological interactions. J. Parasit. Dis. 2021, 45, 273–278. [Google Scholar] [CrossRef]
- Wu, R.; Guo, R.; Xi, Q.; Paulay, G.; An, J. Phylogenetic position of Bopyroides hippolytes, with comments on the rearrangement of the mitochondrial genome in isopods (Isopoda: Epicaridea: Bopyridae). BMC Genom. 2022, 23, 253. [Google Scholar] [CrossRef]
- Boyko, C.B.; Moss, J.; Williams, J.D.; Shields, J.D. A molecular phylogeny of Bopyroidea and Cryptoniscoidea (Crustacea: Isopoda). Syst. Biodivers. 2013, 11, 495–506. [Google Scholar] [CrossRef]
- Williams, J.D.; Horch, A.P.; Ceballos, A.; Bracken-Grissom, H. Resurrection of the parasitic isopod genus Cryptione Hansen, 1897 (Epicaridea: Bopyridae) and description of a new species of parasitic isopod (Epicaridea: Bopyridae) from the deep-sea shrimp Notostomus gibbosus (Caridea: Acanthephyridae) with an analysis of its phylogenetic position based on molecular data. Mar. Biodivers. 2024, 54, 86. [Google Scholar] [CrossRef]
- An, J.; Yin, X.; Chen, R.; Boyko, C.B.; Liu, X. Integrative taxonomy of the subfamily Orbioninae Codreanu, 1967 (Crustacea: Isopoda) based on mitochondrial and nuclear data with evidence that supports Epicaridea Latreille, 1825 as a suborder. Mol. Phylogenetics Evol. 2023, 180, 107681. [Google Scholar] [CrossRef]
- Markham, J.C. Six new species of bopyrid isopods parasitic on galatheid crabs of the genus Munida in the western Atlantic. Bull. Mar. Sci. 1974, 23, 613–648. [Google Scholar]
- He, X.; Wang, H.; Xu, T.; Zhang, Y.; Chen, C.; Sun, Y.; Qiu, J.W.; Zhou, Y.; Sun, J. Genomic analysis of a scale worm provides insights into its adaptation to deep-sea hydrothermal vents. Genome Biol. Evol. 2023, 15, evad125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, S.; Liu, J.; Guo, Q.; Meng, L.; Chen, J.; Xiang, X.; Zhou, Y.; Zhang, N.; Liu, H.; et al. The amphipod genome reveals population dynamics and adaptations to hadal environment. Cell 2025, 188, 1378–1392.e1318. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, X.; Kou, Q.; Sun, Y.; Liu, C.; Li, S.; Yu, Y.; Zhang, C.; Jin, S.; Xiang, J.; et al. Genome of a giant isopod, Bathynomus jamesi, provides insights into body size evolution and adaptation to deep-sea environment. BMC Biol. 2022, 20, 113. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Cheng, J.; Xin, Q.; Sha, Z.; Hui, M. A first genome survey sequencing of alvinocaridid shrimp Shinkaicaris leurokolos in deep-sea hydrothermal vent environment. J. Mar. Biol. Assoc. UK 2023, 103, e65. [Google Scholar] [CrossRef]
- Hui, M.; Zhang, Y.; Wang, A.; Sha, Z. The first genome survey of the snail Provanna glabra inhabiting deep-sea hydrothermal vents. Animals 2023, 13, 3313. [Google Scholar] [CrossRef]
- Huang, Y.; Bian, C.; Liu, Z.; Wang, L.; Xue, C.; Huang, H.; Yi, Y.; You, X.; Song, W.; Mao, X.; et al. The first genome survey of the Antarctic krill (Euphausia superba) provides a valuable genetic resource for polar biomedical research. Mar. Drugs 2020, 18, 185. [Google Scholar] [CrossRef] [PubMed]
- Bravo, H.; Baeza, J.A.; van der Meij, S.E.T. Genomic survey sequencing and complete mitochondrial genome of the elkhorn coral crab Domecia acanthophora (Desbonne in Desbonne & Schramm, 1867) (Decapoda: Brachyura: Domeciidae). J. Crust. Biol. 2023, 43, 1–9. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. Available online: http://genomescope.org/ (accessed on 9 January 2025). [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, e18. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. Available online: https://chlorobox.mpimp-golm.mpg.de/geseq.html (accessed on 22 January 2025). [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Markham, J.C. New species and records of Bopyridae (Crustacea: Isopoda) infesting species of the genus Upogebia (Crustacea: Decapoda: Upogebiidae): The genera Orthione Markham, 1988, and Gyge Cornalia & Panceri, 1861. Proc. Biol. Soc. Wash. 2004, 117, 186–198. [Google Scholar]
- Yu, J.; An, J.; Li, Y.; Boyko, C.B. The first complete mitochondrial genome of a parasitic isopod supports Epicaridea Latreille, 1825 as a suborder and reveals the less conservative genome of isopods. Syst. Parasitol. 2018, 95, 465–478. [Google Scholar] [CrossRef]
- Brockerhoff, A.M. Occurrence of the internal parasite Portunion sp. (Isopoda: Entoniscidae) and its effect on reproduction in intertidal crabs (Decapoda: Grapsidae) from New Zealand. J. Parasitol. 2004, 90, 1338–1344. [Google Scholar] [CrossRef]
- Quinn, J.; Lee, S.; Greeley, D.; Gehman, A.; Kuris, A.M.; Wood, C.L. Long-term change in the parasite burden of shore crabs (Hemigrapsus oregonensis and Hemigrapsus nudus) on the northwestern Pacific coast of North America. Proc. R. Soc. B Biol. Sci. 2021, 288, 20203036. [Google Scholar] [CrossRef]
- Hua, C.J.; Li, W.X.; Zhang, D.; Zou, H.; Li, M.; Jakovlić, I.; Wu, S.G.; Wang, G.T. Basal position of two new complete mitochondrial genomes of parasitic Cymothoida (Crustacea: Isopoda) challenges the monophyly of the suborder and phylogeny of the entire order. Parasites Vectors 2018, 11, 628. [Google Scholar] [CrossRef]
- Öktener, A.; Trilles, J.P.; Alaş, A. Elthusa poutassouiensis (Penso, 1939), comb, nov. (Isopoda, Cymothoidae) for Meinertia (Ceratothoa) poutassouiensis, parasite of the blue whiting, Micromesistius poutassou. Bull. Eur. Assoc. Fish. Pathol. 2018, 38, 12–23. [Google Scholar]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. Available online: https://itol.embl.de/ (accessed on 29 January 2025). [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar] [CrossRef]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef]
- Yang, Z.; Wong, W.S.W.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Lü, Z.; Liu, Y.; Zhao, S.; Fang, J.; Zhu, K.; Liu, J.; Gong, L.; Liu, L.; Liu, B. Amblyopinae mitogenomes provide novel insights into the paraphyletic origin of their adaptation to mudflat habitats. Int. J. Mol. Sci. 2023, 24, 4362. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Song, L.; Jin, C.; Li, M.; Gong, S.; Wang, Y. Genome survey and SSR analysis of Apocynum venetum. Biosci. Rep. 2019, 39, BSR20190146. [Google Scholar] [CrossRef]
- Aird, D.; Ross, M.G.; Chen, W.S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.G.E.; Kurland, C.G. Reductive evolution of resident genomes. Trends Microbiol. 1998, 6, 263–268. [Google Scholar] [CrossRef]
- Mira, A.; Ochman, H.; Moran, N.A. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001, 17, 589–596. [Google Scholar] [CrossRef]
- Opperman, C.H.; Bird, D.M.; Williamson, V.M.; Rokhsar, D.S.; Burke, M.; Cohn, J.; Cromer, J.; Diener, S.; Gajan, J.; Graham, S.; et al. Sequence and genetic map of Meloidogyne hapla: A compact nematode genome for plant parasitism. Proc. Natl. Acad. Sci. USA 2008, 105, 14802–14807. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Shoura, M.J.; Artiles, K.L.; Jeong, D.-E.; Owa, C.; Kobayashi, H.; Suzuki, Y.; Kanamori, M.; Toyoshima, Y.; Iino, Y.; et al. CGC1, a new reference genome for Caenorhabditis elegans. bioRxiv 2024. 2024.2012.2004.626850. [Google Scholar] [CrossRef]
- Slyusarev, G.S.; Starunov, V.V.; Bondarenko, A.S.; Zorina, N.A.; Bondarenko, N.I. Extreme genome and nervous system streamlining in the invertebrate parasite Intoshia variabili. Curr. Biol. 2020, 30, 1292–1298.e1293. [Google Scholar] [CrossRef]
- Ryan Gregory, T. Genome size and developmental complexity. Genetica 2002, 115, 131–146. [Google Scholar] [CrossRef]
- Leckenby, A.; Hall, N. Genomic changes during evolution of animal parasitism in eukaryotes. Curr. Opin. Genet. Dev. 2015, 35, 86–92. [Google Scholar] [CrossRef]
- Totikov, A.; Tomarovsky, A.; Prokopov, D.; Yakupova, A.; Bulyonkova, T.; Derezanin, L.; Rasskazov, D.; Wolfsberger, W.W.; Koepfli, K.-P.; Oleksyk, T.K.; et al. Chromosome-level genome assemblies expand capabilities of genomics for conservation biology. Genes 2021, 12, 1336. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.D.; Boyko, C.B.; Marin, I.N. A new species and depth record of bopyrid (Crustacea, Isopoda) from a squat lobster in the Kuril-Kamchatka Trench. Eur. J. Taxon. 2020, 724, 122–133. [Google Scholar] [CrossRef]
- Shiino, S.M. Phylogeny of the genera within the family Bopyridae. Bull. Du Muséum Natl. d’Hist. Nat. 1965, 37, 462–465. [Google Scholar]
- Rodríguez-Flores, P.C.; Macpherson, E.; Schnabel, K.E.; Ahyong, S.T.; Corbari, L.; Machordom, A. Depth as a driver of evolution and diversification of ancient squat lobsters (Decapoda, Galatheoidea, Phylladiorhynchus). Mol. Phylogenetics Evol. 2022, 171, 107467. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.e.; Sha, Z.; Wang, Y. Phylogenetic position of Alvinocarididae (Crustacea: Decapoda: Caridea): New insights into the origin and evolutionary history of the hydrothermal vent alvinocarid shrimps. Deep. Sea Res. Part I 2018, 141, 93–105. [Google Scholar] [CrossRef]
- Lindner, A.; Cairns, S.D.; Cunningham, C.W. From offshore to onshore: Multiple origins of shallow-water corals from deep-sea ancestors. PLoS ONE 2008, 3, e2429. [Google Scholar] [CrossRef]
- Bribiesca-Contreras, G.; Verbruggen, H.; Hugall, A.F.; O’Hara, T.D. The importance of offshore origination revealed through ophiuroid phylogenomics. Proc. R. Soc. B-Biol. Sci. 2017, 284, 20170160. [Google Scholar] [CrossRef]
- Hourdez, S.; Lallier, F.H. Adaptations to hypoxia in hydrothermal-vent and cold-seep invertebrates. Rev. Environ. Sci. Bio 2007, 6, 143–159. [Google Scholar] [CrossRef]
- Dong, X.; Rattray, J.E.; Campbell, D.C.; Webb, J.; Chakraborty, A.; Adebayo, O.; Matthews, S.; Li, C.; Fowler, M.; Morrison, N.M.; et al. Thermogenic hydrocarbon biodegradation by diverse depth-stratified microbial populations at a Scotian Basin cold seep. Nat. Commun. 2020, 11, 5825. [Google Scholar] [CrossRef]
- Orphan, V.J.; House, C.H.; Hinrichs, K.-U.; McKeegan, K.D.; DeLong, E.F. Multiple archaeal groups mediate methane oxidation in anoxic cold seep sediments. Proc. Natl. Acad. Sci. USA 2002, 99, 7663–7668. [Google Scholar] [CrossRef]
- Yu, L.; Wang, X.; Ting, N.; Zhang, Y. Mitogenomic analysis of Chinese snub-nosed monkeys: Evidence of positive selection in NADH dehydrogenase genes in high-altitude adaptation. Mitochondrion 2011, 11, 497–503. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, R.R.; Johnson, W.E.; O’Brien, S.J.; Ramos, M.J.; Antunes, A. The adaptive evolution of the mammalian mitochondrial genome. BMC Genom. 2008, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, A.; Ropiquet, A.; Couloux, A.; Cruaud, C. Evolution of the mitochondrial genome in mammals living at high altitude: New insights from a study of the tribe caprini (Bovidae, Antilopinae). J. Mol. Evol. 2009, 68, 293–310. [Google Scholar] [CrossRef]
- Yang, M.; Dong, D.; Li, X. The complete mitogenome of Phymorhynchus sp. (Neogastropoda, Conoidea, Raphitomidae) provides insights into the deep-sea adaptive evolution of Conoidea. Ecol. Evol. 2021, 11, 7518–7531. [Google Scholar] [CrossRef]
- Zhang, K.; Sun, J.; Xu, T.; Qiu, J.W.; Qian, P.Y. Phylogenetic relationships and adaptation in deep-sea mussels: Insights from mitochondrial genomes. Int. J. Mol. Sci. 2021, 22, 1900. [Google Scholar] [CrossRef]
- Yang, M.; Gong, L.; Sui, J.; Li, X. The complete mitochondrial genome of Calyptogena marissinica (Heterodonta: Veneroida: Vesicomyidae): Insight into the deep-sea adaptive evolution of vesicomyids. PLoS ONE 2019, 14, e0217952. [Google Scholar] [CrossRef]
- Sun, S.e.; Sha, Z.; Wang, Y. The complete mitochondrial genomes of two vent squat lobsters, Munidopsis lauensis and M. verrilli: Novel gene arrangements and phylogenetic implications. Ecol. Evol. 2019, 9, 12390–12407. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.e.; Hui, M.; Wang, M.; Sha, Z. The complete mitochondrial genome of the alvinocaridid shrimp Shinkaicaris leurokolos (Decapoda, Caridea): Insight into the mitochondrial genetic basis of deep-sea hydrothermal vent adaptation in the shrimp. Comp. Biochem. Physiol. D-Genom. Proteom. 2018, 25, 42–52. [Google Scholar] [CrossRef]
- Luo, Y.; Gao, W.; Gao, Y.; Tang, S.; Huang, Q.; Tan, X.; Chen, J.; Huang, T. Mitochondrial genome analysis of Ochotona curzoniae and implication of cytochrome c oxidase in hypoxic adaptation. Mitochondrion 2008, 8, 352–357. [Google Scholar] [CrossRef]
- Ramos, N.I.; DeLeo, D.M.; Horowitz, J.; McFadden, C.S.; Quattrini, A.M. Selection in coral mitogenomes, with insights into adaptations in the deep sea. Sci. Rep. 2023, 13, 6016. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Host | Origin | Taxonomic Status | Length (bp) | Accession Number |
|---|---|---|---|---|---|
| Pleurocryptella fimbriata | Munidopsidae [10] | Deep-sea cold seep | Pleurocryptellinae | 14,711 | PV243289 |
| Orthione mesoamericana | Upogebiidae [29] | Shallow sea | Pseudioninae | 14,377 | MG729627 |
| Gyge ovalis | Upogebiidae [30] | Shallow sea | Pseudioninae | 14,268 | KY038053 |
| Aparapenaeon japonica | Penaeoidea [9] | Shallow sea | Orbioninae | 14,360 | MK886810 |
| Parapenaeon diatropa | Penaeoidea [9] | Shallow sea | Orbioninae | 14,405 | MG753993 |
| Parasymmetrorbione bicauda | Penaeoidea [9] | Shallow sea | Orbioninae | 14,284 | MK886808 |
| Parapenaeonella distincta | Penaeoidea [9] | Shallow sea | Orbioninae | 14,369 | MH084345 |
| Parapenaeonella expansa | Penaeoidea [9] | Shallow sea | Orbioninae | 14,384 | MG807478 |
| Portunion sp. | Brachyura [2,31] | Shallow sea | Entioninae | 14,604 | OL677861 |
| Portunion conformis | Brachyura [32] | Shallow sea | Entioninae | 14,210 | OP829302 |
| Ichthyoxenos japonensis | Teleostei [33] | Fresh water | Cymothoidae | 15,440 | NC039713 |
| Elthusa poutassouiensis | Teleostei [34] | Shallow and deep sea | Cymothoidae | 15,223 | OR800751 |
| Total Length (bp) | Total Number | Maximum Length (bp) | N50 Length (bp) | GC Content (%) | |
|---|---|---|---|---|---|
| Contig | 888,848,407 | 2,289,701 | 137,550 | 726 | 35.09 |
| Scaffold | 927,035,547 | 1,960,944 | 266,030 | 989 | 35.09 |
| Gene | Start Position | Stop Position | Intergenic Length | Start Code | Stop Code | Size (bp) | Strand |
|---|---|---|---|---|---|---|---|
| COX1 | 1 | 1539 | 0 | ACG | TTG | 1539 | + |
| tRNA-Leu (TAA) | 1538 | 1595 | −2 | 58 | + | ||
| COX2 | 1596 | 2276 | 0 | ATT | GGT | 681 | + |
| tRNA-Lys (TTT) | 2277 | 2340 | 0 | 64 | + | ||
| tRNA-Asp (GTC) | 2339 | 2397 | −2 | 59 | + | ||
| ATP8 | 2398 | 2553 | 0 | GTG | TAA | 156 | + |
| ATP6 | 2547 | 3218 | −7 | GTG | TAA | 672 | + |
| COX3 | 3200 | 4000 | −19 | ATA | AGT | 801 | + |
| tRNA-Gly (TCC) | 4001 | 4058 | 0 | 58 | + | ||
| tRNA-Arg (TCG) | 4061 | 4119 | 2 | 59 | + | ||
| NAD3 | 4150 | 4497 | 30 | TTG | GAA | 348 | + |
| 4498 | 4557 | 0 | 60 | + | |||
| NAD1 | 4572 | 5492 | 14 | GTG | TAA | 921 | _ |
| tRNA-Leu (TAG) | 5493 | 5554 | 0 | 62 | _ | ||
| tRNA-Asn (GTT) | 5550 | 5613 | −5 | 64 | + | ||
| 12S rRNA | 5614 | 6347 | 0 | 734 | + | ||
| tRNA-Trp (TCA) | 6348 | 6407 | 0 | 60 | _ | ||
| Control region | 6408 | 6838 | 0 | 431 | + | ||
| CYTB | 6839 | 7975 | 0 | ATT | TAG | 1137 | _ |
| tRNA-Thr (TGT) | 7976 | 8034 | 0 | 59 | _ | ||
| NAD5 | 8039 | 9763 | 4 | ATA | TAG | 1725 | + |
| tRNA-Phe (GAA) | 9756 | 9819 | −8 | 64 | + | ||
| tRNA-His (GTG) | 9818 | 9876 | −2 | 59 | _ | ||
| NAD4 | 9827 | 11,218 | −50 | ATG | TAA | 1392 | _ |
| NAD4L | 11,331 | 11,633 | 112 | TTG | TAA | 303 | _ |
| tRNA-Pro (TGG) | 11,634 | 11,697 | 0 | 64 | _ | ||
| NAD6 | 11,700 | 12,185 | 2 | ATA | TAA | 486 | + |
| tRNA-Ser (TGA) | 12,184 | 12,245 | −2 | 62 | + | ||
| 16S rRNA | 12,247 | 13,430 | 1 | 1184 | _ | ||
| tRNA-Val (TAC) | 13,429 | 13,492 | −2 | 64 | _ | ||
| tRNA-Gln (TTG) | 13,491 | 13,551 | −2 | 61 | _ | ||
| tRNA-Met (CAT) | 13,557 | 13,617 | 5 | 61 | + | ||
| NAD2 | 13,618 | 14,616 | 0 | ATC | TAA | 999 | + |
| tRNA-Cys (GCA) | 14,602 | 14,652 | −15 | 51 | _ | ||
| tRNA-Tyr (GTA) | 14,652 | 14,711 | −1 | 60 | _ |
| Gene | Length (bp) | Transition | Transversion | Mutation Rates (%) | Amino Acid Change |
|---|---|---|---|---|---|
| COX1 | 1559 | 0 | 0 | 0 | |
| COX2 | 726 | 2 | 1 | 0.44 | G→W |
| ATP8 | 156 | 0 | 0 | 0 | |
| ATP6 | 672 | 1 | 2 | 0.45 | K→S; M→L |
| COX3 | 840 | 1 | 0 | 0.13 | |
| NAD3 | 396 | 1 | 0 | 0.29 | |
| NAD1 | 991 | 0 | 0 | 0 | |
| CYTB | 1137 | 1 | 1 | 0.18 | |
| NAD5 | 1725 | 3 | 1 | 0.24 | I→T |
| NAD4 | 1392 | 4 | 0 | 0.30 | |
| NAD4L | 303 | 1 | 1 | 0.66 | L→I |
| NAD6 | 486 | 1 | 0 | 0.21 | |
| NAD2 | 999 | 3 | 0 | 0.30 |
| Gene | Model | lnL | 2ΔlnL | Parameter |
|---|---|---|---|---|
| COX1 | M0 | −7860.520 | ω0 = 0.019 | |
| M2 | −7858.590 | 3.859 * | ω0 = 0.020; ω1 = 0.070 | |
| COX2 | M0 | −4311.744 | ω0 = 0.029 | |
| M2 | −4311.742 | 0.003 | ω0 = 0.029; ω1 = 0.031 | |
| ATP8 | M0 | −1127.946 | ω0 = 0.207 | |
| M2 | −1126.767 | 2.358 | ω0 = 0.224; ω1 = 0.003 | |
| ATP6 | M0 | −4851.615 | ω0 = 0.072 | |
| M2 | −4851.612 | 0.006 | ω0 = 0.072; ω1 = 0.076 | |
| COX3 | M0 | −5126.943 | ω0 = 0.057 | |
| M2 | −5125.836 | 2.214 | ω0 = 0.060; ω1 = 0.017 | |
| NAD3 | M0 | −2275.007 | ω0 = 0.080 | |
| M2 | −2274.638 | 0.738 | ω0 = 0.077; ω1 = 0.204 | |
| NAD1 | M0 | −6009.838 | ω0 = 0.068 | |
| M2 | −6008.601 | 2.473 | ω0 = 0.067; ω1 = 0.155 | |
| CYTB | M0 | −6327.929 | ω0 = 0.048 | |
| M2 | −6325.360 | 5.138 * | ω0 = 0.045; ω1 = 0.110 | |
| NAD5 | M0 | −9665.997 | ω0 = 0.0453 | |
| M2 | −9665.198 | 1.598 | ω0 = 0.044; ω1 = 0.095 | |
| NAD4 | M0 | −9551.749 | ω0 = 0.052 | |
| M2 | −9551.525 | 0.448 | ω0 = 0.052; ω1 = 0.011 | |
| NAD4L | M0 | −2204.944 | ω0 = 0.062 | |
| M2 | −2204.944 | 0.001 | ω0 = 0.061; ω1 = 0.065 | |
| NAD6 | M0 | −3834.889 | ω0 = 0.071 | |
| M2 | −3834.578 | 0.621 | ω0 = 0.071; ω1 = 0.005 | |
| NAD2 | M0 | −7556.800 | ω0 = 0.085 | |
| M2 | −7556.540 | 0.521 | ω0 = 0.086; ω1 = 0.045 |
| Gene | Model | lnL | 2ΔlnL | Parameter | Positively Selected Site |
|---|---|---|---|---|---|
| COX1 | Null model | −7676.200 | P0 = 0.865; P1 = 0.073; P2a = 0.057; P2b = 0.005; ω0 = 0.012; ω1 = 1.000; ω2a = 1.000; ω2b = 1.000 | ||
| Model A | −7679.190 | 5.982 * | P0 = 0.877; P1 = 0.076; P2a = 0.043; P2b = 0.004; ω0 = 0.012; ω1 = 1.000; ω2a = 20.550; ω2b = 20.550 | 397 K (0.992); 456 Y (0.994) | |
| NAD2 | Null model | −2405.140 | P0 = 0.530; P1 = 0.191; P2a = 0.205; P2b = 0.074; ω0 = 0.070; ω1 = 1.000; ω2a = 1.000; ω2b = 1.000 | ||
| Model A | −2405.390 | 4.502 * | P0 = 0.526; P1 = 0.192; P2a = 0.207; P2b = 0.075; ω0 = 0.072; ω1 = 1.000; ω2a = 6.577; ω2b = 6.577 | 42 G (0.996); 54 G (0.974); 100 M (0.975); 114 N (0.985); 124 I (0.973); 153 G (0.965); 174 V (0.991); 207 K (0.997); 243 T (0.984); 266 M (0.978); 278 L (0.964) | |
| NAD4 | Null model | −8918.260 | P0 = 0.844; P1 = 0.156; P2a = 0.000; P2b = 0.000; ω0 = 0.053; ω1 = 1.000; ω2a = 1.000; ω2b = 1.000 | ||
| Model A | −8916.400 | 43.675 ** | P0 = 0.672; P1 = 0.110; P2a = 0.187; P2b = 0.031; ω0 = 0.050; ω1 = 1.000; ω2a = 154.839; ω2b = 154.839 | 45 S (0.994); 68 N (0.989); 70 L (0.956); 176 S (0.993); 225 S (0.968); 230 H (0.961); 285 W (0.970); 358 G (0.981); 368 Y (0.981); 369 M (0.952); 401 W (0.988) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, A.; Hui, M.; Sha, Z. First Genomic Survey of Pleurocryptella fimbriata Provides Preliminary Insights into Genome Characteristics and Evolution of a Deep-Sea Parasitic Isopod. Diversity 2025, 17, 297. https://doi.org/10.3390/d17040297
Wang A, Hui M, Sha Z. First Genomic Survey of Pleurocryptella fimbriata Provides Preliminary Insights into Genome Characteristics and Evolution of a Deep-Sea Parasitic Isopod. Diversity. 2025; 17(4):297. https://doi.org/10.3390/d17040297
Chicago/Turabian StyleWang, Aiyang, Min Hui, and Zhongli Sha. 2025. "First Genomic Survey of Pleurocryptella fimbriata Provides Preliminary Insights into Genome Characteristics and Evolution of a Deep-Sea Parasitic Isopod" Diversity 17, no. 4: 297. https://doi.org/10.3390/d17040297
APA StyleWang, A., Hui, M., & Sha, Z. (2025). First Genomic Survey of Pleurocryptella fimbriata Provides Preliminary Insights into Genome Characteristics and Evolution of a Deep-Sea Parasitic Isopod. Diversity, 17(4), 297. https://doi.org/10.3390/d17040297