
Targeting SHP2 with Natural Products: Exploring Saponin-Based Allosteric Inhibitors and Their Therapeutic Potential

Abstract
1. Introduction
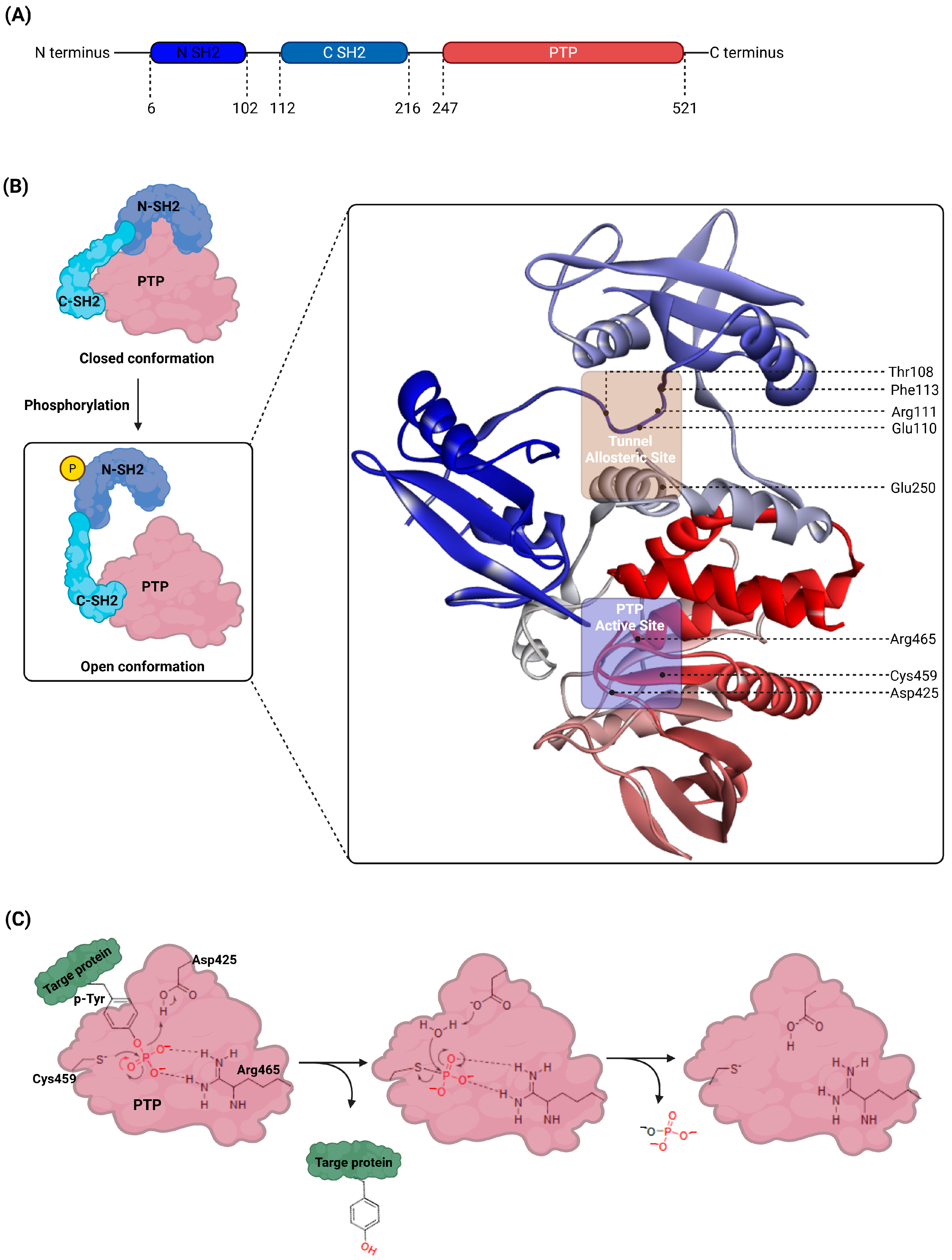
2. Structural and Functional Dynamics of SHP2
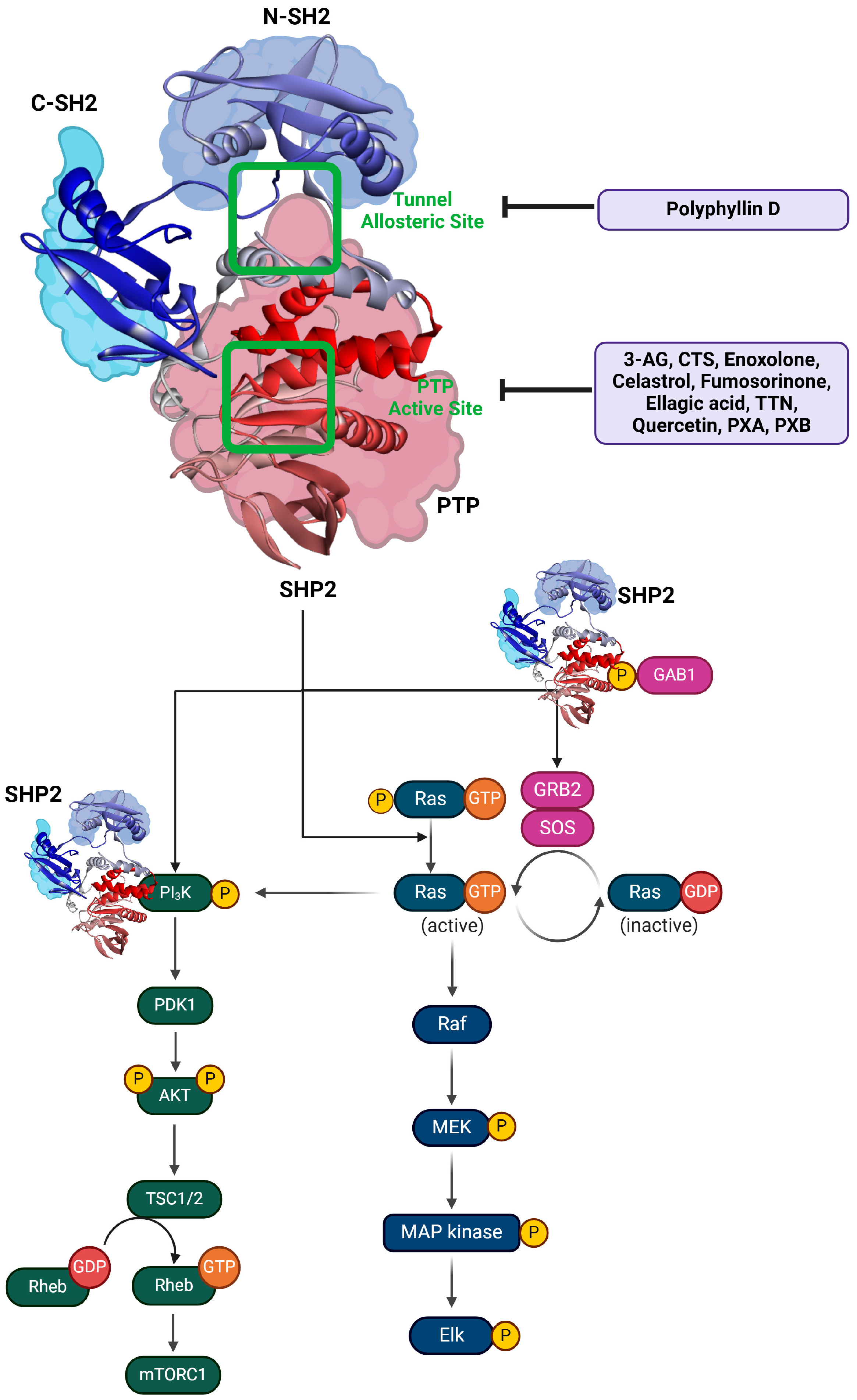
3. Natural Products as Promising SHP2 Inhibitors
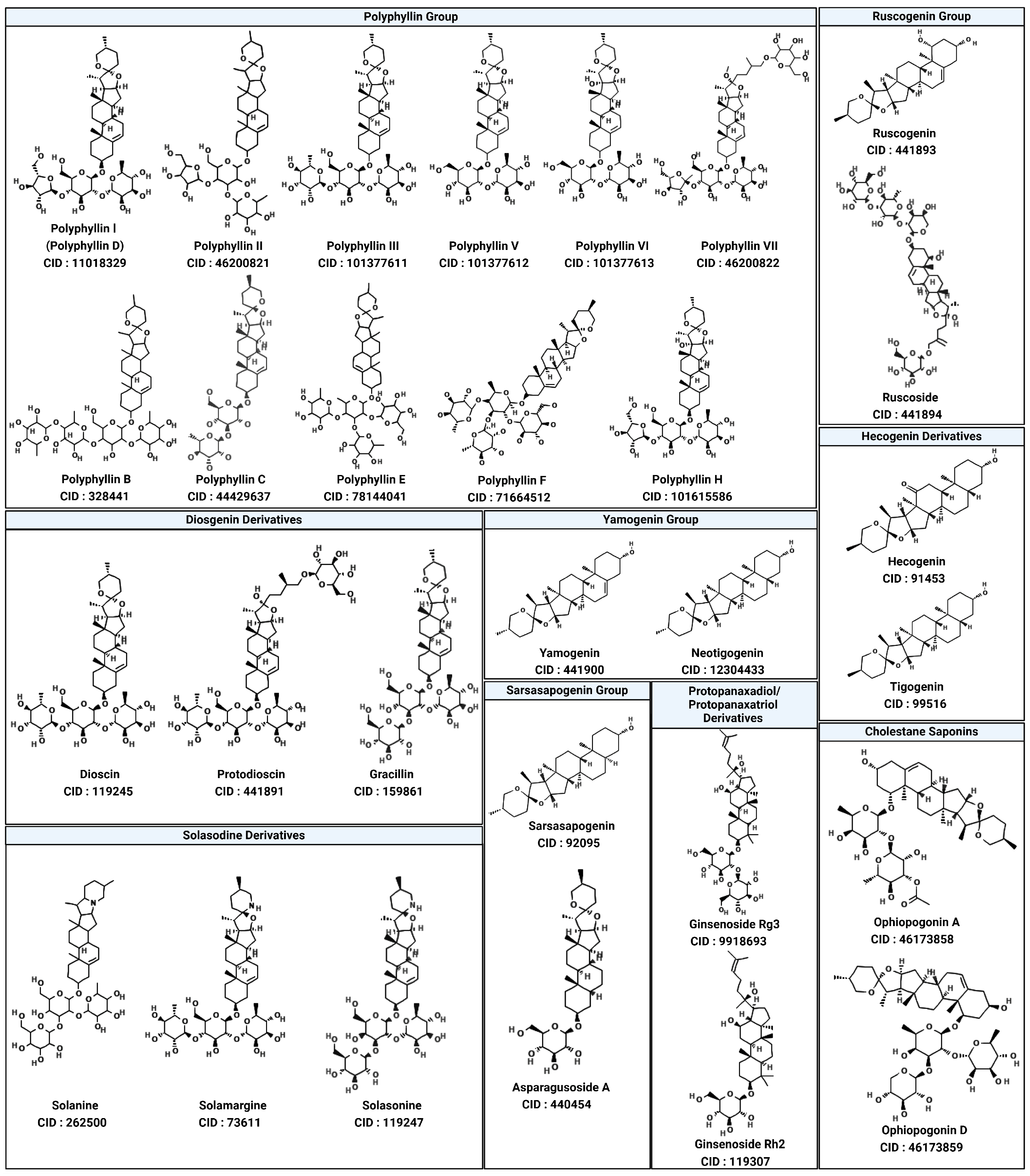
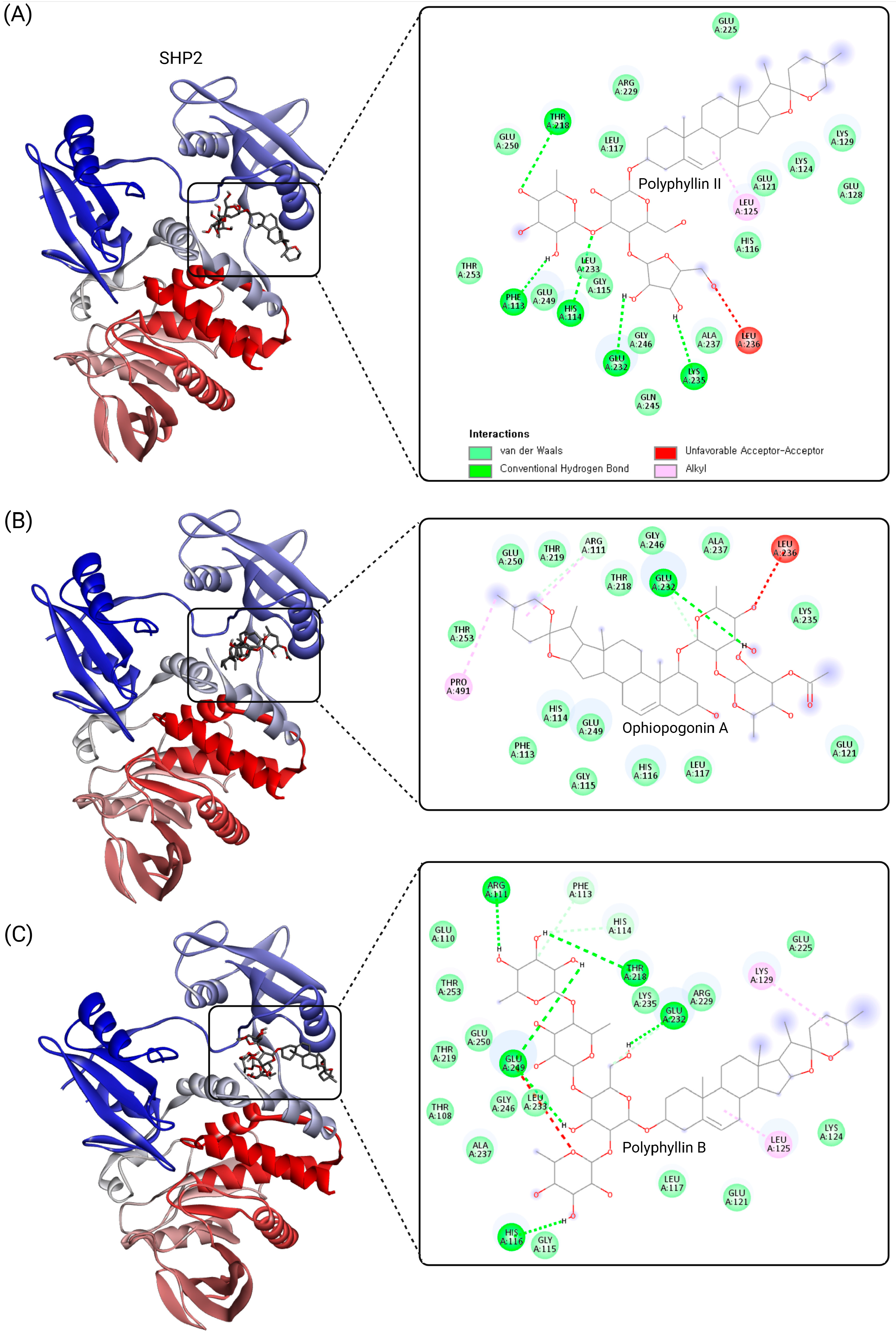
4. Exploring Saponins as Allosteric Inhibitors of SHP2
5. Future Strategies for SHP2 Inhibition Using Saponin-Based Natural Products
5.1. Combination Strategies for Enhanced Therapeutic Outcomes
5.2. Enhancing Immune-Mediated Cancer Clearance
5.3. Optimizing Saponin-Based Therapeutics
5.4. Enhancing Binding Affinity
5.5. Developing PROTAC-Based Therapies
6. Conclusions
Funding
Conflicts of Interest
References
- Welsh, C.L.; Allen, S.; Madan, L.K. Setting sail: Maneuvering SHP2 activity and its effects in cancer. Adv. Cancer Res. 2023, 160, 17–60. [Google Scholar] [PubMed]
- Asmamaw, M.D.; Shi, X.J.; Zhang, L.R.; Liu, H.M. A comprehensive review of SHP2 and its role in cancer. Cell. Oncol. 2022, 45, 729–753. [Google Scholar] [CrossRef]
- Feng, G.S.; Hui, C.C.; Pawson, T. SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science 1993, 259, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Hof, P.; Pluskey, S.; Dhe-Paganon, S.; Eck, M.J.; Shoelson, S.E. Crystal structure of the tyrosine phosphatase SHP-2. Cell 1998, 92, 441–450. [Google Scholar] [CrossRef]
- Wu, X.; Xu, G.; Li, X.; Xu, W.; Li, Q.; Liu, W.; Kirby, K.A.; Loh, M.L.; Li, J.; Sarafianos, S.G.; et al. Small Molecule Inhibitor that Stabilizes the Autoinhibited Conformation of the Oncogenic Tyrosine Phosphatase SHP2. J. Med. Chem. 2019, 62, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Bobone, S.; Pannone, L.; Biondi, B.; Solman, M.; Flex, E.; Canale, V.C.; Calligari, P.; De Faveri, C.; Gandini, T.; Quercioli, A.; et al. Targeting Oncogenic Src Homology 2 Domain-Containing Phosphatase 2 (SHP2) by Inhibiting Its Protein-Protein Interactions. J. Med. Chem. 2021, 64, 15973–15990. [Google Scholar] [CrossRef]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- Liotti, F.; Kumar, N.; Prevete, N.; Marotta, M.; Sorriento, D.; Ieranò, C.; Ronchi, A.; Marino, F.Z.; Moretti, S.; Colella, R.; et al. PD-1 blockade delays tumor growth by inhibiting an intrinsic SHP2/Ras/MAPK signalling in thyroid cancer cells. J. Exp. Clin. Cancer Res. 2021, 40, 22. [Google Scholar] [CrossRef]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Tsiaras, W.G.; Araki, T.; Wen, G.; Minichiello, L.; Klein, R.; Neel, B.G. Receptor-specific regulation of phosphatidylinositol 3′-kinase activation by the protein tyrosine phosphatase Shp2. Mol. Cell Biol. 2002, 22, 4062–4072. [Google Scholar] [CrossRef]
- Alhumaid, M.S.; Dasouki, M.J.; Ahmed, S.O.; AbalKhail, H.; Hagos, S.; Wakil, S.; Hashmi, S.K. Comprehensive Genomic Analysis of Noonan Syndrome and Acute Myeloid Leukemia in Adults: A Review and Future Directions. Acta Haematol. 2020, 143, 583–593. [Google Scholar] [CrossRef]
- Araki, T.; Mohi, M.G.; Ismat, F.A.; Bronson, R.T.; Williams, I.R.; Kutok, J.L.; Yang, W.; Pao, L.I.; Gilliland, D.G.; Epstein, J.A.; et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat. Med. 2004, 10, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, X.; Fang, H.; Liu, Y.; Chen, D.; Zhang, Q.; Liu, X.; Wei, D.; Qu, C.; Wang, S. A tyrosine phosphatase SHP2 gain-of-function mutation enhances malignancy of breast carcinoma. Oncotarget 2016, 7, 5664–5676. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Wang, M.; Ge, Y.; Chen, X.P.; Xu, Z.; Sun, Y.; Xiong, X.F. Tyrosine phosphatase SHP2 inhibitors in tumor-targeted therapies. Acta Pharm. Sin. B 2021, 11, 13–29. [Google Scholar] [CrossRef]
- Huang, C.F.; Mrksich, M. Profiling Protein Tyrosine Phosphatase Specificity with Self-Assembled Monolayers for Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry and Peptide Arrays. ACS Comb. Sci. 2019, 21, 760–769. [Google Scholar] [CrossRef]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; de Wit, N.; Gonçalves-Ribeiro, S.; et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, R.; Ran, H.; Neel, B.G. Off-target inhibition by active site-targeting SHP2 inhibitors. FEBS Open Bio. 2018, 8, 1405–1411. [Google Scholar] [CrossRef]
- Kerr, D.L.; Haderk, F.; Bivona, T.G. Allosteric SHP2 inhibitors in cancer: Targeting the intersection of RAS, resistance, and the immune microenvironment. Curr. Opin. Chem. Biol. 2021, 62, 1–12. [Google Scholar] [CrossRef]
- Guo, Z.; Duan, Y.; Sun, K.; Zheng, T.; Liu, J.; Xu, S.; Xu, J. Advances in SHP2 tunnel allosteric inhibitors and bifunctional molecules. Eur. J. Med. Chem. 2024, 275, 116579. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, H.; Zhao, G.; Wang, R. Allosteric Inhibitors of SHP2: An Updated Patent Review (2015–2020). Curr. Med. Chem. 2021, 28, 3825–3842. [Google Scholar] [CrossRef]
- Garcia Fortanet, J.; Chen, C.H.; Chen, Y.N.; Chen, Z.; Deng, Z.; Firestone, B.; Fekkes, P.; Fodor, M.; Fortin, P.D.; Fridrich, C.; et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J. Med. Chem. 2016, 59, 7773–7782. [Google Scholar] [CrossRef]
- Song, Y.; Zhao, M.; Zhang, H.; Yu, B. Double-edged roles of protein tyrosine phosphatase SHP2 in cancer and its inhibitors in clinical trials. Pharmacol. Ther. 2022, 230, 107966. [Google Scholar] [CrossRef] [PubMed]
- LaMarche, M.J.; Acker, M.; Argintaru, A.; Bauer, D.; Boisclair, J.; Chan, H.; Chen, C.H.; Chen, Y.N.; Chen, Z.; Deng, Z.; et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 13578–13594. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, S.; Zhao, M.; Yang, X.; Yu, B. Strategies Targeting Protein Tyrosine Phosphatase SHP2 for Cancer Therapy. J. Med. Chem. 2022, 65, 3066–3079. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yu, D.; Li, H.; Qin, P.; Chen, H.; Chen, L. Promising natural products targeting protein tyrosine phosphatase SHP2 for cancer therapy. Phytother. Res. 2024, 39, 1735–1757. [Google Scholar] [CrossRef]
- Kwon, S.J.; Ahn, D.; Yang, H.M.; Kang, H.J.; Chung, S.J. Polyphyllin D Shows Anticancer Effect through a Selective Inhibition of Src Homology Region 2-Containing Protein Tyrosine Phosphatase-2 (SHP2). Molecules 2021, 26, 848. [Google Scholar] [CrossRef]
- Song, Y.; Zhao, M.; Wu, Y.; Yu, B.; Liu, H.M. A multifunctional cross-validation high-throughput screening protocol enabling the discovery of new SHP2 inhibitors. Acta Pharm. Sin. B 2021, 11, 750–762. [Google Scholar] [CrossRef]
- Neel, B.G.; Gu, H.; Pao, L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef]
- Pádua, R.A.P.; Sun, Y.; Marko, I.; Pitsawong, W.; Stiller, J.B.; Otten, R.; Kern, D. Mechanism of activating mutations and allosteric drug inhibition of the phosphatase SHP2. Nat. Commun. 2018, 9, 4507. [Google Scholar] [CrossRef]
- Araki, T.; Nawa, H.; Neel, B.G. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J. Biol. Chem. 2003, 278, 41677–41684. [Google Scholar] [CrossRef]
- LaRochelle, J.R.; Fodor, M.; Vemulapalli, V.; Mohseni, M.; Wang, P.; Stams, T.; LaMarche, M.J.; Chopra, R.; Acker, M.G.; Blacklow, S.C. Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat. Commun. 2018, 9, 4508. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.G.; Dubé, N.; Hardy, S.; Tremblay, M.L. Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 2011, 11, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.Y.; Yu, Z.H.; Chen, L.; Walls, C.D.; Zhang, S.; Wu, L.; Zhang, Z.Y. Mechanistic insights explain the transforming potential of the T507K substitution in the protein-tyrosine phosphatase SHP2. J. Biol. Chem. 2020, 295, 6187–6201. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.G.; Tonks, N.K. Protein tyrosine phosphatases in signal transduction. Curr. Opin. Cell Biol. 1997, 9, 193–204. [Google Scholar] [CrossRef]
- Pannifer, A.D.; Flint, A.J.; Tonks, N.K.; Barford, D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography. J. Biol. Chem. 1998, 273, 10454–10462. [Google Scholar] [CrossRef]
- Rehman, A.U.; Rahman, M.U.; Khan, M.T.; Saud, S.; Liu, H.; Song, D.; Sultana, P.; Wadood, A.; Chen, H.F. The Landscape of Protein Tyrosine Phosphatase (Shp2) and Cancer. Curr. Pharm. Des. 2018, 24, 3767–3777. [Google Scholar] [CrossRef]
- Agazie, Y.M.; Hayman, M.J. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol. Cell Biol. 2003, 23, 7875–7886. [Google Scholar] [CrossRef]
- Dance, M.; Montagner, A.; Salles, J.P.; Yart, A.; Raynal, P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal 2008, 20, 453–459. [Google Scholar] [CrossRef]
- Li, J.; Reed, S.A.; Johnson, S.E. Hepatocyte growth factor (HGF) signals through SHP2 to regulate primary mouse myoblast proliferation. Exp. Cell Res. 2009, 315, 2284–2292. [Google Scholar] [CrossRef]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef]
- Matalkah, F.; Martin, E.; Zhao, H.; Agazie, Y.M. SHP2 acts both upstream and downstream of multiple receptor tyrosine kinases to promote basal-like and triple-negative breast cancer. Breast Cancer Res. 2016, 18, 2. [Google Scholar] [CrossRef]
- Daly, R.J.; Gu, H.; Parmar, J.; Malaney, S.; Lyons, R.J.; Kairouz, R.; Head, D.R.; Henshall, S.M.; Neel, B.G.; Sutherland, R.L. The docking protein Gab2 is overexpressed and estrogen regulated in human breast cancer. Oncogene 2002, 21, 5175–5181. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Montagner, A.; Lee, W.H.; Miteva, M.; Vidal, M.; Vidaud, M.; Parfait, B.; Raynal, P. Reduced phosphatase activity of SHP-2 in LEOPARD syndrome: Consequences for PI3K binding on Gab1. FEBS Lett. 2006, 580, 2477–2482. [Google Scholar] [CrossRef]
- Montagner, A.; Yart, A.; Dance, M.; Perret, B.; Salles, J.P.; Raynal, P. A novel role for Gab1 and SHP2 in epidermal growth factor-induced Ras activation. J. Biol. Chem. 2005, 280, 5350–5360. [Google Scholar] [CrossRef] [PubMed]
- Yart, A.; Laffargue, M.; Mayeux, P.; Chretien, S.; Peres, C.; Tonks, N.; Roche, S.; Payrastre, B.; Chap, H.; Raynal, P. A critical role for phosphoinositide 3-kinase upstream of Gab1 and SHP2 in the activation of ras and mitogen-activated protein kinases by epidermal growth factor. J. Biol. Chem. 2001, 276, 8856–8864. [Google Scholar] [CrossRef]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Zhang, L.; Song, J.; Kong, L.; Yuan, T.; Li, W.; Zhang, W.; Hou, B.; Lu, Y.; Du, G. The strategies and techniques of drug discovery from natural products. Pharmacol. Ther. 2020, 216, 107686. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 2022, 80, 1–17. [Google Scholar] [CrossRef]
- Liu, D.M.; Cao, Z.X.; Yan, H.L.; Li, W.; Yang, F.; Zhao, W.J.; Diao, Q.C.; Tan, Y.Z. A new abietane diterpenoid from Ajuga ovalifolia var. calantha induces human lung epithelial A549 cell apoptosis by inhibiting SHP2. Fitoterapia 2020, 141, 104484. [Google Scholar] [CrossRef]
- Liu, W.; Yu, B.; Xu, G.; Xu, W.R.; Loh, M.L.; Tang, L.D.; Qu, C.K. Identification of cryptotanshinone as an inhibitor of oncogenic protein tyrosine phosphatase SHP2 (PTPN11). J. Med. Chem. 2013, 56, 7212–7221. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Chen, L.; Daniel, K.G.; Brooks, W.H.; Guida, W.C.; Lawrence, H.R.; Sebti, S.M.; Lawrence, N.J.; Wu, J. Shp2 protein tyrosine phosphatase inhibitor activity of estramustine phosphate and its triterpenoid analogs. Bioorg Med. Chem. Lett. 2011, 21, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, X.; Meng, Q.; Li, Y.; Shen, H.; Fu, Y.; Zhang, F.; Chen, J.; Zhang, W.; Chang, W.; et al. SHP2 inhibition improves celastrol-induced growth suppression of colorectal cancer. Front. Pharmacol. 2022, 13, 929087. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cao, M.; Zhu, S.; Wang, C.; Liang, F.; Yan, L.; Luo, D. Discovery of a Novel Inhibitor of the Protein Tyrosine Phosphatase Shp2. Sci. Rep. 2015, 5, 17626. [Google Scholar] [CrossRef]
- Ma, C.H.; Zhao, J.F.; Zhang, X.G.; Ding, C.H.; Hao, H.H.; Ji, Y.H.; Li, L.P.; Guo, Z.T.; Liu, W.S. Discovery of ellagic acid as a competitive inhibitor of Src homology phosphotyrosyl phosphatase 2 (SHP2) for cancer treatment: In vitro and in silico study. Int. J. Biol. Macromol. 2024, 254 Pt 2, 127845. [Google Scholar] [CrossRef]
- Liu, S.; Yu, Z.; Yu, X.; Huang, S.X.; Luo, Y.; Wu, L.; Shen, W.; Yang, Z.; Wang, L.; Gunawan, A.M.; et al. SHP2 is a target of the immunosuppressant tautomycetin. Chem. Biol. 2011, 18, 101–110. [Google Scholar] [CrossRef]
- Igbe, I.; Shen, X.F.; Jiao, W.; Qiang, Z.; Deng, T.; Li, S.; Liu, W.L.; Liu, H.W.; Zhang, G.L.; Wang, F. Dietary quercetin potentiates the antiproliferative effect of interferon-α in hepatocellular carcinoma cells through activation of JAK/STAT pathway signaling by inhibition of SHP2 phosphatase. Oncotarget 2017, 8, 113734–113748. [Google Scholar] [CrossRef]
- Yang, R.; Dong, Q.; Xu, H.; Gao, X.; Zhao, Z.; Qin, J.; Chen, C.; Luo, D. Identification of Phomoxanthone A and B as Protein Tyrosine Phosphatase Inhibitors. ACS Omega 2020, 5, 25927–25935. [Google Scholar] [CrossRef]
- He, R.; Yu, Z.H.; Zhang, R.Y.; Wu, L.; Gunawan, A.M.; Lane, B.S.; Shim, J.S.; Zeng, L.F.; He, Y.; Chen, L.; et al. Exploring the Existing Drug Space for Novel pTyr Mimetic and SHP2 Inhibitors. ACS Med. Chem. Lett. 2015, 6, 782–786. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Dong, Z.J.; Li, Z.H.; Yin, X.; Yang, X.Y.; Feng, T.; Wei, K.; Liu, J.K.; Zhao, B.H. Two new ylangene-type sesquiterpenoids from cultures of the fungus Postia sp. J. Asian Nat. Prod. Res. 2014, 16, 254–258. [Google Scholar] [CrossRef]
- Zhang, W.; Hong, D.; Zhou, Y.; Zhang, Y.; Shen, Q.; Li, J.Y.; Hu, L.H.; Li, J. Ursolic acid and its derivative inhibit protein tyrosine phosphatase 1B, enhancing insulin receptor phosphorylation and stimulating glucose uptake. Biochim. Biophys. Acta 2006, 1760, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Xiong, J.; Zang, Y.; Li, J.; Osman, E.E.A.; Li, J.Y.; Zhou, Y.B.; Li, J.; Hu, J.F. Phytochemical and biological studies on rare and endangered plants endemic to China. Part XIV. Structurally diverse terpenoids from the twigs and needles of the endangered plant Picea brachytyla. Phytochemistry 2020, 169, 112161. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wei, J.; Chen, D.Y.; Wang, M.J.; Sun, Y.; Jiao, F.W.; Jiao, R.H.; Tan, R.X.; Ge, H.M. Study on the secondary metabolites of grasshopper-derived fungi Arthrinium sp. NF2410. Chin. J. Nat. Med. 2020, 18, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Huo, C.; Lu, X.; Zheng, Z.; Li, Y.; Xu, Y.; Zheng, H.; Niu, Y. Azaphilones with protein tyrosine phosphatase inhibitory activity isolated from the fungus Aspergillus deflectus. Phytochemistry 2020, 170, 112224. [Google Scholar] [CrossRef]
- Wu, J.; Deng, Y.; Zhang, X.; Ma, J.; Zheng, X.; Chen, Y. Suchilactone inhibits the growth of acute myeloid leukaemia by inactivating SHP2. Pharm. Biol. 2022, 60, 144–153. [Google Scholar] [CrossRef]
- Chen, C.; Liang, F.; Chen, B.; Sun, Z.; Xue, T.; Yang, R.; Luo, D. Identification of demethylincisterol A(3) as a selective inhibitor of protein tyrosine phosphatase Shp2. Eur. J. Pharmacol. 2017, 795, 124–133. [Google Scholar] [CrossRef]
- Raju, J.; Mehta, R. Cancer chemopreventive and therapeutic effects of diosgenin, a food saponin. Nutr. Cancer 2009, 61, 27–35. [Google Scholar] [CrossRef]
- Elekofehinti, O.O.; Iwaloye, O.; Olawale, F.; Ariyo, E.O. Saponins in Cancer Treatment: Current Progress and Future Prospects. Pathophysiology 2021, 28, 250–272. [Google Scholar] [CrossRef]
- Dong, J.; Liang, W.; Wang, T.; Sui, J.; Wang, J.; Deng, Z.; Chen, D. Saponins regulate intestinal inflammation in colon cancer and IBD. Pharmacol. Res. 2019, 144, 66–72. [Google Scholar] [CrossRef]
- MacDonald, R.S.; Guo, J.; Copeland, J.; Browning, J.D., Jr.; Sleper, D.; Rottinghaus, G.E.; Berhow, M.A. Environmental influences on isoflavones and saponins in soybeans and their role in colon cancer. J. Nutr. 2005, 135, 1239–1242. [Google Scholar] [CrossRef]
- Moses, T.; Papadopoulou, K.K.; Osbourn, A. Metabolic and functional diversity of saponins, biosynthetic intermediates and semi-synthetic derivatives. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 439–462. [Google Scholar] [CrossRef] [PubMed]
- Bouabdallah, S.; Al-Maktoum, A.; Amin, A. Steroidal Saponins: Naturally Occurring Compounds as Inhibitors of the Hallmarks of Cancer. Cancers 2023, 15, 3900. [Google Scholar] [CrossRef] [PubMed]
- Osbourn, A.E. Saponins in cereals. Phytochemistry 2003, 62, 1–4. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y. Protein-Ligand Blind Docking Using CB-Dock2. Methods Mol. Biol. 2024, 2714, 113–125. [Google Scholar]
- Liu, M.; Gao, S.; Elhassan, R.M.; Hou, X.; Fang, H. Strategies to overcome drug resistance using SHP2 inhibitors. Acta Pharm. Sin. B 2021, 11, 3908–3924. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Qu, J.; Zhao, M.; Xu, Q.; Sun, Y. Targeting SHP2 as a promising strategy for cancer immunotherapy. Pharmacol. Res. 2020, 152, 104595. [Google Scholar] [CrossRef]
- Li, J.; Jie, H.B.; Lei, Y.; Gildener-Leapman, N.; Trivedi, S.; Green, T.; Kane, L.P.; Ferris, R.L. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res. 2015, 75, 508–518. [Google Scholar] [CrossRef]
- Sun, Y.; Ding, N.; Song, Y.; Yang, Z.; Liu, W.; Zhu, J.; Rao, Y. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia 2019, 33, 2105–2110. [Google Scholar] [CrossRef]
- Wang, M.; Lu, J.; Wang, M.; Yang, C.Y.; Wang, S. Discovery of SHP2-D26 as a First, Potent, and Effective PROTAC Degrader of SHP2 Protein. J. Med. Chem. 2020, 63, 7510–7528. [Google Scholar] [CrossRef]
- Zhu, Z.Y.; Yao, Q.; Liu, Y.; Si, C.L.; Chen, J.; Liu, N.; Lian, H.Y.; Ding, L.N.; Zhang, Y.M. Highly efficient synthesis and antitumor activity of monosaccharide saponins mimicking components of Chinese folk medicine Cordyceps sinensis. J. Asian Nat. Prod. Res. 2012, 14, 429–435. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Compound | IC50 (µM) | Inhibitory Site | Contact Residues | Ref. |
|---|---|---|---|---|---|
| Terpenoids | 3-acetoxylteuvincenone G (3-AG) 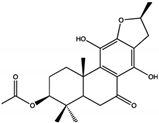 | 10.79 ± 0.14 | PTP Active Site | Lys260, Lys492, Gln256, Gln257, Cys259, Tyr263, Glu313, Gln495, Met496, Arg498, Ser499 | [50] |
Cryptotanshinone (CTS)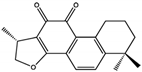 | 22.50 | PTP Active Site | Lys358, Arg362, Lys364, Ser 365 | [51] | |
Enoxolone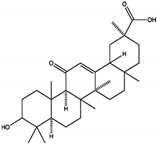 | 9.6 ± 3.2 | PTP Active Site | Arg465, Gln510 | [52] | |
Celastrol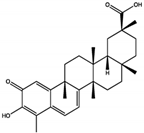 | 3.3 ± 0.6 | PTP Active Site | Arg465, Gln510 | [52] | |
| Alkaloids | Fumosorinone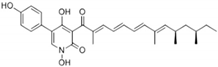 | 6.31 | PTP Active Site (Non-Competitive) | Cys459, Gly427, Ala461, Ser460, Arg465, Tyr279, Lys366 | [54] |
| Polyphenols | Ellagic acid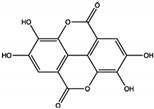 | 0.69 ± 0.07 | PTP Active Site | Trp423, Gly427, Ser460, Gln506, Gln510 | [55] |
| Polyketide | Tautomycetin (TTN) | 2.9 ± 0.2 | PTP Active Site | Arg465, Ala461, Ser460, Gln510, Lys366 | [56] |
| Flavonoids | Quercetin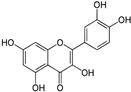 | 10.17 ± 0.21 | PTP Active Site | Arg362, Trp423, Asp425, Arg465, Gln506, Gln510 | [57] |
| Xanthones | Phomoxanthone A (PXA)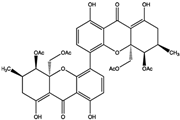 | 20.47 ± 4.45 | PTP Active Site (Non-Competitive) | Lys366, Asp425, Gly427, Ser460, Gly464, Arg465, Gln506, Gln510 | [58] |
Phomoxanthone B (PXB)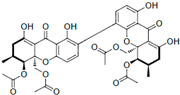 | 11.86 ± 3.02 | PTP Active Site (Non-Competitive) | Lys366, Asp425, Gly427, Ser460, Gly464, Arg465, Gln506, Gln510 | [58] | |
| β-Lactam Antibiotic | Cefsulodin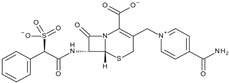 | 16.8 ± 2.0 | PTP Active Site | Ser460, Ala461, Ile463, Gly464, Arg465, Lys366, Tyr279, Gln506 | [59] |
| Saponins | Polyphyllin D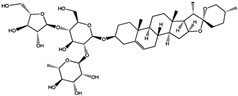 | 15.3 ± 0.8 | Tunnel Allosteric Site | Arg111, Phe113, His114 | [26] |
| Compounds | Binding Site | Vina Score (kcal/mol) | Contact Residues |
|---|---|---|---|
| SHP099 (positive control) 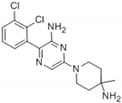 | Tunnel site | −10.4 | Conventional Hydrogen Bond: Thr218, Arg229, Glu249, Thr253, His114, Arg111, Glu110 Van der Waals Interactions: Leu125, Glu232, Glu225, Lys129, Thr219, Glu250 Alkyl Interaction: Lys129 |
| Polyphyllin I (Polyphyllin D) | Tunnel site | −9.6 | Conventional Hydrogen Bond: Thr218, Arg229, Glu249, Thr253, His114, Arg111, Glu110 Van der Waals Interactions: Phe113, Leu125, Glu232, Glu225, Lys129, Thr219, Glu250 Alkyl Interaction: Lys129 |
| Polyphyllin II | Tunnel site | −11.5 | Conventional Hydrogen Bond: Thr 218, Arg 229, Glu 249, Thr 253, His 114 Van der Waals Interactions: Leu 125, Glu 232, Glu 225, Lys 129, Thr 219, Glu 250 Alkyl Interaction: Leu 125 Unfavorable Acceptor–Acceptor: Leu 236 |
| Polyphyllin III | Tunnel site | −10.1 | Conventional Hydrogen Bond: Thr 218, Glu 249, Thr 219, His 114 Van der Waals Interactions: Leu 233, Glu 225, Lys 129, Leu 125, Gly 115 Alkyl Interaction: Lys 129 Unfavorable Donor–Donor: Arg 229 |
| Polyphyllin V | Tunnel site | −7.8 | Conventional Hydrogen Bond: Arg 220, Asn 217, Arg 111 Van der Waals Interactions: Ser 499, Tyr 263, Phe 314, Met 496, Gln 495 Unfavorable Donor–Donor: Lys 131 |
| Polyphyllin VI | Tunnel site | −8.7 | Conventional Hydrogen Bond: Thr 218, Arg 229, Glu 249, Thr 253, His 114, Arg 111, Glu 121, Ala 237, Leu 236 Van der Waals Interactions: Leu 233, Glu 250, Gly 246, Ser 118, His 116 Alkyl Interaction: Arg 111 |
| Polyphyllin VII (Polyphyllin G) | Tunnel site | −8.9 | Conventional Hydrogen Bond: His114, Arg229, Glu249, Glu128, Thr218 Carbon Hydrogen Bond: Gly115, Ala237 Van der Waals Interactions: Arg231, Glu121, Leu117, Leu125, Leu233, Leu236, Gly246, Glu250 Unfavorable Interactions: Lys124 (Donor–Donor), Ala237 (Acceptor–Acceptor) |
| Polyphyllin B | Tunnel site | −10.8 | Conventional Hydrogen Bond: Arg111, Glu110, Glu249, Thr218, Thr253 Carbon Hydrogen Bond: Ala237 Van der Waals Interactions: His114, Gly115, Leu233, Gly246, Glu250 Pi-Donor Hydrogen Bond: His116 |
| Polyphyllin C | Tunnel site | −9.0 | Conventional Hydrogen Bond: Arg229, His114, Glu249 Carbon Hydrogen Bond: Gly115, Gly246 Van der Waals Interactions: Glu225, Glu232, His116, Phe113, Thr108, Glu139 |
| Polyphyllin E | Tunnel site | −8.9 | Conventional Hydrogen Bond: His116, Glu121, Arg229, Glu232, Glu249 Carbon Hydrogen Bond: Ala237 Van der Waals Interactions: Leu117, Lys124, Glu128, Glu139, Leu233, Leu236, Lys235, Gly246 Pi-Alkyl Interaction: Leu117 |
| Polyphyllin F | Tunnel site | −8.1 | Conventional Hydrogen Bond: Asn 336, Asn 339, Glu 299, Asp 296, Asp 340 Van der Waals Interactions: Ser 302, Gly 295, Val 301, Val 368, Val 382 Alkyl Interaction: Lys 369 Unfavorable Donor–Donor: Arg 343 |
| Polyphyllin H | Tunnel site | −9.2 | Conventional Hydrogen Bond: Arg 111, Thr 218, Arg 229, Thr 253, Glu 249, His 114, Glu 232 Van der Waals Interactions: Pro 491, Leu 125, Leu 117, Glu 110, Phe 113, Gly 246 |
| Dioscin | Tunnel site | −9.2 | Conventional Hydrogen Bond: Glu249, Glu250, Thr218, Thr219 Carbon Hydrogen Bond: Gly115, Ala237 Van der Waals Interactions: His114, His116, Leu117, Leu125, Leu233, Leu236, Gly246 Unfavorable Interaction: Arg229 (Donor–Donor) |
| Protodioscin | Tunnel site | −9.4 | Conventional Hydrogen Bond: Glu249, Glu250, Thr218, Thr253, Arg111, Glu110, Thr108, Phe113 Van der Waals Interactions: Gly115, His116, Leu117, Arg220, Leu125, Glu121, Ala237 Unfavorable Interaction: Arg229 (Donor–Donor) |
| Gracillin | Tunnel site | −8.1 | Conventional Hydrogen Bond: Asn336, Asn339, Asp340, Asp296 Van der Waals Interactions: Tyr380, Gly381, Val301, Val382, Pro300 |
| Ruscogenin | Tunnel site | −9.3 | Van der Waals Interactions: Glu121, Glu232, Glu249, His114, Thr218, Thr219, Glu250 Alkyl Interaction: Leu117, Leu125 |
| Ruscoside | Tunnel site | −8.8 | Conventional Hydrogen Bond: Arg111, Arg229, Arg220, Glu110, Glu249, Glu250, Lys124, Lys129, Asn222, Asp487 Van der Waals Interactions: Phe113, Trp112, Thr108, Ser109, Thr253 Unfavorable Interaction: Asp487 (Donor–Donor) |
| Hecogenin | Tunnel site | −8.9 | Conventional Hydrogen Bond: Thr218, Glu250 Van der Waals Interactions: His114, Glu249, Thr219, Thr253, Gly246, Leu233 Alkyl Interaction: Leu125 |
| Tigogenin | Tunnel site | −8.9 | Conventional Hydrogen Bond: Thr218, Glu121 Van der Waals Interactions: Thr219, Thr253, Glu249, Glu250, His114, His116 Alkyl Interaction: Leu117 |
| Solanine | Tunnel site | −8.4 | Conventional Hydrogen Bond: Thr218, Thr219, Glu249, Ala237 Van der Waals Interactions: His116, Glu250, Thr253, Gly115, Gly246, Leu254 Alkyl Interaction: Arg111, Pro491 Unfavorable Interaction: His114 (Donor–Donor) |
| Solamargine | Tunnel site | −9.5 | Conventional Hydrogen Bond: Glu232, Glu249, Thr218, Thr219 Van der Waals Interactions: His114, His116, Glu250, Gly246, Leu233, Ala237 Alkyl Interaction: Leu125, Lys124, Lys129 |
| Solasonine | Tunnel site | −9.0 | Conventional Hydrogen Bond: Glu232, Thr218, Ser228, Arg284 Van der Waals Interactions: His114, Glu249, Thr219, Thr253, Leu117, Leu233 Alkyl Interaction: Arg111 Unfavorable Interaction: Arg284 (Donor–Donor) |
| Yamogenin | Tunnel site | −9.3 | Van der Waals Interactions: Leu125, Leu233, Glu249, Glu250, Thr218, Thr219 Pi-Alkyl Interaction: Leu117, His114 |
| Neotigogenin | Tunnel site | −8.9 | Conventional Hydrogen Bond: Glu121, Thr218 Van der Waals Interactions: Thr219, Thr253, Glu249, Glu250 Pi-Alkyl Interaction: Leu117 Alkyl Interaction: Arg111 |
| Ophiopogonin A | Tunnel site | −11.0 | Conventional Hydrogen Bond: Glu232, Thr218 Van der Waals Interactions: Gly246, Glu249, Glu250, Thr253, His116, Ala237 Pi-Alkyl Interaction: Pro491 Unfavorable Interaction: Leu236 (Acceptor–Acceptor) |
| Ophiopogonin D | Tunnel site | −10.1 | Conventional Hydrogen Bond: Glu121, His114, Thr218 Van der Waals Interactions: Ala237, Leu236, Leu233, Glu249, Glu250, Thr219 Alkyl Interaction: Arg111 Carbon Hydrogen Bond: His116 |
| Sarsasapogenin | Tunnelsite | −8.9 | Conventional Hydrogen Bond: Thr218 Van der Waals Interactions: Thr219, Thr253, Glu249, Glu250, His114 Alkyl Interaction: Leu117, Arg111 Carbon Hydrogen Bond: His116 |
| Asparagusoside A | Tunnel site | −8.4 | Conventional Hydrogen Bond: Glu225, Arg229 Van der Waals Interactions: Glu232, Glu249, Gly246, Ala237 Alkyl Interaction: Leu117, Leu125 Unfavorable Interaction: Leu236 (Acceptor–Acceptor) |
| Ginsenoside Rg3 | Tunnel site | −8.8 | Conventional Hydrogen Bond: Glu121, Thr218, Arg229 Van der Waals Interactions: Leu125, Leu236, Glu249, Thr219, Ala237 Alkyl Interaction: Arg111, Pro491, Leu254 Unfavorable Interaction: Lys235 (Donor–Donor), Leu236 (Acceptor–Acceptor) |
| Ginsenoside Rh2 | Tunnel site | −8.8 | Conventional Hydrogen Bond: Thr218, Arg229, Glu249, Thr253, His114, Arg111, Glu110 Van der Waals Interactions: Leu125, Glu232, Glu225, Lys129, Thr219, Glu250 Alkyl Interaction: Lys129 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, D.-O. Targeting SHP2 with Natural Products: Exploring Saponin-Based Allosteric Inhibitors and Their Therapeutic Potential. Curr. Issues Mol. Biol. 2025, 47, 309. https://doi.org/10.3390/cimb47050309
Moon D-O. Targeting SHP2 with Natural Products: Exploring Saponin-Based Allosteric Inhibitors and Their Therapeutic Potential. Current Issues in Molecular Biology. 2025; 47(5):309. https://doi.org/10.3390/cimb47050309
Chicago/Turabian StyleMoon, Dong-Oh. 2025. "Targeting SHP2 with Natural Products: Exploring Saponin-Based Allosteric Inhibitors and Their Therapeutic Potential" Current Issues in Molecular Biology 47, no. 5: 309. https://doi.org/10.3390/cimb47050309
APA StyleMoon, D.-O. (2025). Targeting SHP2 with Natural Products: Exploring Saponin-Based Allosteric Inhibitors and Their Therapeutic Potential. Current Issues in Molecular Biology, 47(5), 309. https://doi.org/10.3390/cimb47050309