
Autophagy: Are Amino Acid Signals Dependent on the mTORC1 Pathway or Independent?

 , , and
, , and
Abstract
:1. Introduction
2. Autophagy
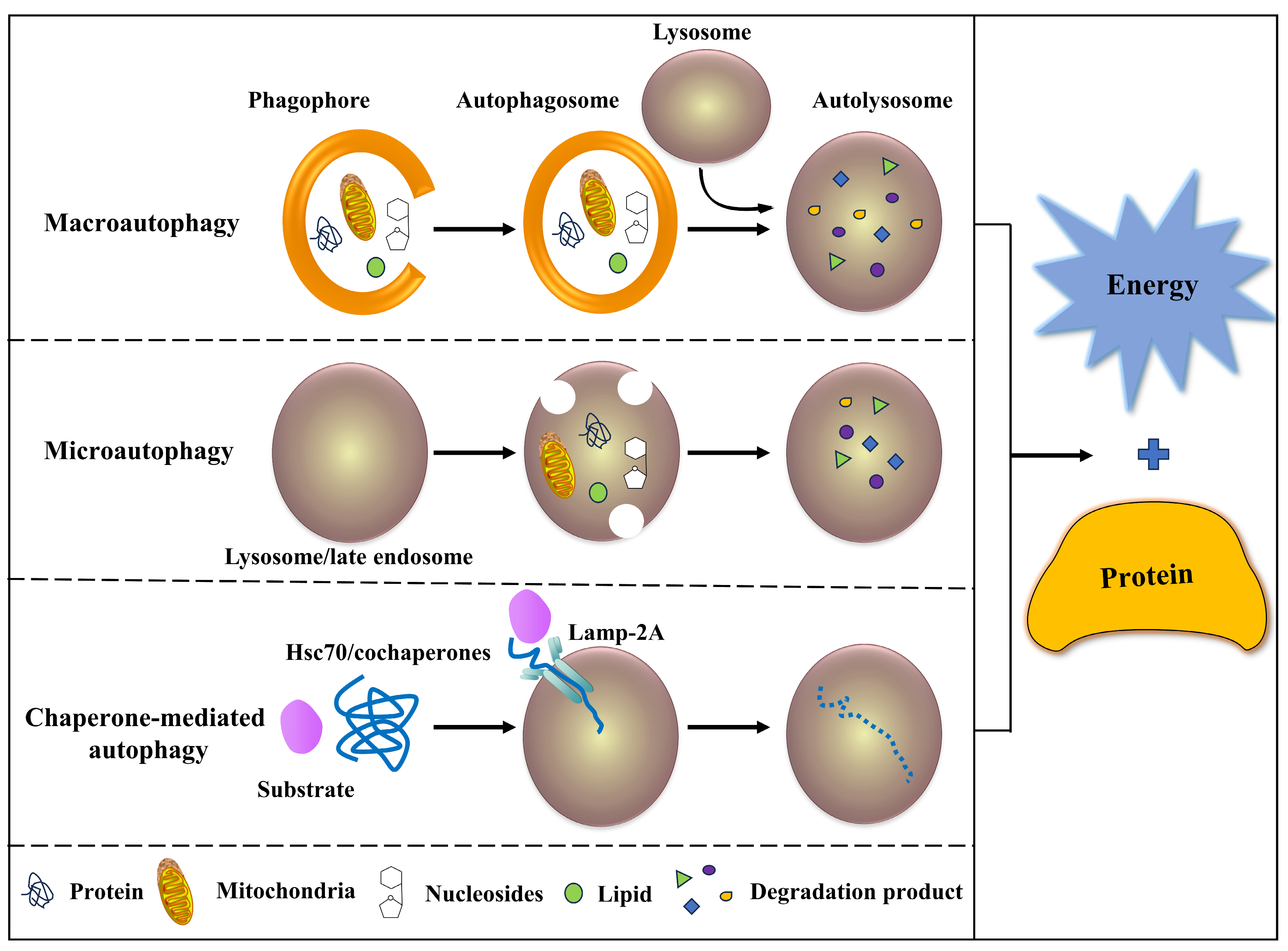
2.1. Classification of Autophagy
2.2. Characteristics of Autophagy
2.3. Process of Autophagy
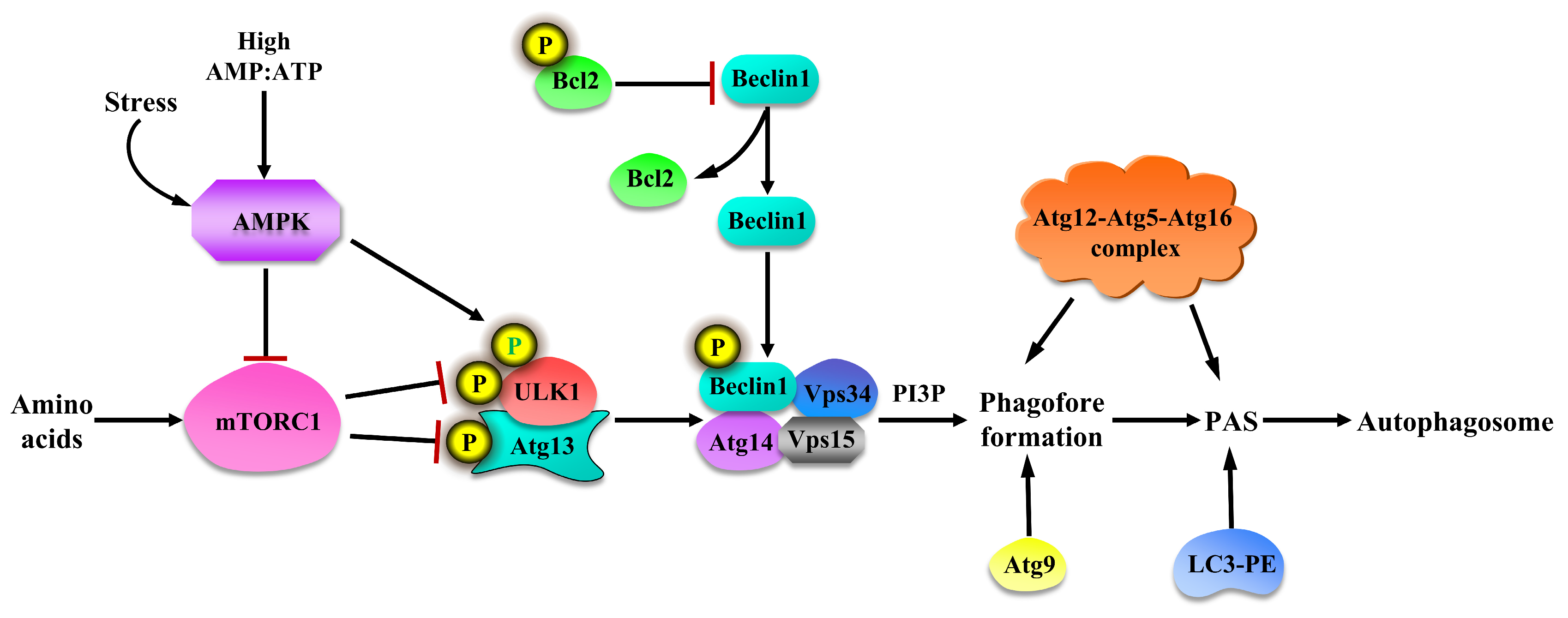
2.4. Regulation of Autophagy
3. Autophagy: Amino Acid Signals Dependent on mTORC1 Pathway
3.1. Lys in Regulating Autophagy with mTORC1 Pathway
3.2. Leu in Regulating Autophagy with mTORC1 Pathway
3.3. Arg in Regulating Autophagy with mTORC1 Pathway
3.4. Gln in Regulating Autophagy with mTORC1 Pathway

{kind=link}
{kind=link}
| Amino Acids | Treatment | Samples | Description | References |
|---|---|---|---|---|
| Lys | 10 mM Lys treated for 30 min | C2C12 | Suppressed autophagy, activated mTORC1 | [60,63] |
| Rats fed with 10% casein diet supplemented by Lys | Rat muscle | Suppressed autophagy, activated mTORC1 | [79] | |
| Leu | 10 mM Leu treated for 30 min | C2C12 | Suppressed autophagy, activated mTORC1 | [60,63] |
| Fasted mice administered an oral gavage of Leu (1.35 g/kg BW) | Mouse muscle | Increased protein synthesis and mTORC1 | [64] | |
| Fasted pigs were infused intra-arterially with Leu (0, 200, or 400 μmol/kg/h) | Pig muscle | Increased protein synthesis and mTORC1 | [70] | |
| Oral administration of Leu in fasted rats | Rat muscle | Increased protein synthesis and mTORC1 | [71,80] | |
| Arg | Medium Arg deficiency | Melanoma cells | Induced autophagy, restricted mTORC1 | [73] |
| Diet Arg supplementation in heat-stress-treated rats | Rat intestine | Induced autophagy, restricted mTORC1 | [74] | |
| Medium Arg supplementation in heat-stress-treated IEC-6 | IEC-6 | Induced autophagy, restricted mTORC1 | [74] | |
| Arg-treated skin fibroblasts isolated from GM2 gangliosidosis | Skin fibroblasts | Recovered autophagy and mTORC1 | [75] | |
| Gln | Gln-treated MEFs and HepG2 cells under amino acid starvation | MEFs and HepG2 cells | Restored autophagy and mTORC1 | [77] |
| Gln deprivation treated IPEC-1 | IPEC-1 | Induced autophagy, restricted mTORC1 | [78] |
4. Autophagy: Amino Acid Signals Independent of mTORC1 Pathway
4.1. Lys in Regulating Autophagy without mTORC1 Pathway
4.2. Leu in Regulating Autophagy without mTORC1 Pathway
| Amino Acids | Treatment | Samples | Description | References |
|---|---|---|---|---|
| Lys | Oral administration of Lys (114 mg/100 g BW, sufficient) in fasted rats | Rat muscle | Suppressed autophagy, unchanged mTORC1 | [59] |
| 10 mM Lys treated for 30 min | C2C12 | Suppressed autophagy and AMPK | [60] | |
| SAMP8 fed with 3.0% Lys diet | SAMP8 | Suppressed autophagy, unchanged mTORC1 | [61] | |
| Leu | 10 mM-Leu-treated for 30 min | C2C12 | Suppressed autophagy and AMPK | [60] |
| MiR-20a- and miR-106b-treated Leu deficiency medium pre-cultured C2C12 | C2C12 | Suppressed autophagy, unchanged mTORC1 | [67] | |
| Old rats fed a 15% protein diet supplemented with 4.5% leucine | Rat muscle | Suppressed autophagy, unchanged mTORC1 | [82] | |
| Leu-deficiency-treated C2C12 | C2C12 | Induced autophagy, unchanged mTORC1 | [84] | |
| Arg | Medium Arg deficiency | Melanoma cells | Induced autophagy, activated AMPK | [73] |
| IFN-γ treated BMECs | BMECs | Decreased Arg content, increased GCN2 pathway and autophagy | [85] | |
| Arg-deficiency-treated H4-II-E and Hep G2 cells | H4-II-E and Hep G2 cells | Induced autophagy, activated NO pathway | [86,87] | |
| Gln | Atg5 or Atg7 knockdown HeLa cells with Gln supplementation | HeLa cells | Suppressed autophagy, unchanged mTORC1 | [88] |
| Gln-deficiency-treated porcine-circovirus-type-2-infected mice and PK 15 cells | Mice and PK 15 cells | Induced autophagy, activated JAK2/STAT3 | [89] |
4.3. Arg in Regulating Autophagy without mTORC1 Pathway
4.4. Gln in Regulating Autophagy without mTORC1 Pathway
5. Defects in Studies on Amino-Acid-Regulated Autophagy
6. Summary and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paulusma, C.C.; Lamers, W.H.; Broer, S.; van de Graaf, S.F.J. Amino acid metabolism, transport and signalling in the liver revisited. Biochem. Pharmacol. 2022, 201, 115074. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.L.; Ye, M.; Song, Z.W.; Zhang, Z.M.; Gao, C.Q.; Yan, H.C.; Wang, X.Q. Lysine interacts with frizzled7 to activate β-catenin in satellite cell-participated skeletal muscle growth. J. Agric. Food Chem. 2022, 70, 3745–3756. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cui, L.; Lu, S.; Xu, S. Amino acid metabolism in tumor biology and therapy. Cell Death Dis. 2024, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Shiozaki, K. Multiplexed suppression of TOR complex 1 induces autophagy during starvation. Autophagy 2021, 17, 1794–1795. [Google Scholar] [CrossRef] [PubMed]
- Bodineau, C.; Tomé, M.; Murdoch, P.D.S.; Durán, R.V. Glutamine, MTOR and autophagy: A multiconnection relationship. Autophagy 2022, 18, 2749–2750. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.L.; Zhang, Z.M.; Ye, J.L.; Gao, C.Q.; Yan, H.C.; Li, H.C.; Wang, X.Q. Lysine-induced swine satellite cell migration is mediated by the FAK pathway. Food Funct. 2019, 10, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Q.; Jeon, J.; Li, Y.; Huang, Y.; Xiong, J.; Wang, Q.; Xu, T.L.; Li, Y.; Ji, F.H.; Du, G.; et al. CAMK2/CaMKII activates MLKL in short-term starvation to facilitate autophagic flux. Autophagy 2022, 18, 726–744. [Google Scholar] [CrossRef] [PubMed]
- Corral-Ramos, C.; Barrios, R.; Ayté, J.; Hidalgo, E. TOR and MAP kinase pathways synergistically regulate autophagy in response to nutrient depletion in fission yeast. Autophagy 2022, 18, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yao, S.; Yang, H.; Liu, S.; Wang, Y. Autophagy: Regulator of cell death. Cell Death Dis. 2023, 14, 648. [Google Scholar] [CrossRef]
- Eapen, V.V.; Swarup, S.; Hoyer, M.J.; Paulo, J.A.; Harper, J.W. Quantitative proteomics reveals the selectivity of ubiquitin-binding autophagy receptors in the turnover of damaged lysosomes by lysophagy. eLife 2021, 10, e72328. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2023, 24, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Miyahara, H.; Shiraishi, H.; Shimizu, N.; Tsumori, M.; Kiyota, K.; Maeda, M.; Umeda, R.; Ishitani, T.; Hanada, R.; et al. Leucyl-tRNA synthetase deficiency systemically induces excessive autophagy in zebrafish. Sci. Rep. 2021, 11, 8392. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef] [PubMed]
- Al-Bari, M.A.A.; Xu, P. Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann. N. Y. Acad. Sci. 2020, 1467, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Mohanasundaram, P.; Coelho-Rato, L.S.; Modi, M.K.; Urbanska, M.; Lautenschläger, F.; Cheng, F.; Eriksson, J.E. Cytoskeletal vimentin regulates cell size and autophagy through mTORC1 signaling. PLoS Biol. 2022, 20, e3001737. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lu, Y.; Wang, H.; Wu, Y.; Xu, X.; Li, Y. High ATF4 expression is associated with poor prognosis, amino acid metabolism, and autophagy in gastric cancer. Front. Oncol. 2021, 11, 740120. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Iwata, J. Amino acid metabolism and autophagy in skeletal development and homeostasis. Bone 2021, 146, 115881. [Google Scholar] [CrossRef] [PubMed]
- Sadria, M.; Layton, A.T. Interactions among mTORC, AMPK and SIRT: A computational model for cell energy balance and metabolism. Cell Commun. Signal 2021, 19, 57. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Wang, Q.; Kao, Y.R.; Diaz, A.; Tasset, I.; Kaushik, S.; Thiruthuvanathan, V.; Zintiridou, A.; Nieves, E.; Dzieciatkowska, M.; et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 2021, 591, 117–123. [Google Scholar] [CrossRef]
- Yao, R.; Shen, J. Chaperone-mediated autophagy: Molecular mechanisms, biological functions, and diseases. MedComm 2023, 4, e347. [Google Scholar] [CrossRef]
- Chen, C.; Wang, D.; Yu, Y.; Zhao, T.; Min, N.; Wu, Y.; Kang, L.; Zhao, Y.; Du, L.; Zhang, M.; et al. Legumain promotes tubular ferroptosis by facilitating chaperone-mediated autophagy of GPX4 in AKI. Cell Death Dis. 2021, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cai, J.; Zhao, X.; Ma, L.; Zeng, P.; Zhou, L.; Liu, Y.; Yang, S.; Cai, Z.; Zhang, S.; et al. Palmitoylation prevents sustained inflammation by limiting NLRP3 inflammasome activation through chaperone-mediated autophagy. Mol. Cell 2023, 83, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Cayo, A.; Segovia, R.; Venturini, W.; Moore-Carrasco, R.; Valenzuela, C.; Brown, N. mTOR activity and autophagy in senescent cells, a complex partnership. Int. J. Mol. Sci. 2021, 22, 8149. [Google Scholar] [CrossRef] [PubMed]
- Foerster, E.G.; Mukherjee, T.; Cabral-Fernandes, L.; Rocha, J.D.B.; Girardin, S.E.; Philpott, D.J. How autophagy controls the intestinal epithelial barrier. Autophagy 2022, 18, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, A.; Tryon, L.D.; Pauly, M.; Hood, D.A. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am. J. Physiol. Cell Physiol. 2015, 308, C710–C719. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ji, L.; Hu, J.; Zhao, Y.; Johnston, L.J.; Zhang, X.; Ma, X. Functional amino acids and autophagy: Diverse signal transduction and application. Int. J. Mol. Sci. 2021, 22, 11427. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Codogno, P. The mechanism and physiological function of macroautophagy. J. Innate Immun. 2013, 5, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Mizushima, N. At the end of the autophagic road: An emerging understanding of lysosomal functions in autophagy. Trends Biochem. Sci. 2014, 39, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Yin, C.; Fan, Q.; Yan, G.; Wang, Z.; Li, X.; Chen, C.; Yang, X.; Liu, L.; Zheng, Z.; et al. Global liver proteome analysis using iTRAQ reveals AMPK-mTOR-autophagy signaling is altered by intrauterine growth restriction in newborn piglets. J. Proteome Res. 2016, 15, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Ke, Z.Q.; Guo, S.; Yang, X.S.; Zhang, F.X.; Liu, X.F.; Chen, X.; Chen, H.G.; Ke, H.Y.; Liu, C. Curcumin protects against diabetic cardiomyopathy by promoting autophagy and alleviating apoptosis. J. Mol. Cell. Cardiol. 2018, 124, 26–34. [Google Scholar] [CrossRef]
- Jin, C.L.; Ye, J.L.; Yang, J.; Gao, C.Q.; Yan, H.C.; Li, H.C.; Wang, X.Q. mTORC1 mediates lysine-induced satellite cell activation to promote skeletal muscle growth. Cells 2019, 8, 1549. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wang, X.Q. Regulation of mTORC1 by Small GTPases in response to nutrients. J. Nutr. 2020, 150, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.Y.; Neufeld, T.P. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol. Biol. Cell 2009, 20, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Espert, L.; Biard-Piechaczyk, M.; Codogno, P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 2008, 90, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.; Gomez-Sanchez, C.E.; Plonczynski, M.; Naray-Fejes-Toth, A.; Fejes-Toth, G.; Gomez-Sanchez, E.P. mTOR Regulates mineralocorticoid receptor transcriptional activity by ULK1-dependent and -independent mechanisms. Endocrinology 2024, 165, bqae015. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Lee, D.H.; Kim, D.H. Redefining the role of AMPK in autophagy and the energy stress response. Nat. Commun. 2023, 14, 2994. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn II, G.W.; Kang, J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef]
- Yen, W.L.; Legakis, J.E.; Nair, U.; Klionsky, D.J. Atg27 is required for autophagy-dependent cycling of Atg9. Mol. Biol. Cell 2007, 18, 581–593. [Google Scholar] [CrossRef]
- Binotti, B.; Ninov, M.; Cepeda, A.P.; Ganzella, M.; Matti, U.; Riedel, D.; Urlaub, H.; Sambandan, S.; Jahn, R. ATG9 resides on a unique population of small vesicles in presynaptic nerve terminals. Autophagy 2023, 26, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Walczak, M.; Martens, S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 2013, 9, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.M.; Maruyama, T.; Noshiro, D.; Kakuta, C.; Kotani, T.; Nakatogawa, H.; Noda, N.N. Complete set of the Atg8-E1-E2-E3 conjugation machinery forms an interaction web that mediates membrane shaping. Nat. Struct. Mol. Biol. 2024, 31, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Fracchiolla, D. Activation and targeting of ATG8 protein lipidation. Cell Discov. 2020, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Popelka, H.; Reinhart, E.F.; Metur, S.P.; Leary, K.A.; Ragusa, M.J.; Klionsky, D.J. Membrane binding and homodimerization of atg16 via two distinct protein regions is essential for autophagy in yeast. J. Mol. Biol. 2021, 433, 166809. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Noda, N.N.; Nakatogawa, H.; Ohsumi, Y.; Inagaki, F. Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J. Biol. Chem. 2010, 285, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Noda, N.N.; Nakatogawa, H.; Kumeta, H.; Ohsumi, Y.; Inagaki, F. Autophagy-related protein 8 (Atg8) family interacting motif in Atg3 mediates the Atg3-Atg8 interaction and is crucial for the cytoplasm-to-vacuole targeting pathway. J. Biol. Chem. 2010, 285, 29599–29607. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ouyang, Y.; Yin, H.; Cui, H.; Deng, H.; Liu, H.; Jian, Z.; Fang, J.; Zuo, Z.; Wang, X.; et al. Induction of autophagy via the ROS-dependent AMPK-mTOR pathway protects copper-induced spermatogenesis disorder. Redox Biol. 2022, 49, 102227. [Google Scholar] [CrossRef]
- Lu, Q.B.; Ding, Y.; Liu, Y.; Wang, Z.C.; Wu, Y.J.; Niu, K.M.; Li, K.X.; Zhang, J.R.; Sun, H.J. Metrnl ameliorates diabetic cardiomyopathy via inactivation of cGAS/STING signaling dependent on LKB1/AMPK/ULK1-mediated autophagy. J. Adv. Res. 2023, 51, 161–179. [Google Scholar] [CrossRef]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Tan, V.P.; Miyamoto, S. Nutrient-sensing mTORC1: Integration of metabolic and autophagic signals. J. Mol. Cell Cardiol. 2016, 95, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Mortimore, G.E.; Schworer, C.M. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature 1977, 270, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Park, S.J.; Stamatakou, E.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat. Commun. 2020, 11, 3148. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Lee, E.X.; Gordon, K.L.; Paz, E.A.; Shen, W.C.; Ohnishi, K.; Meisenhelder, J.; Hunter, T.; La Spada, A.R. MAP4K3 mediates amino acid-dependent regulation of autophagy via phosphorylation of TFEB. Nat. Commun. 2018, 9, 942. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Li, F.; Tan, K.; Liu, H.; Li, Y.; Liu, Y.; Kong, X.; Tang, Y.; Wu, G.; Yin, Y. Key mediators of intracellular amino acids signaling to mTORC1 activation. Amino Acids 2015, 47, 857–867. [Google Scholar] [CrossRef]
- Jin, C.L.; Zhang, Z.M.; Song, Z.W.; Gao, C.Q.; Yan, H.C.; Wang, X.Q. mTORC1-Mediated Satellite cell differentiation is required for lysine-induced skeletal muscle growth. J. Agric. Food Chem. 2020, 68, 4884–4892. [Google Scholar] [CrossRef] [PubMed]
- Palma-Granados, P.; Haro, A.; Seiquer, I.; Lara, L.; Aguilera, J.F.; Nieto, R. Similar effects of lysine deficiency in muscle biochemical characteristics of fatty and lean piglets. J. Anim. Sci. 2017, 95, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ito, Y.; Nagasawa, T. Regulation of skeletal muscle protein degradation and synthesis by oral administration of lysine in rats. J. Nutr. Sci. Vitaminol. 2013, 59, 412–419. [Google Scholar] [CrossRef]
- Sato, T.; Ito, Y.; Nagasawa, T. Lysine suppresses myofibrillar protein degradation by regulating the autophagic-lysosomal system through phosphorylation of Akt in C2C12 cells. Springerplus 2014, 3, 584. [Google Scholar] [CrossRef]
- Sato, T.; Ito, Y.; Nagasawa, T. L-Lysine suppresses myofibrillar protein degradation and autophagy in skeletal muscles of senescence-accelerated mouse prone 8. Biogerontology 2017, 18, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Seibert, M.; Kurrle, N.; Schnütgen, F.; Serve, H. Amino acid sensory complex proteins in mTORC1 and macroautophagy regulation. Matrix Biol. 2021, 100–101, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ito, Y.; Nedachi, T.; Nagasawa, T. Lysine suppresses protein degradation through autophagic-lysosomal system in C2C12 myotubes. Mol. Cell. Biochem. 2014, 391, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.M.; Kazi, A.A.; Hong-Brown, L.; Lang, C.H. Delayed recovery of skeletal muscle mass following hindlimb immobilization in mTOR heterozygous mice. PLoS ONE 2012, 7, e38910. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Ding, C.; Tong, H.; Yan, Y.; Li, S.; Li, S.; Cao, Y. Leu promotes C2C12 cell differentiation by regulating the GSK3β/β-catenin signaling pathway through facilitating the interaction between SESN2 and RPN2. J. Sci. Food Agric. 2024, 104, 6696–6705. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, F.; Hu, S.; Yin, C.; Li, X.; Zhao, S.; Wang, J.; Yan, X. MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell. Signal. 2012, 24, 2179–2186. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhong, Q.; Yan, X. YWHAE/14-3-3ε crotonylation regulates leucine deprivation-induced autophagy. Autophagy 2023, 19, 2401–2402. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.M.; Tee, A.R. Leucine and mTORC1: A complex relationship. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1329–E1342. [Google Scholar] [CrossRef]
- Escobar, J.; Frank, J.W.; Suryawan, A.; Nguyen, H.V.; Kimball, S.R.; Jefferson, L.S.; Davis, T.A. Physiological rise in plasma leucine stimulates muscle protein synthesis in neonatal pigs by enhancing translation initiation factor activation. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E914–E921. [Google Scholar] [CrossRef]
- Yoshizawa, F. Regulation of protein synthesis by branched-chain amino acids in vivo. Biochem. Biophys. Res. Commun. 2004, 313, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Nüse, B.; Holland, T.; Rauh, M.; Gerlach, R.G.; Mattner, J. L-arginine metabolism as pivotal interface of mutual host-microbe interactions in the gut. Gut Microbes 2023, 15, 2222961. [Google Scholar] [CrossRef] [PubMed]
- Savaraj, N.; You, M.; Wu, C.; Wangpaichitr, M.; Kuo, M.T.; Feun, L.G. Arginine deprivation, autophagy, apoptosis (AAA) for the treatment of melanoma. Curr. Mol. Med. 2010, 10, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yin, P.; Liu, F.; Liu, Y.; Liu, Y.; Xia, Z. Protective effects of L-arginine on the intestinal epithelial barrier under heat stress conditions in rats and IEC-6 cell line. J. Anim. Physiol. Anim. Nutr. 2020, 104, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Castejón-Vega, B.; Rubio, A.; Pérez-Pulido, A.J.; Quiles, J.L.; Lane, J.D.; Fernández-Domínguez, B.; Cachón-González, M.B.; Martín-Ruiz, C.; Sanz, A.; Cox, T.M.; et al. L-Arginine ameliorates defective autophagy in GM2 gangliosidoses by mTOR modulation. Cells 2021, 10, 3122. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Bazer, F.W.; Johnson, G.A.; Knabe, D.A.; Burghardt, R.C.; Spencer, T.E.; Li, X.L.; Wang, J.J. Triennial growth symposium: Important roles for L-glutamine in swine nutrition and production. J. Anim. Sci. 2011, 89, 2017–2030. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.W.S.; Sim, A.Y.L.; Long, Y.C. Glutamine metabolism regulates autophagy-dependent mTORC1 reactivation during amino acid starvation. Nat. Commun. 2017, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Lin, G.; Dai, Z.; Zhou, T.; Li, T.; Yuan, T.; Wu, L.; Wu, J.; Wang, J. L-Glutamine deprivation induces autophagy and alters the mTOR and MAPK signaling pathways in porcine intestinal epithelial cells. Amino Acids 2015, 47, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ito, Y.; Nagasawa, T. Dietary L-lysine suppresses autophagic proteolysis and stimulates Akt/mTOR signaling in the skeletal muscle of rats fed a low-protein diet. J. Agric. Food Chem. 2015, 63, 8192–8198. [Google Scholar] [CrossRef]
- Crozier, S.J.; Kimball, S.R.; Emmert, S.W.; Anthony, J.C.; Jefferson, L.S. Oral leucine administration stimulates protein synthesis in rat skeletal muscle. J. Nutr. 2005, 135, 376–382. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.T.; Lisi, R.D.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Zeanandin, G.; Balage, M.; Schneider, S.M.; Dupont, J.; Hébuterne, X.; Mothe-Satney, I.; Dardevet, D. Differential effect of long-term leucine supplementation on skeletal muscle and adipose tissue in old rats: An insulin signaling pathway approach. Age 2012, 34, 371–387. [Google Scholar] [CrossRef]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Mordier, S.; Deval, C.; Béchet, D.; Tassa, A.; Ferrara, M. Leucine limitation induces autophagy and activation of lysosome-dependent proteolysis in C2C12 myotubes through a mammalian target of rapamycin-independent signaling pathway. J. Biol. Chem. 2000, 275, 29900–29906. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.J.; Gao, Y.Y.; Zhang, J.; Wang, L.; Zhao, S.; Che, Y.Y.; Ao, C.J.; Yang, H.J.; Wang, J.Q.; Lei, L.C. Autophagy mediated by arginine depletion activation of the nutrient sensor GCN2 contributes to interferon-γ-induced malignant transformation of primary bovine mammary epithelial cells. Cell Death Discov. 2016, 2, 15065. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Korolchuk, V.I.; Renna, M.; Imarisio, S.; Fleming, A.; Williams, A.; Garcia-Arencibia, M.; Rose, C.; Luo, S.; Underwood, B.R.; et al. Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell 2011, 43, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Angcajas, A.B.; Hirai, N.; Kaneshiro, K.; Karim, M.R.; Horii, Y.; Kubota, M.; Fujimura, S.; Kadowaki, M. Diversity of amino acid signaling pathways on autophagy regulation: A novel pathway for arginine. Biochem. Biophys. Res. Commun. 2014, 446, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Wilden, A.R.; Molina, J.A.; Feuerborn, M.; Boyle, D.; Lee, S.Y. Glutamine-dependent lysosome homeostatic changes induced by starvation and lysosome inhibition. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1356–1367. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Lin, J.; Su, J.; Chen, X.; Jiang, P.; Huang, K. Glutamine deficiency promotes PCV2 infection through induction of autophagy via activation of ROS-mediated JAK2/STAT3 signaling pathway. J. Agric. Food Chem. 2018, 66, 11757–11766. [Google Scholar] [CrossRef]
- Vernizzi, L.; Paiardi, C.; Licata, G.; Vitali, T.; Santarelli, S.; Raneli, M.; Manelli, V.; Rizzetto, M.; Gioria, M.; Pasini, M.E.; et al. Glutamine synthetase 1 increases autophagy lysosomal degradation of mutant huntingtin aggregates in neurons, ameliorating motility in a drosophila model for Huntington’s disease. Cells 2020, 9, 196. [Google Scholar] [CrossRef]
- van der Vos, K.E.; Coffer, P.J. Glutamine metabolism links growth factor signaling to the regulation of autophagy. Autophagy 2012, 8, 1862–1864. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; de Meyer, G.R.; Herman, A.G.; Kockx, M.M. Amino acid deprivation induces both apoptosis and autophagy in murine C2C12 muscle cells. Biotechnol. Lett. 2005, 27, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, C.; Zhu, M.; Ye, J.; Song, Z.; Zheng, C.; Chen, W. Autophagy: Are Amino Acid Signals Dependent on the mTORC1 Pathway or Independent? Curr. Issues Mol. Biol. 2024, 46, 8780-8793. https://doi.org/10.3390/cimb46080519
Jin C, Zhu M, Ye J, Song Z, Zheng C, Chen W. Autophagy: Are Amino Acid Signals Dependent on the mTORC1 Pathway or Independent? Current Issues in Molecular Biology. 2024; 46(8):8780-8793. https://doi.org/10.3390/cimb46080519
Chicago/Turabian StyleJin, Chenglong, Min Zhu, Jinling Ye, Zhiwen Song, Chuntian Zheng, and Wei Chen. 2024. "Autophagy: Are Amino Acid Signals Dependent on the mTORC1 Pathway or Independent?" Current Issues in Molecular Biology 46, no. 8: 8780-8793. https://doi.org/10.3390/cimb46080519