
Application of Transcriptome-Based Gene Set Featurization for Machine Learning Model to Predict the Origin of Metastatic Cancer

 , , ,
, , ,
Abstract
1. Introduction
2. Methods and Materials
2.1. Data Source
2.1.1. RNA Sequencing and Gene Expression Profiling
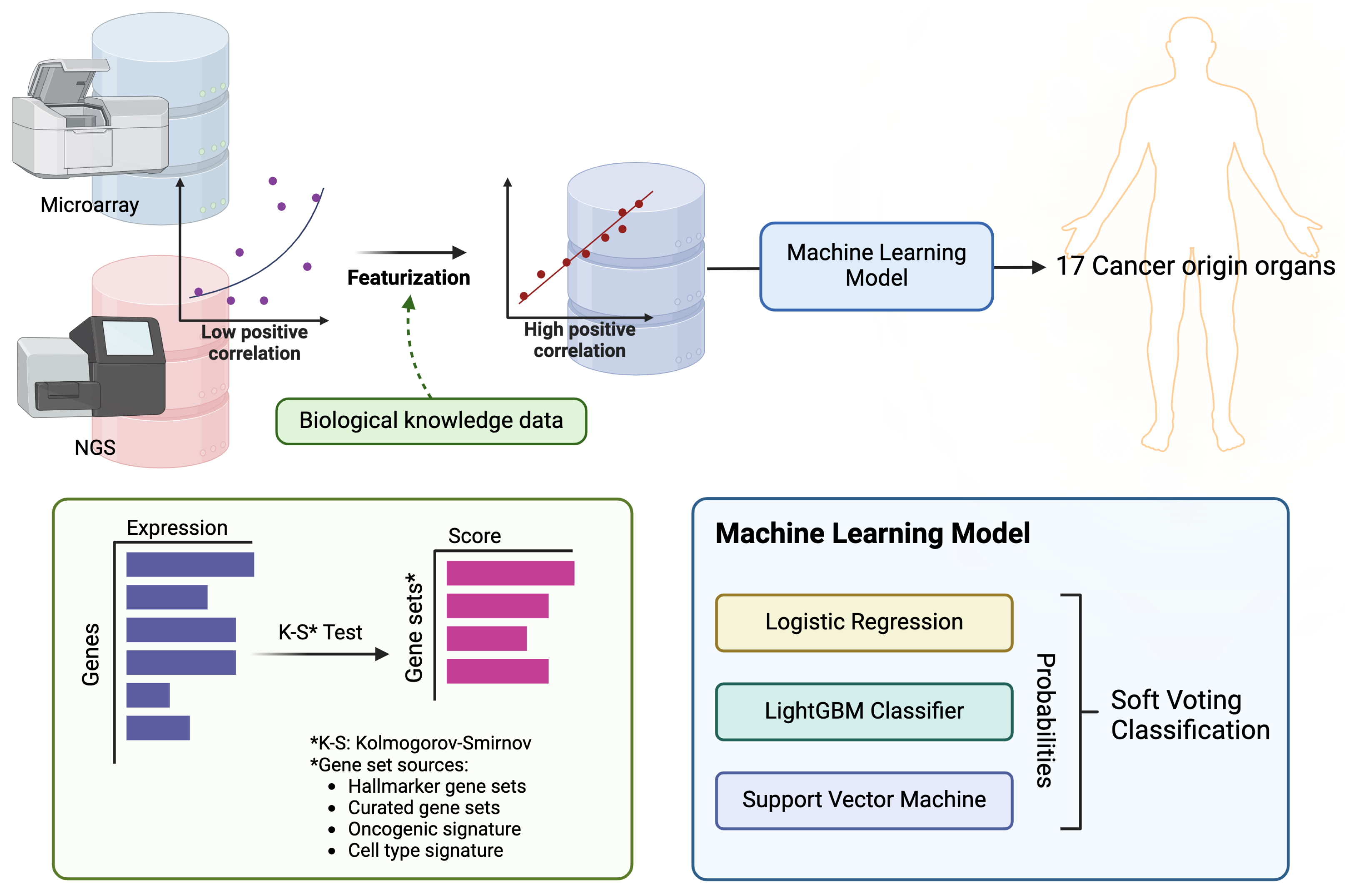
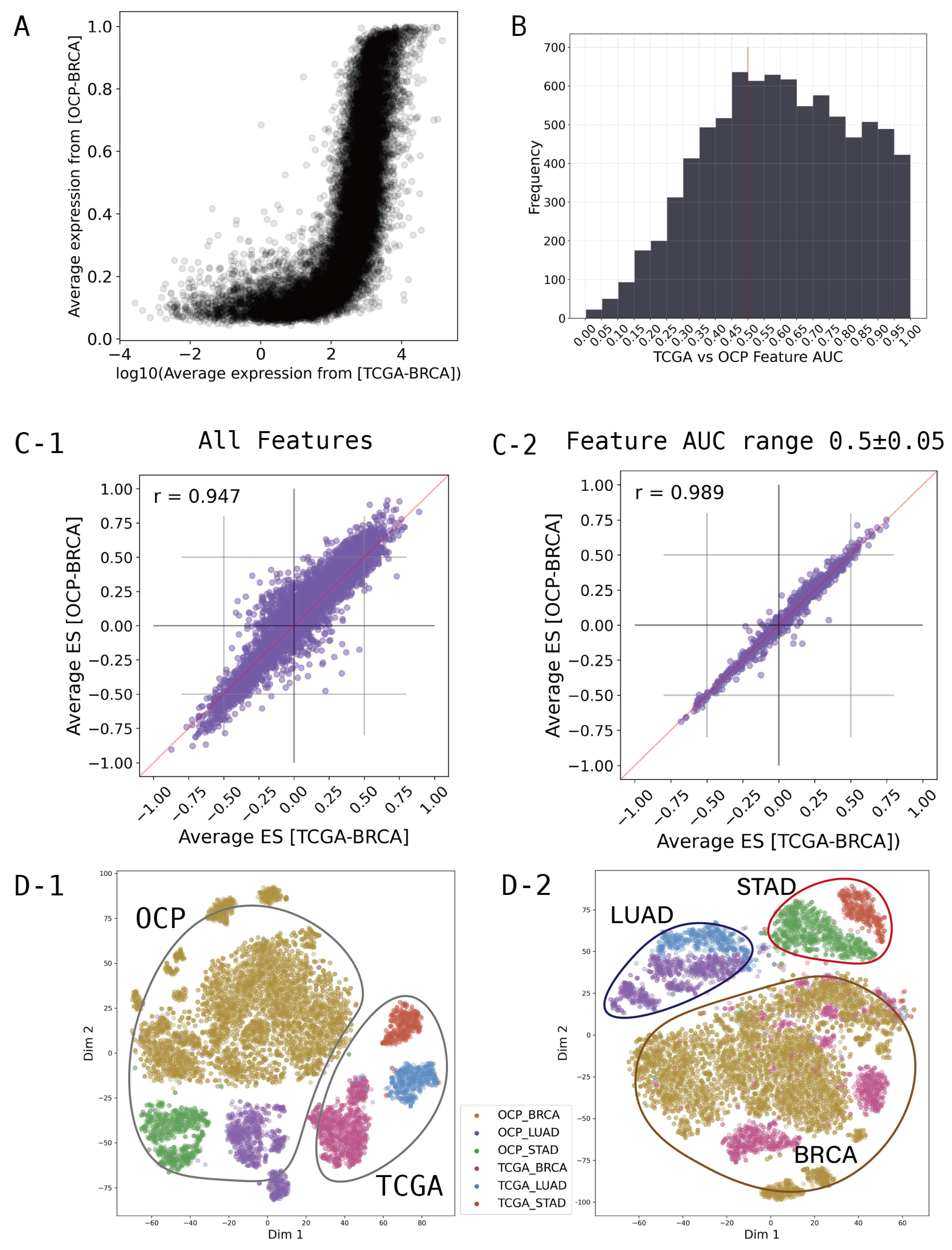
2.2. Featurization and Feature Selection
2.3. Cancer Primary Site Classification Model
2.4. External Validation
3. Results and Discussion
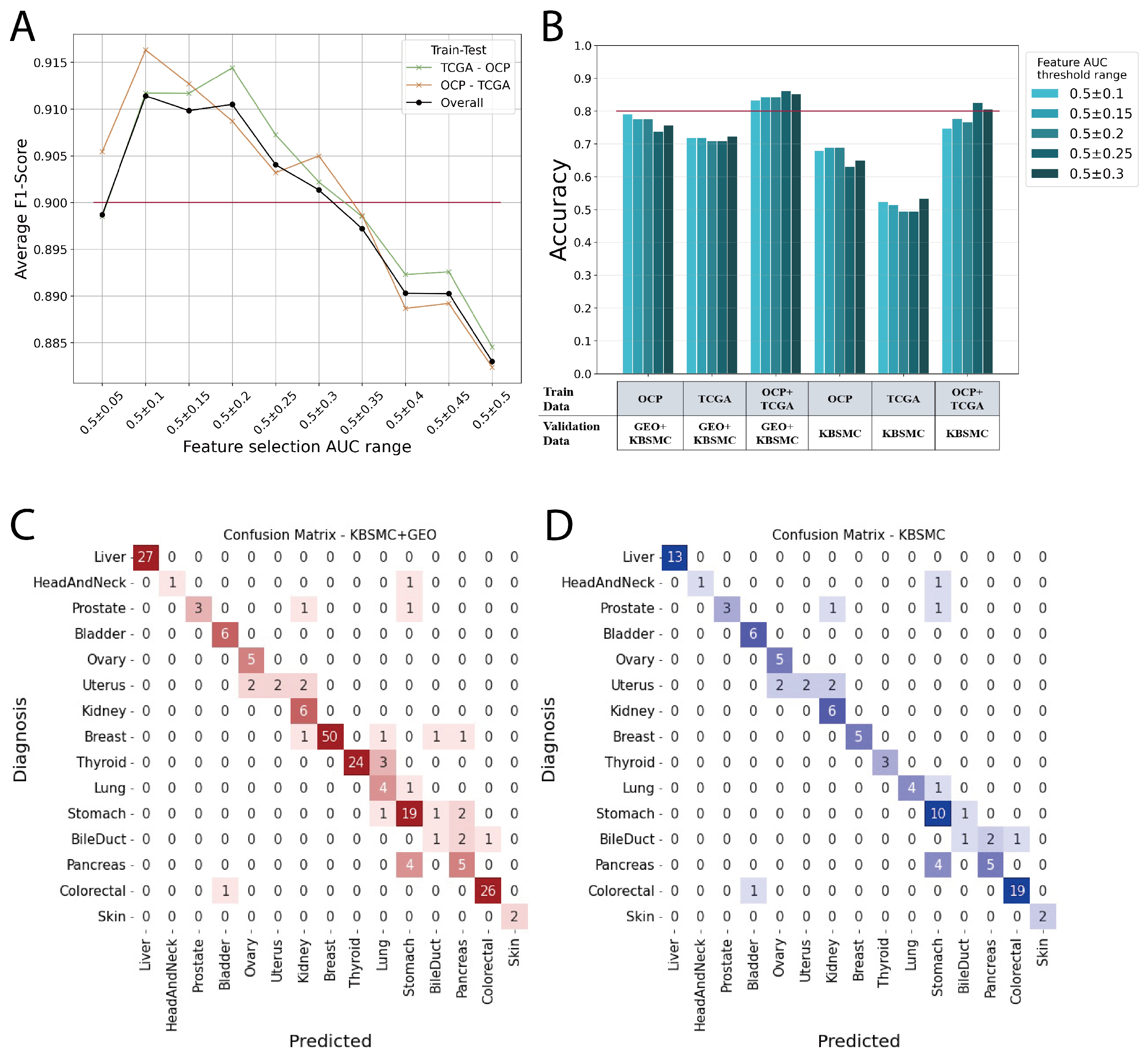
3.1. Performance of ONCOfind-AI
3.2. Limitation and Future Work
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pavlidis, N.; Pentheroudakis, G. Cancer of unknown primary site. Lancet 2012, 379, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Varadhachary, G.; Abbruzzese, J.L. Carcinoma of unknown primary. In Abeloff’s Clinical Oncology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1694–1702. [Google Scholar]
- Qaseem, A.; Usman, N.; Jayaraj, J.S.; Janapala, R.N.; Kashif, T. Cancer of unknown primary: A review on clinical guidelines in the development and targeted management of patients with the unknown primary site. Cureus 2019, 11, e5552. [Google Scholar] [CrossRef]
- Hyphantis, T.; Papadimitriou, I.; Petrakis, D.; Fountzilas, G.; Repana, D.; Assimakopoulos, K.; Carvalho, A.F.; Pavlidis, N. Psychiatric manifestations, personality traits and health-related quality of life in cancer of unknown primary site. Psycho-Oncology 2013, 22, 2009–2015. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wu, H.; Chen, Y.; Xu, H.; Jiang, J.; Du, B.; Wan, M.; Ma, X.; Chen, X.; Lin, L.; et al. New techniques to identify the tissue of origin for cancer of unknown primary in the era of precision medicine: Progress and challenges. Briefings Bioinform. 2024, 25, bbae028. [Google Scholar] [CrossRef] [PubMed]
- Rassy, E.; Pavlidis, N. Progress in refining the clinical management of cancer of unknown primary in the molecular era. Nat. Rev. Clin. Oncol. 2020, 17, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Shuel, S.L. Targeted cancer therapies: Clinical pearls for primary care. Can. Fam. Physician 2022, 68, 515. [Google Scholar] [CrossRef]
- Ding, Y.; Jiang, J.; Xu, J.; Chen, Y.; Zheng, Y.; Jiang, W.; Mao, C.; Jiang, H.; Bao, X.; Shen, Y.; et al. Site-specific therapy in cancers of unknown primary site: A systematic review and meta-analysis. ESMO Open 2022, 7, 100407. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Loriot, Y.; Fizazi, K. Carcinomas of an unknown primary origin—Diagnosis and treatment. Nat. Rev. Clin. Oncol. 2011, 8, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Varghese, A.; Arora, A.; Capanu, M.; Camacho, N.; Won, H.; Zehir, A.; Gao, J.; Chakravarty, D.; Schultz, N.; Klimstra, D.; et al. Clinical and molecular characterization of patients with cancer of unknown primary in the modern era. Ann. Oncol. 2017, 28, 3015–3021. [Google Scholar] [CrossRef]
- Mai, J.; Lu, M.; Gao, Q.; Zeng, J.; Xiao, J. Transcriptome-wide association studies: Recent advances in methods, applications and available databases. Commun. Biol. 2023, 6, 899. [Google Scholar] [CrossRef]
- Cao, C.; Kwok, D.; Edie, S.; Li, Q.; Ding, B.; Kossinna, P.; Campbell, S.; Wu, J.; Greenberg, M.; Long, Q. kTWAS: Integrating kernel machine with transcriptome-wide association studies improves statistical power and reveals novel genes. Briefings Bioinform. 2021, 22, bbaa270. [Google Scholar] [CrossRef] [PubMed]
- Petinrin, O.O.; Saeed, F.; Toseef, M.; Liu, Z.; Basurra, S.; Muyide, I.O.; Li, X.; Lin, Q.; Wong, K.C. Machine learning in metastatic cancer research: Potentials, possibilities, and prospects. Comput. Struct. Biotechnol. J. 2023, 21, 2454–2470. [Google Scholar] [CrossRef]
- Divate, M.; Tyagi, A.; Richard, D.J.; Prasad, P.A.; Gowda, H.; Nagaraj, S.H. Deep learning-based pan-cancer classification model reveals tissue-of-origin specific gene expression signatures. Cancers 2022, 14, 1185. [Google Scholar] [CrossRef]
- Zheng, Y.; Ding, Y.; Wang, Q.; Sun, Y.; Teng, X.; Gao, Q.; Zhong, W.; Lou, X.; Xiao, C.; Chen, C.; et al. 90-gene signature assay for tissue origin diagnosis of brain metastases. J. Transl. Med. 2019, 17, 331. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Shen, Y.; Ding, Y.; Ye, C.; Zheng, Y.; Zhao, P.; Liu, L.; Tong, Z.; Zhou, L.; Sun, S.; et al. A naive Bayes algorithm for tissue origin diagnosis (TOD-Bayes) of synchronous multifocal tumors in the hepatobiliary and pancreatic system. Int. J. Cancer 2018, 142, 357–368. [Google Scholar] [CrossRef]
- Grewal, J.K.; Tessier-Cloutier, B.; Jones, M.; Gakkhar, S.; Ma, Y.; Moore, R.; Mungall, A.J.; Zhao, Y.; Taylor, M.D.; Gelmon, K.; et al. Application of a neural network whole transcriptome–based pan-cancer method for diagnosis of primary and metastatic cancers. JAMA Netw. Open 2019, 2, e192597. [Google Scholar] [CrossRef]
- Zhao, Y.; Pan, Z.; Namburi, S.; Pattison, A.; Posner, A.; Balachander, S.; Paisie, C.A.; Reddi, H.V.; Rueter, J.; Gill, A.J.; et al. CUP-AI-Dx: A tool for inferring cancer tissue of origin and molecular subtype using RNA gene-expression data and artificial intelligence. EBioMedicine 2020, 61, 103030. [Google Scholar] [CrossRef]
- Moon, I.; LoPiccolo, J.; Baca, S.C.; Sholl, L.M.; Kehl, K.L.; Hassett, M.J.; Liu, D.; Schrag, D.; Gusev, A. Machine learning for genetics-based classification and treatment response prediction in cancer of unknown primary. Nat. Med. 2023, 29, 2057–2067. [Google Scholar] [CrossRef]
- van der Kloet, F.M.; Buurmans, J.; Jonker, M.J.; Smilde, A.K.; Westerhuis, J.A. Increased comparability between RNA-Seq and microarray data by utilization of gene sets. PLoS Comput. Biol. 2020, 16, e1008295. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Hu, Z.; Mahal, B.A.; Zhao, S.D.; Kensler, K.H.; Pi, J.; Hu, X.; Zhang, Y.; Wang, Y.; Jiang, J.; et al. Integrated analysis of genetic ancestry and genomic alterations across cancers. Cancer Cell 2018, 34, 549–560. [Google Scholar] [CrossRef]
- Lee, J.; Choi, C. Oncopression: Gene expression compendium for cancer with matched normal tissues. Bioinformatics 2017, 33, 2068–2070. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Gomez, C.D.; Kapoor, N.; Considine, J.M.; Grams, C.; Gao, Y.; Naba, A. MatrisomeDB 2.0: 2023 updates to the ECM-protein knowledge database. Nucleic Acids Res. 2023, 51, D1519–D1530. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Weiner, A.M. L2L: A simple tool for discovering the hidden significance in microarray expression data. Genome Biol. 2005, 6, R81. [Google Scholar] [CrossRef]
- Zeller, K.I.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. An integrated database of genes responsive to the Myc oncogenic transcription factor: Identification of direct genomic targets. Genome Biol. 2003, 4, R69. [Google Scholar] [CrossRef]
- Nishimura, D. BioCarta. Biotech Softw. Internet Rep. Comput. Softw. J. Sci. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The pathway interaction database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Pico, A.R.; Kelder, T.; Van Iersel, M.P.; Hanspers, K.; Conklin, B.R.; Evelo, C. WikiPathways: Pathway editing for the people. PLoS Biol. 2008, 6, e184. [Google Scholar] [CrossRef]
- Sun, L.; Wang, J.; Wei, J. AVC: Selecting discriminative features on basis of AUC by maximizing variable complementarity. BMC Bioinform. 2017, 18, 73–89. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Data Source | Sample No. |
|---|---|---|
| Adrenal Gland | OCP | 311 |
| TCGA | 261 | |
| Bile Duct | OCP | 184 |
| TCGA | 36 | |
| Bladder | OCP | 310 |
| TCGA | 408 | |
| Brain | OCP | 3054 |
| TCGA | 696 | |
| Breast | OCP | 5543 |
| TCGA | 1093 | |
| Colorectal | OCP | 3074 |
| TCGA | 381 | |
| Head And Neck | OCP | 622 |
| TCGA | 520 | |
| Kidney | OCP | 356 |
| TCGA | 891 | |
| Liver | OCP | 413 |
| TCGA | 373 | |
| Lung | OCP | 2243 |
| TCGA | 1018 | |
| Ovary | OCP | 1143 |
| TCGA | 307 | |
| Pancreas | OCP | 207 |
| TCGA | 178 | |
| Prostate | OCP | 247 |
| TCGA | 497 | |
| Skin | OCP | 294 |
| TCGA | 103 | |
| Stomach | OCP | 920 |
| TCGA | 599 | |
| Thyroid | OCP | 298 |
| TCGA | 501 | |
| Uterus | OCP | 322 |
| TCGA | 538 | |
| Total | OCP | 19,541 |
| TCGA | 8400 | |
| OCP + TCGA | 27,941 |
| Cancer | Data Source | Sample No. |
|---|---|---|
| Bile Duct | KBSMC | 4 |
| Bladder | KBSMC | 6 |
| Breast | GSE14017 | 29 |
| GSE147995 | 13 | |
| GSE191230 | 7 | |
| KBSMC | 5 | |
| Colorectal | KBSMC | 20 |
| GSE40367 | 7 | |
| Head And Neck | KBSMC | 2 |
| Kidney | KBSMC | 6 |
| Liver | GSE40367 | 15 |
| KBSMC | 13 | |
| Lung | KBSMC | 5 |
| Ovary | KBSMC | 5 |
| Pancreas | KBSMC | 10 |
| Prostate | KBSMC | 5 |
| Skin | KBSMC | 2 |
| Stomach | KBSMC | 11 |
| GSE246963 | 8 | |
| GSE191139 | 4 | |
| Thyroid | GSE60542 | 24 |
| KBSMC | 3 | |
| Uterus | KBSMC | 6 |
| Total | KBSMC | 103 |
| GEO | 107 | |
| Total | 210 |
| Training Data | Test Data | Feature AUC Range 0.5± | Weighted Accuracy |
|---|---|---|---|
| OCP + TCGA | GEO + KBSMC | 0.25 | 0.862 |
| OCP + TCGA | GEO + KBSMC | 0.3 | 0.852 |
| OCP + TCGA | GEO + KBSMC | 0.15 | 0.843 |
| OCP + TCGA | GEO + KBSMC | 0.2 | 0.843 |
| OCP + TCGA | GEO + KBSMC | 0.1 | 0.833 |
| OCP + TCGA | KBSMC | 0.25 | 0.825 |
| OCP + TCGA | KBSMC | 0.3 | 0.806 |
| OCP | GEO + KBSMC | 0.1 | 0.79 |
| OCP + TCGA | KBSMC | 0.15 | 0.777 |
| OCP | GEO + KBSMC | 0.2 | 0.776 |
| OCP | GEO + KBSMC | 0.15 | 0.776 |
| OCP + TCGA | KBSMC | 0.2 | 0.767 |
| OCP | GEO + KBSMC | 0.3 | 0.757 |
| OCP + TCGA | KBSMC | 0.1 | 0.748 |
| OCP | GEO + KBSMC | 0.25 | 0.738 |
| TCGA | GEO + KBSMC | 0.3 | 0.724 |
| TCGA | GEO + KBSMC | 0.15 | 0.719 |
| TCGA | GEO + KBSMC | 0.1 | 0.719 |
| TCGA | GEO + KBSMC | 0.2 | 0.71 |
| TCGA | GEO + KBSMC | 0.25 | 0.71 |
| OCP | KBSMC | 0.15 | 0.689 |
| OCP | KBSMC | 0.2 | 0.689 |
| OCP | KBSMC | 0.1 | 0.68 |
| OCP | KBSMC | 0.3 | 0.65 |
| OCP | KBSMC | 0.25 | 0.631 |
| TCGA | KBSMC | 0.3 | 0.534 |
| TCGA | KBSMC | 0.1 | 0.524 |
| TCGA | KBSMC | 0.15 | 0.515 |
| TCGA | KBSMC | 0.25 | 0.495 |
| TCGA | KBSMC | 0.2 | 0.495 |
| Cancer Type | Top-2 Accuracy for GEO + KBSMC Validation |
|---|---|
| BileDuct | 0.250 |
| Bladder | 1.000 |
| Breast | 0.926 |
| Colorectal | 0.963 |
| HeadAndNeck | 0.500 |
| Kidney | 1.000 |
| Liver | 0.964 |
| Lung | 0.800 |
| Ovary | 1.000 |
| Pancreas | 0.900 |
| Prostate | 0.800 |
| Skin | 1.000 |
| Stomach | 0.870 |
| Thyroid | 0.889 |
| Uterus | 0.667 |
| Weighted Average | 0.900 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, Y.; Chu, J.; Kang, J.; Baek, S.; Lee, J.-H.; Jung, D.-S.; Kim, W.-W.; Kim, Y.-R.; Kang, J.; Do, I.-G. Application of Transcriptome-Based Gene Set Featurization for Machine Learning Model to Predict the Origin of Metastatic Cancer. Curr. Issues Mol. Biol. 2024, 46, 7291-7302. https://doi.org/10.3390/cimb46070432
Jeong Y, Chu J, Kang J, Baek S, Lee J-H, Jung D-S, Kim W-W, Kim Y-R, Kang J, Do I-G. Application of Transcriptome-Based Gene Set Featurization for Machine Learning Model to Predict the Origin of Metastatic Cancer. Current Issues in Molecular Biology. 2024; 46(7):7291-7302. https://doi.org/10.3390/cimb46070432
Chicago/Turabian StyleJeong, Yeonuk, Jinah Chu, Juwon Kang, Seungjun Baek, Jae-Hak Lee, Dong-Sub Jung, Won-Woo Kim, Yi-Rang Kim, Jihoon Kang, and In-Gu Do. 2024. "Application of Transcriptome-Based Gene Set Featurization for Machine Learning Model to Predict the Origin of Metastatic Cancer" Current Issues in Molecular Biology 46, no. 7: 7291-7302. https://doi.org/10.3390/cimb46070432
APA StyleJeong, Y., Chu, J., Kang, J., Baek, S., Lee, J.-H., Jung, D.-S., Kim, W.-W., Kim, Y.-R., Kang, J., & Do, I.-G. (2024). Application of Transcriptome-Based Gene Set Featurization for Machine Learning Model to Predict the Origin of Metastatic Cancer. Current Issues in Molecular Biology, 46(7), 7291-7302. https://doi.org/10.3390/cimb46070432