
Using Copy Number Variation Data and Neural Networks to Predict Cancer Metastasis Origin Achieves High Area under the Curve Value with a Trade-Off in Precision

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Methods
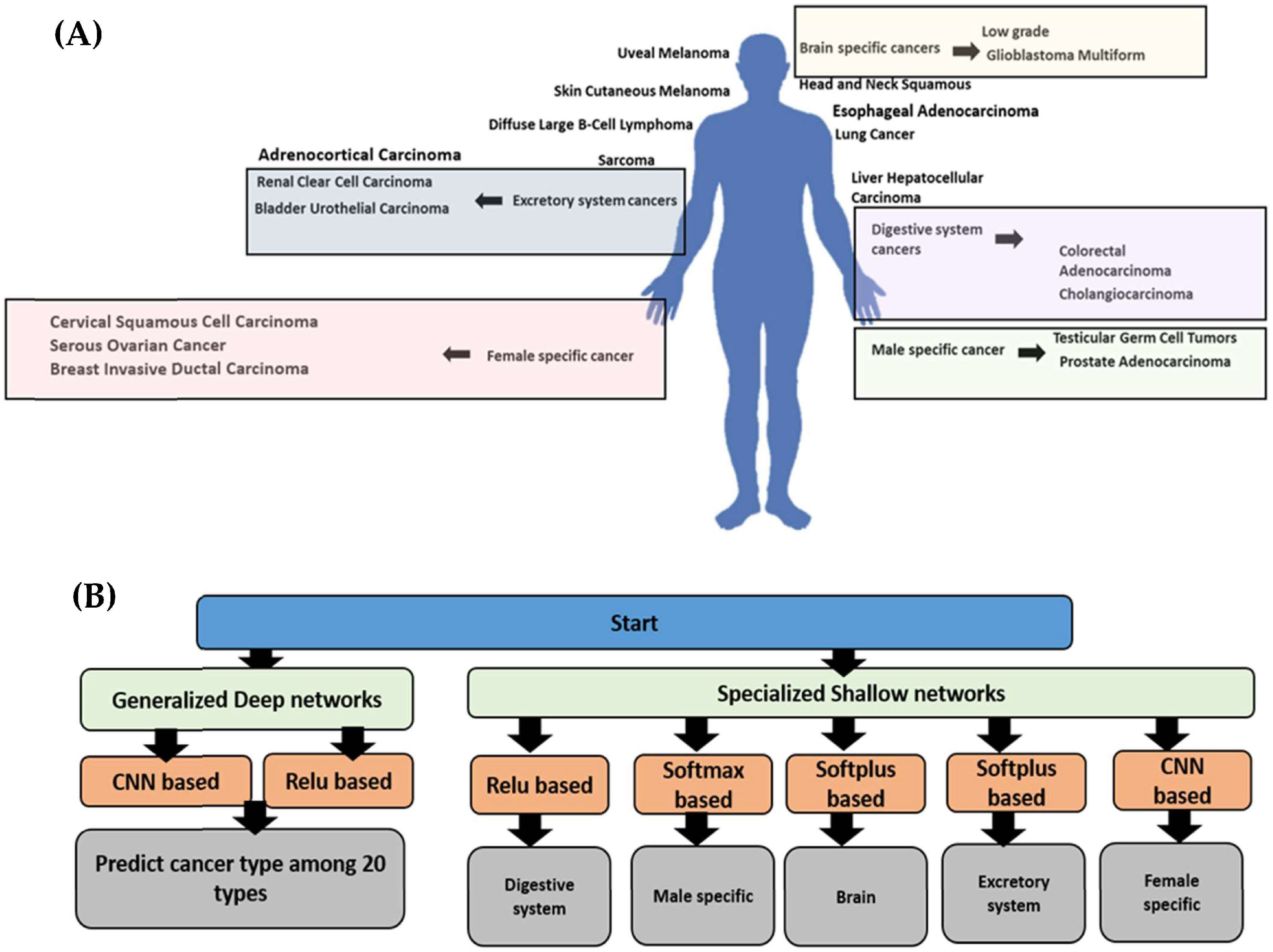
2.1. Database Construction and Preprocessing
2.2. Model Construction
2.2.1. The Generalized Model
2.2.2. The Specialized Models
2.3. Hyperparameter Tuning
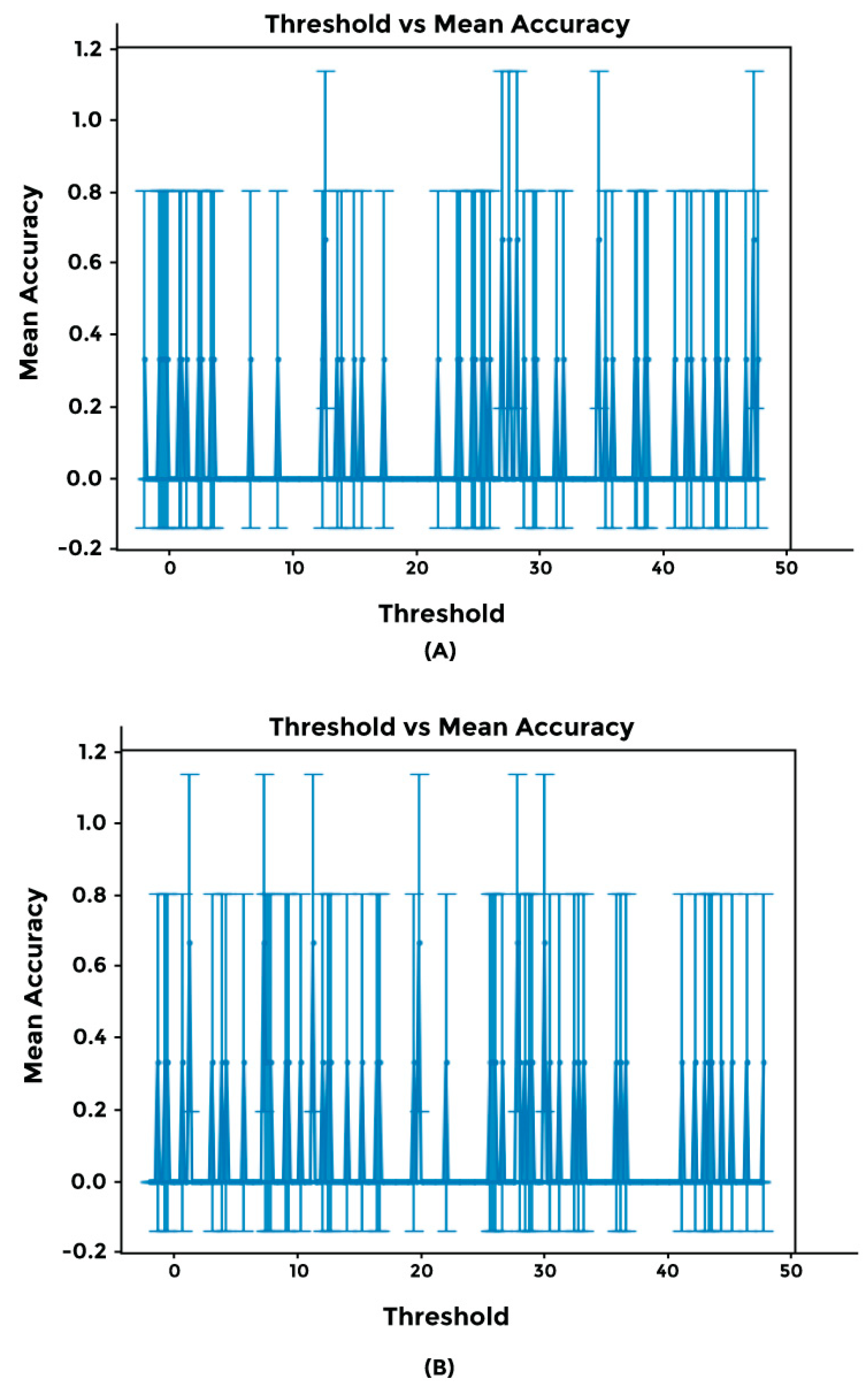
2.4. Cross-Validation Using Synthetic Dataset and Sensitivity Threshold Setting
2.5. Biological Explainability Analysis
2.6. Explainability Analysis Using SHAP in TensorFlow
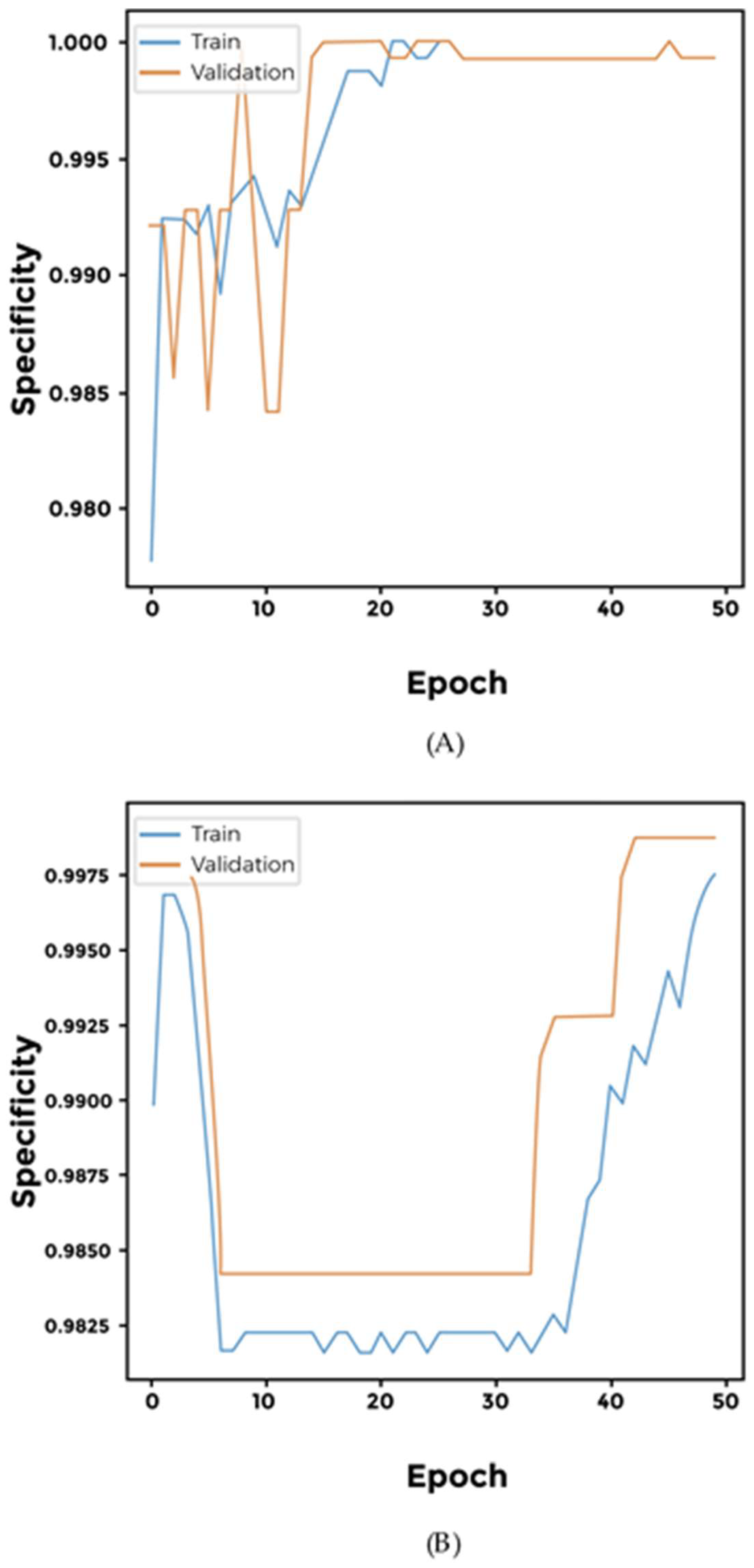
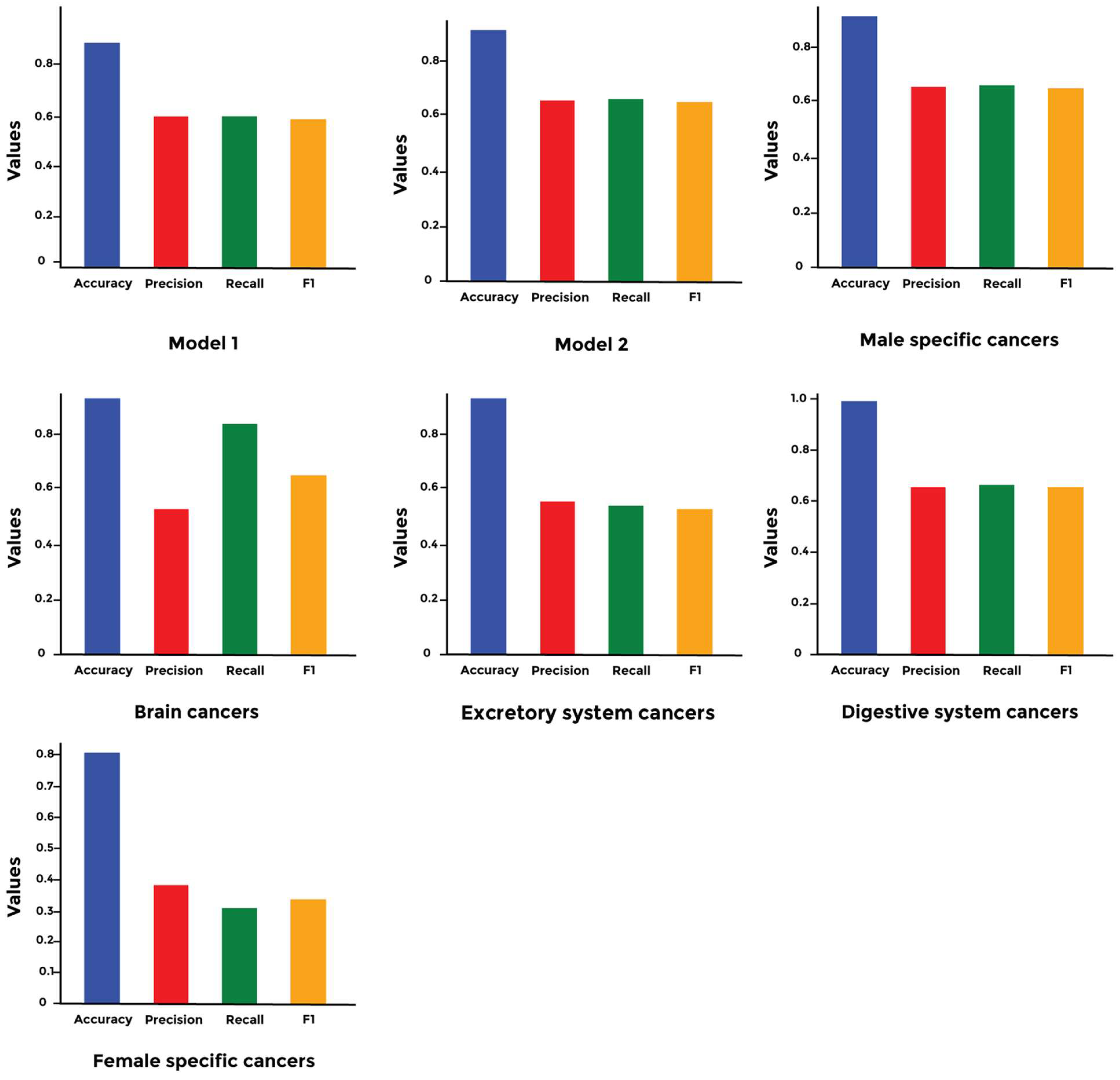
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Łazarczyk, M.; Mickael, M.E.; Skiba, D.; Kurzejamska, E.; Ławiński, M.; Horbańczuk, J.O.; Radziszewski, J.; Fraczek, K.; Wolinska, R.; Paszkiewicz, J.; et al. The Journey of Cancer Cells to the Brain: Challenges and Opportunities. Int. J. Mol. Sci. 2023, 24, 3854. [Google Scholar] [CrossRef] [PubMed]
- Lyden, D.; Ghajar, C.M.; Correia, A.L.; Aguirre-Ghiso, J.A.; Cai, S.; Rescigno, M.; Zhang, P.; Hu, G.; Fendt, S.M.; Boire, A.; et al. Metastasis. Cancer Cell 2022, 40, 787–791. [Google Scholar] [CrossRef]
- Nimmakayala, R.K.; Batra, S.K.; Ponnusamy, M.P. Unraveling the Journey of Cancer Stem Cells from Origin to Metastasis. Biochim. Biophys. Acta (BBA) Rev. Cancer 2019, 1871, 50–63. [Google Scholar] [CrossRef]
- Kato, S.; Alsafar, A.; Walavalkar, V.; Hainsworth, J.; Kurzrock, R. Cancer of Unknown Primary in the Molecular Era. Trends Cancer 2021, 7, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Karim, M.R.; Rahman, A.; Jares, J.B.; Decker, S.; Beyan, O. A Snapshot Neural Ensemble Method for Cancer-Type Prediction Based on Copy Number Variations. Neural Comput. Appl. 2020, 32, 15281–15299. [Google Scholar] [CrossRef]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour Evolution Inferred by Single-Cell Sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Nicoś, M.; Krawczyk, P. Genetic Clonality as the Hallmark Driving Evolution of Non-Small Cell Lung Cancer. Cancers 2022, 14, 1813. [Google Scholar] [CrossRef]
- Wu, H.-J.; Temko, D.; Maliga, Z.; Moreira, A.L.; Sei, E.; Minussi, D.C.; Dean, J.; Lee, C.; Xu, Q.; Hochart, G.; et al. Spatial Intra-Tumor Heterogeneity Is Associated with Survival of Lung Adenocarcinoma Patients. Cell Genom. 2022, 2, 100165. [Google Scholar] [CrossRef]
- Ding, X.; Tsang, S.-Y.; Ng, S.-K.; Xue, H. Application of Machine Learning to Development of Copy Number Variation-Based Prediction of Cancer Risk. Genom. Insights 2014, 7, GEI.S15002–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, M.; Zhang, P.; Huang, T. Classification of Cancers Based on Copy Number Variation Landscapes. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1860, 2750–2755. [Google Scholar] [CrossRef]
- Elsadek, S.F.A.; Makhlouf, M.A.A.; Aldeen, M.A. Supervised Classification of Cancers Based on Copy Number Variation. In Proceedings of the Advances in Intelligent Systems and Computing, Cairo, Egypt, 1–3 September 2018. [Google Scholar]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Bonner, E.R.; Dawood, A.; Gordish-Dressman, H.; Eze, A.; Bhattacharya, S.; Yadavilli, S.; Mueller, S.; Waszak, S.M.; Nazarian, J. Pan-Cancer Atlas of Somatic Core and Linker Histone Mutations. Npj Genom. Med. 2023, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; Fitzhugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Kubick, N.; Pajares, M.; Enache, I.; Manda, G.; Mickael, M.-E. Repurposing Zileuton as a Depression Drug Using an AI and In Vitro Approach. Molecules 2020, 25, 2155. [Google Scholar] [CrossRef]
- Lazarczyk, M.; Duda, K.; Mickael, M.E.; Ak, O.; Paszkiewicz, J.; Kowalczyk, A.; Horbańczuk, J.O.; Sacharczuk, M. Adera2.0: A Drug Repurposing Workflow for Neuroimmunological Investigations Using Neural Networks. Molecules 2022, 27, 6453. [Google Scholar] [CrossRef]
- Agarwal, M.; Gupta, S.; Biswas, K.K. A New Conv2D Model with Modified ReLU Activation Function for Identification of Disease Type and Severity in Cucumber Plant. Sustain. Comput. Inform. Syst. 2021, 30, 100473. [Google Scholar] [CrossRef]
- Glessner, J.T.; Hou, X.; Zhong, C.; Zhang, J.; Khan, M.; Brand, F.; Krawitz, P.; Sleiman, P.M.A.; Hakonarson, H.; Wei, Z. DeepCNV: A Deep Learning Approach for Authenticating Copy Number Variations. Brief. Bioinform. 2021, 22, bbaa381. [Google Scholar] [CrossRef]
- Lu, Y.; Gould, S.; Ajanthan, T. Bidirectionally Self-Normalizing Neural Networks. Neural Netw. 2023, 167, 283–291. [Google Scholar] [CrossRef]
- Mostavi, M.; Chiu, Y.-C.; Huang, Y.; Chen, Y. Convolutional Neural Network Models for Cancer Type Prediction Based on Gene Expression. BMC Med. Genom. 2020, 13, 44. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shi, Y.; Su, X.; Zou, X.; Luo, Q.; Feng, D.D.; Cai, W.; Han, Z.-G. Cancer Type Prediction Based on Copy Number Aberration and Chromatin 3D Structure with Convolutional Neural Networks. BMC Genom. 2018, 19, 565. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review the Cancer Genome Atlas (TCGA): An Immeasurable Source of Knowledge. Współczesna Onkol. 2015, 1A, 68–77. [Google Scholar] [CrossRef]
- Anderson, G.G.; Weiss, L.M. Determining Tissue of Origin for Metastatic Cancers: Meta-Analysis and Literature Review of Immunohistochemistry Performance. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Pös, O.; Radvanszky, J.; Buglyó, G.; Pös, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA Copy Number Variation: Main Characteristics, Evolutionary Significance, and Pathological Aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.-F.; Wu, M.; Bao, H.; Xu, Y.; Lin, J.-S.; Liang, Y.; Zhang, Y.; Chu, X.-P.; Qiu, Z.-B.; Su, J.; et al. Timing and Origins of Local and Distant Metastases in Lung Cancer. J. Thorac. Oncol. 2021, 16, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Bense, R.D.; Urzúa-Traslaviña, C.G.; de Vries, E.G.E.; van Vugt, M.A.T.M.; Fehrmann, R.S.N. Transcriptional Effects of Copy Number Alterations in a Large Set of Human Cancers. Nat. Commun. 2020, 11, 715. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Ge, J.-Y.; Zhu, S.-Y.; Shao, Z.-M.; Yu, K.-D. Copy Number Amplification of ENSA Promotes the Progression of Triple-Negative Breast Cancer via Cholesterol Biosynthesis. Nat. Commun. 2022, 13, 791. [Google Scholar] [CrossRef] [PubMed]
- Rockman, M.V.; Skrovanek, S.S.; Kruglyak, L. Selection at Linked Sites Shapes Heritable Phenotypic Variation in C. elegans. Science 2010, 330, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Leshner, M.; Devine, M.; Roloff, G.W.; True, L.D.; Misteli, T.; Meaburn, K.J. Locus-Specific Gene Repositioning in Prostate Cancer. Mol. Biol. Cell 2016, 27, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Bishop, D.T.; Ford, D.; Crockford, G.P. Genetic Linkage Analysis in Familial Breast and Ovarian Cancer: Results from 214 Families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1993, 52, 678–701. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Organ | Number of Samples | |
|---|---|---|---|
| 1 | Adrenocortical Carcinoma | Adrenal gland | 132 |
| 2 | Bladder Urothelial Carcinoma | Bladder | 612 |
| 3 | Brain Lower-Grade Glioma | Brain | 3 |
| 4 | Breast Invasive Ductal Carcinoma | Breast | 1007 |
| 5 | Cervical Squamous Cell Carcinoma | Cervix | 475 |
| 6 | Cholangiocarcinoma | Bile duct | 212 |
| 7 | Colorectal Adenocarcinoma | Large intestine | 405 |
| 8 | Diffuse Large B cell lymphoma | B Cells | 858 |
| 9 | Esophageal Adenocarcinoma | Esophagus | 905 |
| 10 | Glioblastoma Multiforme | Brain | 39 |
| 11 | Head and Neck Squamous Cell Carcinoma | Oral cavity, pharynx, and larynx | 3 |
| 12 | Kidney Renal Clear Cell Carcinoma | Kidney | 277 |
| 13 | Liver Hepatocellular Carcinoma | Liver | 183 |
| 14 | Lung Adenocarcinoma | Lung | 53 |
| 15 | Ovarian Serous Cystadenocarcinoma | Ovary | 1071 |
| 16 | Prostate Adenocarcinoma | Prostate | 171 |
| 17 | Sarcoma | Connective tissue | 286 |
| 18 | Skin Cutaneous Melanoma | Skin | 2 |
| 19 | Testicular Germ Cell Tumors | Testicles | 258 |
| 20 | Uveal Melanoma | Eye | 106 |
| Model | Layer | Activation | Description |
|---|---|---|---|
| 1A | Dense | ReLU | 256 |
| Dense | ReLU | 256 | |
| Conv2D | filters = 20, kernel size = 8, strides = 1, padding = same | ||
| Dense | ReLU | 256 | |
| Dropout | 0.5 | ||
| Dense | Softmax | 20 | |
| 1B | Dense | ReLU | 256 |
| BatchNormalization | |||
| Dropout | 0.002 | ||
| Dense | ReLU | 256 | |
| Dense | ELU | 128 | |
| Dropout | 0.05 | ||
| Dense | SELU | 64 | |
| Dense | ELU | 64 | |
| Dense | ELU | 32 | |
| Dense | ReLU | 32 | |
| Dense | Softmax | 20 | |
| Cancer Types | Model | Layer | Activation | Description |
|---|---|---|---|---|
| Male-specific cancer | 2.A | Dense | Softmax | 2 |
| 2.B | Dense | Sigmoid | 2 | |
| 2.C | Dense | Softplus | 2 | |
| Brain cancer | 3.A | Dense | Softmax | 2 |
| 3.B | Dense | Sigmoid | 2 | |
| 3.C | Dense | Softplus | 2 | |
| Excretory system cancer | 4.A | Dense | Softmax | 2 |
| 4.B | Dense | Sigmoid | 2 | |
| 4.C | Dense | Softplus | 2 | |
| Digestive system cancer | 5.A | Dense | ReLU | 128 |
| Dense | Sigmoid | 2 | ||
| 5.B | Dense | ReLU | 128 | |
| Dense | Softmax | 2 | ||
| 5.C | Dense | ReLU | 128 | |
| Dense | Softplus | 2 | ||
| Female-specific cancer | 6.A | Dense | ReLU | 128 |
| Dense | Sigmoid | 2 | ||
| 6.B | Dense | ReLU | 128 | |
| Dense | Softmax | 2 | ||
| 6.C | Dense | ReLU | 128 | |
| Dense | Softplus | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mickael, M.-E.; Kubick, N.; Atanasov, A.G.; Martinek, P.; Horbańczuk, J.O.; Floretes, N.; Michal, M.; Vanecek, T.; Paszkiewicz, J.; Sacharczuk, M.; et al. Using Copy Number Variation Data and Neural Networks to Predict Cancer Metastasis Origin Achieves High Area under the Curve Value with a Trade-Off in Precision. Curr. Issues Mol. Biol. 2024, 46, 8301-8319. https://doi.org/10.3390/cimb46080490
Mickael M-E, Kubick N, Atanasov AG, Martinek P, Horbańczuk JO, Floretes N, Michal M, Vanecek T, Paszkiewicz J, Sacharczuk M, et al. Using Copy Number Variation Data and Neural Networks to Predict Cancer Metastasis Origin Achieves High Area under the Curve Value with a Trade-Off in Precision. Current Issues in Molecular Biology. 2024; 46(8):8301-8319. https://doi.org/10.3390/cimb46080490
Chicago/Turabian StyleMickael, Michel-Edwar, Norwin Kubick, Atanas G. Atanasov, Petr Martinek, Jarosław Olav Horbańczuk, Nikko Floretes, Michael Michal, Tomas Vanecek, Justyna Paszkiewicz, Mariusz Sacharczuk, and et al. 2024. "Using Copy Number Variation Data and Neural Networks to Predict Cancer Metastasis Origin Achieves High Area under the Curve Value with a Trade-Off in Precision" Current Issues in Molecular Biology 46, no. 8: 8301-8319. https://doi.org/10.3390/cimb46080490
APA StyleMickael, M.-E., Kubick, N., Atanasov, A. G., Martinek, P., Horbańczuk, J. O., Floretes, N., Michal, M., Vanecek, T., Paszkiewicz, J., Sacharczuk, M., & Religa, P. (2024). Using Copy Number Variation Data and Neural Networks to Predict Cancer Metastasis Origin Achieves High Area under the Curve Value with a Trade-Off in Precision. Current Issues in Molecular Biology, 46(8), 8301-8319. https://doi.org/10.3390/cimb46080490