
The Relationship of Cholesterol Responses to Mitochondrial Dysfunction and Lung Inflammation in Chronic Obstructive Pulmonary Disease



{kind=link}
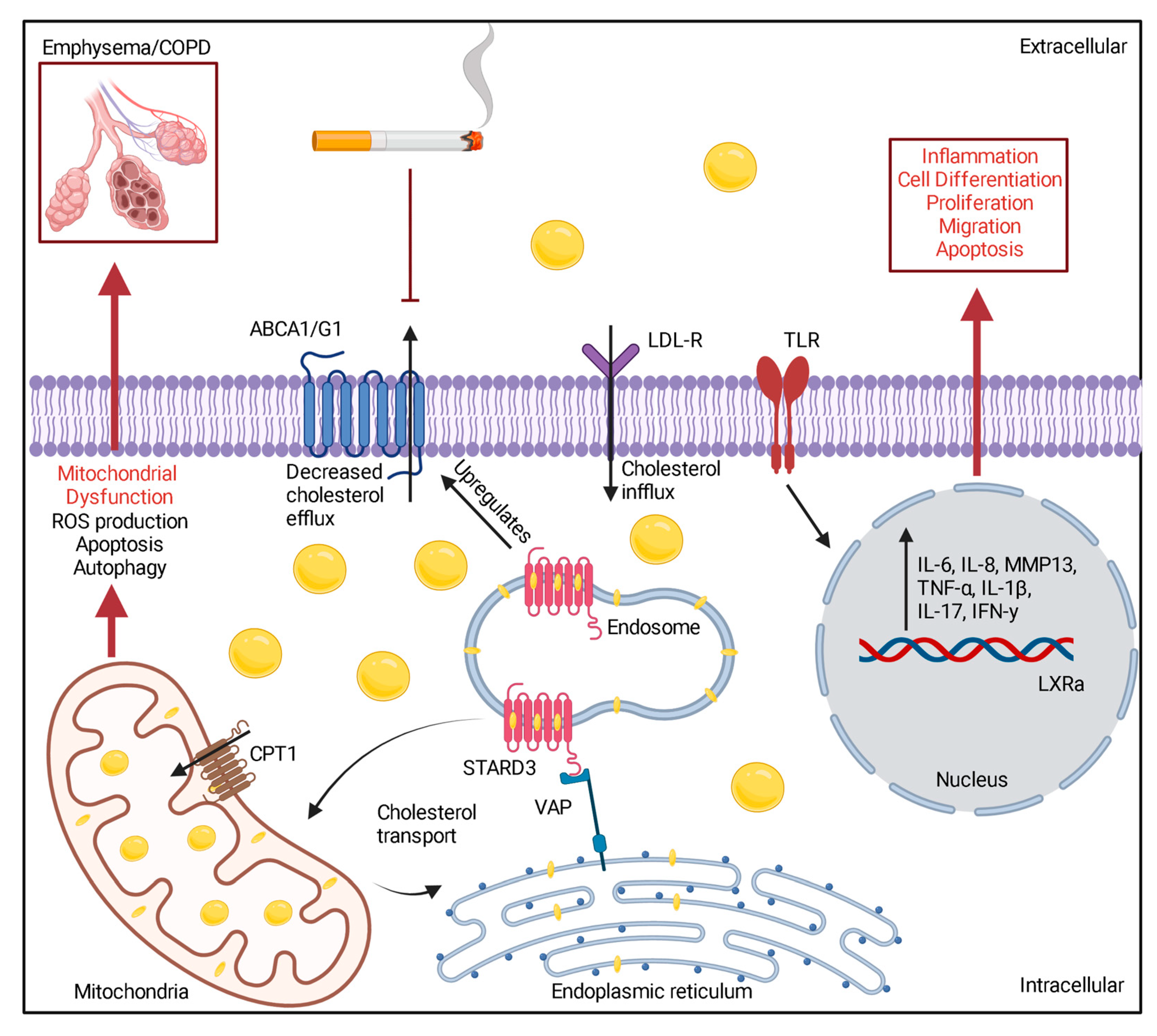
Abstract
:1. Introduction to Hypercholesterolemia in COPD
2. Is There a Relationship between Smoking Status and Lung Cholesterol Levels?
3. Cholesterol Levels Can Influence Systemic and Local Lung Inflammation in COPD
4. Hypercholesterolemia and Mitochondrial Dysfunction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kahnert, K.; Lucke, T.; Huber, R.M.; Behr, J.; Biertz, F.; Vogt, A.; Watz, H.; Alter, P.; Fahndrich, S.; Bals, R.; et al. Relationship of hyperlipidemia to comorbidities and lung function in COPD: Results of the COSYCONET cohort. PLoS ONE 2017, 12, e0177501. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General; U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health: Atlanta, GA, USA, 2014.
- Shi, Y.; Zhang, J.; Huang, Y. Prediction of cardiovascular risk in patients with chronic obstructive pulmonary disease: A study of the National Health and Nutrition Examination Survey database. BMC Cardiovasc. Disord. 2021, 21, 417. [Google Scholar] [CrossRef] [PubMed]
- Zafirova-Ivanovska, B.; Stojkovikj, J.; Dokikj, D.; Anastasova, S.; Debresliovska, A.; Zejnel, S.; Stojkovikj, D. The Level of Cholesterol in COPD Patients with Severe and Very Severe Stage of the Disease. Open Access Maced J. Med. Sci. 2016, 4, 277–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, S.; Jain, R.; Mohan, A.; Tiwari, P.; Guleria, R. Lipid Profile Status in Chronic Obstructive Pulmonary Disease and its Association with Disease Severity. Int. J. Respir. Pulm. Med. 2018, 5, 89. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Mun, S.; Choi, D.P.; Lee, J.Y.; Kim, H.C. Association between high-density lipoprotein cholesterol level and pulmonary function in healthy Korean adolescents: The JS high school study. BMC Pulm. Med. 2017, 17, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirahata, T.; Sato, H.; Yogi, S.; Inoue, K.; Niitsu, M.; Miyazawa, H.; Akagami, T.; Soma, M.; Mio, T.; Nagata, M.; et al. Possible association of high-density lipoprotein cholesterol levels with trunk muscle deficits and decrease in energy expenditure in patients with or at risk for COPD: A pilot study. Respir. Investig. 2022, 60, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Singh, D.; Park, J.H.; Park, Y.B.; Kim, S.I.; Park, B.; Park, J.; Kim, J.H.; Kim, M.A.; Lee, J.H.; et al. Impact of Body Mass Index Change on the Prognosis of Chronic Obstructive Pulmonary Disease. Respiration 2020, 99, 943–953. [Google Scholar] [CrossRef]
- Park, H.J.; Cho, J.H.; Kim, H.J.; Park, J.Y.; Lee, H.S.; Byun, M.K. The effect of low body mass index on the development of chronic obstructive pulmonary disease and mortality. J. Intern. Med. 2019, 286, 573–582. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, T.; Wang, Z.; Yu, F.; Xu, Q.; Guo, W.; Wu, C.; He, J. Body mass index and mortality in chronic obstructive pulmonary disease: A dose-response meta-analysis. Medicine 2016, 95, e4225. [Google Scholar] [CrossRef]
- Iyer, A.S.; Dransfield, M.T. The “Obesity Paradox” in Chronic Obstructive Pulmonary Disease: Can It Be Resolved? Ann. Am. Thorac. Soc. 2018, 15, 158–159. [Google Scholar] [CrossRef]
- Ma, Y.L.; Zhao, H.J.; Su, Y.H. Association between waist circumference change and incident chronic obstructive pulmonary disease among Chinese adults: A 10-year cohort study. Sci. Rep. 2022, 12, 18402. [Google Scholar] [CrossRef] [PubMed]
- Pitta, F.; Troosters, T.; Spruit, M.A.; Probst, V.S.; Decramer, M.; Gosselink, R. Characteristics of physical activities in daily life in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005, 171, 972–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakrania, K.; Edwardson, C.L.; Bodicoat, D.H.; Esliger, D.W.; Gill, J.M.; Kazi, A.; Velayudhan, L.; Sinclair, A.J.; Sattar, N.; Biddle, S.J.; et al. Associations of mutually exclusive categories of physical activity and sedentary time with markers of cardiometabolic health in English adults: A cross-sectional analysis of the Health Survey for England. BMC Public Health 2016, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Young, D.R.; Hivert, M.F.; Alhassan, S.; Camhi, S.M.; Ferguson, J.F.; Katzmarzyk, P.T.; Lewis, C.E.; Owen, N.; Perry, C.K.; Siddique, J.; et al. Sedentary Behavior and Cardiovascular Morbidity and Mortality: A Science Advisory from the American Heart Association. Circulation 2016, 134, e262–e279. [Google Scholar] [CrossRef]
- Kotlyarov, S.; Kotlyarova, A. Molecular Mechanisms of Lipid Metabolism Disorders in Infectious Exacerbations of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 7634. [Google Scholar] [CrossRef] [PubMed]
- Keating, E.; Rahman, L.; Francis, J.; Petersen, A.; Possmayer, F.; Veldhuizen, R.; Petersen, N.O. Effect of cholesterol on the biophysical and physiological properties of a clinical pulmonary surfactant. Biophys. J. 2007, 93, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Murphy, M.J.; MacDonald, T.M.; Schembri, S.; Simpson, W.; Winter, J.; Winter, J.H.; Wei, L. Effect of statins on total cholesterol concentrations, cardiovascular morbidity, and all-cause mortality in chronic obstructive pulmonary disease: A population-based cohort study. Clin. Ther. 2012, 34, 374–384. [Google Scholar] [CrossRef]
- Barnes, P.J.; Celli, B.R. Systemic manifestations and comorbidities of COPD. Eur. Respir. J. 2009, 33, 1165–1185. [Google Scholar] [CrossRef] [Green Version]
- Walsh, A.; Perrem, L.; Khashan, A.S.; Henry, M.T.; Ni Chroinin, M. Statins versus placebo for people with chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2019, 7, CD011959. [Google Scholar] [CrossRef]
- Lu, Y.; Chang, R.; Yao, J.; Xu, X.; Teng, Y.; Cheng, N. Effectiveness of long-term using statins in COPD—A network meta-analysis. Respir. Res. 2019, 20, 17. [Google Scholar] [CrossRef]
- Criner, G.J.; Connett, J.E.; Aaron, S.D.; Albert, R.K.; Bailey, W.C.; Casaburi, R.; Cooper, J.A., Jr.; Curtis, J.L.; Dransfield, M.T.; Han, M.K.; et al. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. N. Engl. J. Med. 2014, 370, 2201–2210. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Conlon, T.M.; Sarker, R.S.; Tasdemir, D.; Smirnova, N.F.; Srivastava, B.; Verleden, S.E.; Gunes, G.; Wu, X.; Prehn, C.; et al. Cholesterol metabolism promotes B-cell positioning during immune pathogenesis of chronic obstructive pulmonary disease. EMBO Mol. Med. 2018, 10, e8349. [Google Scholar] [CrossRef] [PubMed]
- Gepner, A.D.; Piper, M.E.; Johnson, H.M.; Fiore, M.C.; Baker, T.B.; Stein, J.H. Effects of smoking and smoking cessation on lipids and lipoproteins: Outcomes from a randomized clinical trial. Am. Heart J. 2011, 161, 145–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gossett, L.K.; Johnson, H.M.; Piper, M.E.; Fiore, M.C.; Baker, T.B.; Stein, J.H. Smoking intensity and lipoprotein abnormalities in active smokers. J. Clin. Lipidol. 2009, 3, 372–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufeld, E.J.; Mietus-Snyder, M.; Beiser, A.S.; Baker, A.L.; Newburger, J.W. Passive cigarette smoking and reduced HDL cholesterol levels in children with high-risk lipid profiles. Circulation 1997, 96, 1403–1407. [Google Scholar] [CrossRef]
- Li, L.; Liu, Y.; Liu, X.; Zheng, N.; Gu, Y.; Song, Y.; Wang, X. Regulatory roles of external cholesterol in human airway epithelial mitochondrial function through STARD3 signalling. Clin. Transl. Med. 2022, 12, e902. [Google Scholar] [CrossRef] [PubMed]
- Chelland Campbell, S.; Moffatt, R.J.; Stamford, B.A. Smoking and smoking cessation—The relationship between cardiovascular disease and lipoprotein metabolism: A review. Atherosclerosis 2008, 201, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, C.W.; Kumley, B.K.; Area-Gomez, E.; Xu, Y.; Dabo, A.J.; Geraghty, P.; Campos, M.; Foronjy, R.; Garcia-Arcos, I. Decreased surfactant lipids correlate with lung function in chronic obstructive pulmonary disease (COPD). PLoS ONE 2020, 15, e0228279. [Google Scholar] [CrossRef]
- Morissette, M.C.; Shen, P.; Thayaparan, D.; Stampfli, M.R. Disruption of pulmonary lipid homeostasis drives cigarette smoke-induced lung inflammation in mice. Eur. Respir. J. 2015, 46, 1451–1460. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Rodriguez, E.; Perez-Gil, J. Structure-function relationships in pulmonary surfactant membranes: From biophysics to therapy. Biochim. Biophys. Acta 2014, 1838, 1568–1585. [Google Scholar] [CrossRef]
- Hughes, D.A.; Haslam, P.L. Effect of smoking on the lipid composition of lung lining fluid and relationship between immunostimulatory lipids, inflammatory cells and foamy macrophages in extrinsic allergic alveolitis. Eur. Respir. J. 1990, 3, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Jubinville, E.; Talbot, M.; Berube, J.C.; Hamel-Auger, M.; Maranda-Robitaille, M.; Beaulieu, M.J.; Aubin, S.; Pare, M.E.; Kallend, D.G.; Arsenault, B.; et al. Interplay between cigarette smoking and pulmonary reverse lipid transport. Eur. Respir. J. 2017, 50, 1700681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonett, J.; Goldklang, M.; Sklepkiewicz, P.; Gerber, A.; Trischler, J.; Zelonina, T.; Westerterp, M.; Lemaitre, V.; Okada, Y.; D’Armiento, J. A critical role for ABC transporters in persistent lung inflammation in the development of emphysema after smoke exposure. FASEB J. 2018, 32, 6724. [Google Scholar] [CrossRef] [PubMed]
- Anzueto, A.; Jubran, A.; Ohar, J.A.; Piquette, C.A.; Rennard, S.I.; Colice, G.; Pattishall, E.N.; Barrett, J.; Engle, M.; Perret, K.A.; et al. Effects of aerosolized surfactant in patients with stable chronic bronchitis: A prospective randomized controlled trial. JAMA 1997, 278, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, D.J.; Agrawal, Y.; Cassano, P.A. Lipids and pulmonary function in the Third National Health and Nutrition Examination Survey. Am. J. Epidemiol. 2002, 155, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Yvan-Charvet, L.; Ranalletta, M.; Wang, N.; Han, S.; Terasaka, N.; Li, R.; Welch, C.; Tall, A.R. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Investig. 2007, 117, 3900–3908. [Google Scholar] [CrossRef] [Green Version]
- Annema, W.; Nijstad, N.; Tolle, M.; de Boer, J.F.; Buijs, R.V.; Heeringa, P.; van der Giet, M.; Tietge, U.J. Myeloperoxidase and serum amyloid A contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A(2). J. Lipid Res. 2010, 51, 743–754. [Google Scholar] [CrossRef] [Green Version]
- Higham, A.; Lea, S.; Plumb, J.; Maschera, B.; Simpson, K.; Ray, D.; Singh, D. The role of the liver X receptor in chronic obstructive pulmonary disease. Respir. Res. 2013, 14, 106. [Google Scholar] [CrossRef] [Green Version]
- Gowdy, K.M.; Fessler, M.B. Emerging roles for cholesterol and lipoproteins in lung disease. Pulm. Pharmacol. Ther. 2013, 26, 430–437. [Google Scholar] [CrossRef]
- Fujii, W.; Kapellos, T.S.; Bassler, K.; Handler, K.; Holsten, L.; Knoll, R.; Warnat-Herresthal, S.; Oestreich, M.; Hinkley, E.R.; Hasenauer, J.; et al. Alveolar macrophage transcriptomic profiling in COPD shows major lipid metabolism changes. ERJ Open Res. 2021, 7, 00915-2020. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Akhtari, M.; Tolani, S.; Pagler, T.; Bijl, N.; Kuo, C.L.; Wang, M.; Sanson, M.; Abramowicz, S.; Welch, C.; et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Investig. 2011, 121, 4138–4149. [Google Scholar] [CrossRef] [Green Version]
- Tolani, S.; Pagler, T.A.; Murphy, A.J.; Bochem, A.E.; Abramowicz, S.; Welch, C.; Nagareddy, P.R.; Holleran, S.; Hovingh, G.K.; Kuivenhoven, J.A.; et al. Hypercholesterolemia and reduced HDL-C promote hematopoietic stem cell proliferation and monocytosis: Studies in mice and FH children. Atherosclerosis 2013, 229, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, L.P.; Wendling, C.; Vedie, B.; Kobayashi, T.; Chenard, M.P.; Tomasetto, C.; Drin, G.; Alpy, F. STARD3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J. 2017, 36, 1412–1433. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, F.; Allen, A.M.; Taylor, J.M.; Graham, A. Overexpression of STARD3 in human monocyte/macrophages induces an anti-atherogenic lipid phenotype. Clin. Sci. 2010, 119, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, C.; Xueming, H.; Ruihang, D. MLN64 deletion suppresses RANKL-induced osteoclastic differentiation and attenuates diabetic osteoporosis in streptozotocin (STZ)-induced mice. Biochem. Biophys. Res. Commun. 2018, 505, 1228–1235. [Google Scholar] [CrossRef]
- White, M.M.; Geraghty, P.; Hayes, E.; Cox, S.; Leitch, W.; Alfawaz, B.; Lavelle, G.M.; McElvaney, O.J.; Flannery, R.; Keenan, J.; et al. Neutrophil Membrane Cholesterol Content is a Key Factor in Cystic Fibrosis Lung Disease. EBioMedicine 2017, 23, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Fojeda, B.; Gonzalez-Carnicero, Z.; de Lorenzo, A.; Minutti, C.M.; de Tapia, L.; Euba, B.; Iglesias-Ceacero, A.; Castillo-Lluva, S.; Garmendia, J.; Casals, C. Lung Surfactant Lipids Provide Immune Protection Against Haemophilus influenzae Respiratory Infection. Front. Immunol. 2019, 10, 458. [Google Scholar] [CrossRef] [Green Version]
- Turley, S.D.; Andersen, J.M.; Dietschy, J.M. Rates of sterol synthesis and uptake in the major organs of the rat in vivo. J. Lipid Res. 1981, 22, 551–569. [Google Scholar] [CrossRef]
- McDonald, J.G.; Russell, D.W. Editorial: 25-Hydroxycholesterol: A new life in immunology. J. Leukoc. Biol. 2010, 88, 1071–1072. [Google Scholar] [CrossRef]
- Sugiura, H.; Koarai, A.; Ichikawa, T.; Minakata, Y.; Matsunaga, K.; Hirano, T.; Akamatsu, K.; Yanagisawa, S.; Furusawa, M.; Uno, Y.; et al. Increased 25-hydroxycholesterol concentrations in the lungs of patients with chronic obstructive pulmonary disease. Respirology 2012, 17, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Gold, E.S.; Diercks, A.H.; Podolsky, I.; Podyminogin, R.L.; Askovich, P.S.; Treuting, P.M.; Aderem, A. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 10666–10671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Li, W.; Hui, H.; Tiwari, S.K.; Zhang, Q.; Croker, B.A.; Rawlings, S.; Smith, D.; Carlin, A.F.; Rana, T.M. Cholesterol 25-Hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J. 2020, 39, e106057. [Google Scholar] [CrossRef] [PubMed]
- Clayton, S.A.; MacDonald, L.; Kurowska-Stolarska, M.; Clark, A.R. Mitochondria as Key Players in the Pathogenesis and Treatment of Rheumatoid Arthritis. Front. Immunol. 2021, 12, 673916. [Google Scholar] [CrossRef]
- Meyers, A.K.; Zhu, X. The NLRP3 Inflammasome: Metabolic Regulation and Contribution to Inflammaging. Cells 2020, 9, 1808. [Google Scholar] [CrossRef]
- Torres, S.; Solsona-Vilarrasa, E.; Nunez, S.; Matias, N.; Insausti-Urkia, N.; Castro, F.; Casasempere, M.; Fabrias, G.; Casas, J.; Enrich, C.; et al. Acid ceramidase improves mitochondrial function and oxidative stress in Niemann-Pick type C disease by repressing STARD1 expression and mitochondrial cholesterol accumulation. Redox Biol. 2021, 45, 102052. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Gu, R.X.; Baoukina, S.; Tieleman, D.P. Cholesterol Flip-Flop in Heterogeneous Membranes. J. Chem. Theory Comput. 2019, 15, 2064–2070. [Google Scholar] [CrossRef] [Green Version]
- Flaquer, A.; Rospleszcz, S.; Reischl, E.; Zeilinger, S.; Prokisch, H.; Meitinger, T.; Meisinger, C.; Peters, A.; Waldenberger, M.; Grallert, H.; et al. Mitochondrial GWA Analysis of Lipid Profile Identifies Genetic Variants to Be Associated with HDL Cholesterol and Triglyceride Levels. PLoS ONE 2015, 10, e0126294. [Google Scholar] [CrossRef]
- Kang, M.J.; Shadel, G.S. A Mitochondrial Perspective of Chronic Obstructive Pulmonary Disease Pathogenesis. Tuberc. Respir. Dis. 2016, 79, 207–213. [Google Scholar] [CrossRef]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloonan, S.M.; Glass, K.; Laucho-Contreras, M.E.; Bhashyam, A.R.; Cervo, M.; Pabon, M.A.; Konrad, C.; Polverino, F.; Siempos, I.I.; Perez, E.; et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat. Med. 2016, 22, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Belchamber, K.B.R.; Singh, R.; Batista, C.M.; Whyte, M.K.; Dockrell, D.H.; Kilty, I.; Robinson, M.J.; Wedzicha, J.A.; Barnes, P.J.; Donnelly, L.E.; et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur. Respir. J. 2019, 54, 1802244. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhu, Q.; Zeng, J.; Gu, X.; Miao, Y.; Xu, W.; Lv, T.; Song, Y. Extracellular mitochondrial DNA promote NLRP3 inflammasome activation and induce acute lung injury through TLR9 and NF-kappaB. J. Thorac. Dis. 2019, 11, 4816–4828. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.Z.; Rice, M.C.; Hoffman, K.L.; Oromendia, C.; Barjaktarevic, I.Z.; Wells, J.M.; Hastie, A.T.; Labaki, W.W.; Cooper, C.B.; Comellas, A.P.; et al. Association of urine mitochondrial DNA with clinical measures of COPD in the SPIROMICS cohort. JCI Insight 2020, 5, e133984. [Google Scholar] [CrossRef] [Green Version]
- Lloreta, J.; Orozco, M.; Gea, J.; Corominas, J.M.; Serrano, S. Selective diaphragmatic mitochondrial abnormalities in a patient with marked air flow obstruction. Ultrastruct. Pathol. 1996, 20, 67–71. [Google Scholar] [CrossRef]
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.M.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 963. [Google Scholar] [CrossRef] [Green Version]
- Fahndrich, S.; Biertz, F.; Karch, A.; Kleibrink, B.; Koch, A.; Teschler, H.; Welte, T.; Kauczor, H.U.; Janciauskiene, S.; Jorres, R.A.; et al. Cardiovascular risk in patients with alpha-1-antitrypsin deficiency. Respir. Res. 2017, 18, 171. [Google Scholar] [CrossRef]
- Pontzer, H.; Yamada, Y.; Sagayama, H.; Ainslie, P.N.; Andersen, L.F.; Anderson, L.J.; Arab, L.; Baddou, I.; Bedu-Addo, K.; Blaak, E.E.; et al. Daily energy expenditure through the human life course. Science 2021, 373, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Majid, S.; Keith, R.J.; Fetterman, J.L.; Weisbrod, R.M.; Nystoriak, J.; Wilson, T.; Stokes, A.C.; Blaha, M.J.; Srivastava, S.; Robertson, R.M.; et al. Lipid profiles in users of combustible and electronic cigarettes. Vasc. Med. 2021, 26, 483–488. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jundi, B.; Ahmed, H.; Reece, J.; Geraghty, P. The Relationship of Cholesterol Responses to Mitochondrial Dysfunction and Lung Inflammation in Chronic Obstructive Pulmonary Disease. Medicina 2023, 59, 253. https://doi.org/10.3390/medicina59020253
Jundi B, Ahmed H, Reece J, Geraghty P. The Relationship of Cholesterol Responses to Mitochondrial Dysfunction and Lung Inflammation in Chronic Obstructive Pulmonary Disease. Medicina. 2023; 59(2):253. https://doi.org/10.3390/medicina59020253
Chicago/Turabian StyleJundi, Bakr, Huma Ahmed, Joshua Reece, and Patrick Geraghty. 2023. "The Relationship of Cholesterol Responses to Mitochondrial Dysfunction and Lung Inflammation in Chronic Obstructive Pulmonary Disease" Medicina 59, no. 2: 253. https://doi.org/10.3390/medicina59020253
APA StyleJundi, B., Ahmed, H., Reece, J., & Geraghty, P. (2023). The Relationship of Cholesterol Responses to Mitochondrial Dysfunction and Lung Inflammation in Chronic Obstructive Pulmonary Disease. Medicina, 59(2), 253. https://doi.org/10.3390/medicina59020253