
Molecular Dynamic Simulation Analysis of a Novel Missense Variant in CYB5R3 Gene in Patients with Methemoglobinemia
 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval and Study Subjects
2.2. Blood Sampling and Extraction of Genomic DNA
2.3. Whole Exome Sequencing
2.4. Sanger Sequencing
2.5. Determination of Variant Pathogenicity in CYB5R3
2.6. Data Retrieval and Structure Preparation
2.7. Molecular Dynamics Simulation
2.8. Post-Simulation Analysis
3. Results
3.1. Clinical Features
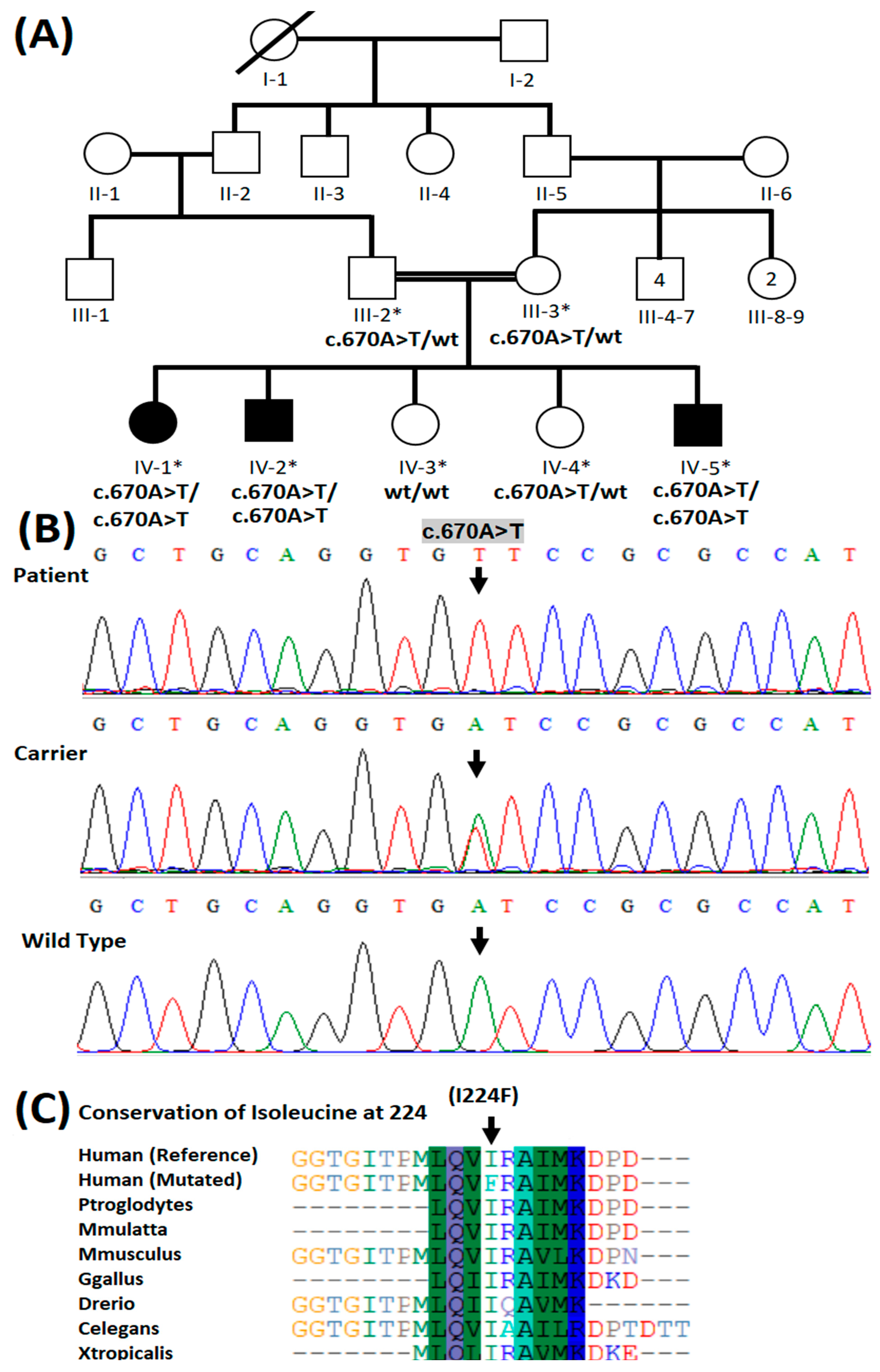
3.2. Molecular Genetic Analysis
3.3. Heme Docking and Predicted Interactions with Native and Mutant CYB5R3
3.4. Molecular Dynamics Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guedri, R.; Missaoui, N.; Essaddam, L.; Ben Becher, S. A rare cause of cyanosis: Congenital methemoglobinemia. Clin. Case Rep. 2021, 9, e04422. [Google Scholar] [CrossRef] [PubMed]
- Molina Herranz, D.; Garcia Escudero, C.; Rite Gracia, S.; Aguilar de la Red, Y.; Martinez Nieto, J.; Izquierdo Alvarez, S.; Montanes Gracia, M.A.; Recasens, V.; Hernandez Mata, C.F. CYB5R3 homozygous pathogenic variant as a rare cause of cyanosis in the newborn. Clin. Biochem. 2022, 102, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Bradberry, S.M. Occupational methaemoglobinaemia. Mechanisms of production, features, diagnosis and management including the use of methylene blue. Toxicol. Rev. 2003, 22, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedar, P.S.; Gupta, V.; Warang, P.; Chiddarwar, A.; Madkaikar, M. Novel mutation (R192C) in CYB5R3 gene causing NADH-cytochrome b5 reductase deficiency in eight Indian patients associated with autosomal recessive congenital methemoglobinemia type-I. Hematology 2018, 23, 567–573. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.; Yuan, S.; Wood, K.; Katona, M.; Straub, A.C. Cytochrome b5 reductases: Redox regulators of cell homeostasis. J. Biol. Chem. 2022, 298, 102654. [Google Scholar] [CrossRef]
- Siendones, E.; Ballesteros, M.; Navas, P. Cellular and Molecular Mechanisms of Recessive Hereditary Methaemoglobinaemia Type II. J. Clin. Med. 2018, 7, 341. [Google Scholar] [CrossRef] [Green Version]
- Borgese, N.; Aggujaro, D.; Carrera, P.; Pietrini, G.; Bassetti, M. A role for N-myristoylation in protein targeting: NADH-cytochrome b5 reductase requires myristic acid for association with outer mitochondrial but not ER membranes. J. Cell Biol. 1996, 135, 1501–1513. [Google Scholar] [CrossRef]
- Borgese, N.; D’Arrigo, A.; De Silvestris, M.; Pietrini, G. NADH-cytochrome b5 reductase and cytochrome b5 isoforms as models for the study of post-translational targeting to the endoplasmic reticulum. FEBS Lett. 1993, 325, 70–75. [Google Scholar] [CrossRef] [Green Version]
- Deorukhkar, A.; Kulkarni, A.; Kedar, P. Three novel mutations in CYB5R3 gene causing NADH-cytochrome b5 reductase enzyme deficiency leads to recessive congenital methaemoglobinemia. Mol. Biol. Rep. 2022, 49, 2141–2147. [Google Scholar] [CrossRef]
- Rehman, H.U. Methemoglobinemia. West J. Med. 2001, 175, 193–196. [Google Scholar] [CrossRef]
- Sheetal, A.; Dhirendra, S.; Ankur, A.; Ridhima, S.; Savitri, S. Congenital Methemoglobinemia with Multiple Limb Anomalies in an 11-Year-Old Boy. Clin. Med. Rev. Case Rep. 2020, 7, 324. [Google Scholar] [CrossRef]
- Gupta, V.; Kulkarni, A.; Warang, P.; Devendra, R.; Chiddarwar, A.; Kedar, P. Mutation update: Variants of the CYB5R3 gene in recessive congenital methemoglobinemia. Hum. Mutat. 2020, 41, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Kalsoom, U.E.; Umair, M.; John, P.; Ansar, M.; Basit, S.; Ahmad, W. Exome sequencing revealed a novel splice site variant in the ALX1 gene underlying frontonasal dysplasia. Clin. Genet. 2017, 91, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genom. 2013, 14 (Suppl. 3), S3. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Hu, Y.; Sun, J.; Cheng, Y.; Cheung, K.-H.; Zhao, H. A Statistical Framework to Predict Functional Non-Coding Regions in the Human Genome Through Integrated Analysis of Annotation Data. Sci. Rep. 2015, 5, 10576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2018, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alirezaie, N.; Kernohan, K.D.; Hartley, T.; Majewski, J.; Hocking, T.D. ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants. Am. J. Hum. Genet. 2018, 103, 474–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.e.a. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Zwanzig, R. Nonlinear generalized Langevin equations. J. Stat. Phys. 1973, 9, 215–220. [Google Scholar] [CrossRef]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Moberly, J.G.; Bernards, M.T.; Waynant, K.V. Key features and updates for Origin 2018. J. Cheminform. 2018, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Carew, N.T.; Schmidt, H.M.; Yuan, S.; Galley, J.C.; Hall., R.; Altmann, H.M.; Hahn, S.A.; Miller, M.P.; Wood, K.C.; Gabris, B.; et al. Loss of cardiomyocyte CYB5R3 impairs redox equilibrium and causes sudden cardiac death. J. Clin. Investig. 2022, 132, e147120. [Google Scholar] [CrossRef]
- Zheng, H.; Li, X.; Shi, L.; Jing, Y.; Song, Q.; Chen, Y.; He, L.; Wang, F.; Gao, J.; Bi, Y. Genome-Wide Identification and Analysis of the Cytochrome B5 Protein Family in Chinese Cabbage (Brassica rapa L. ssp. Pekinensis). Int. J. Genom. 2019, 2019, 2102317. [Google Scholar] [CrossRef] [Green Version]
- Jaffé, E.R. Methemoglobin pathophysiology. Prog. Clin. Biol. Res. 1981, 51, 133–151. [Google Scholar]
- Kimura, S.; Nishida, H.; Iyanagi, T. Effects of Flavin-Binding Motif Amino Acid Mutations in the NADH-Cytochrome b5 Reductase Catalytic Domain on Protein Stability and Catalysis1. J. Biochem. 2001, 130, 481–490. [Google Scholar] [CrossRef]
- Syed, K.; Kattamuri, C.; Thompson, T.B.; Yadav, J.S. Cytochrome b₅ reductase-cytochrome b₅ as an active P450 redox enzyme system in Phanerochaete chrysosporium: Atypical properties and in vivo evidence of electron transfer capability to CYP63A2. Arch. Biochem. Biophys. 2011, 509, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.J.; Lappin, T.R. Recessive congenital methaemoglobinaemia: Cytochrome b5 reductase deficiency. Br. J. Haematol. 2008, 141, 298–308. [Google Scholar] [PubMed]
- Katsube, T.; Sakamoto, N.; Kobayashi, Y.; Seki, R.; Hirano, M.; Tanishima, K.; Tomoda, A.; Takazakura, E.; Yubisui, T.; Takeshita, M.; et al. Exonic point mutations in NADH-cytochrome B5 reductase genes of homozygotes for hereditary methemoglobinemia, types I and III: Putative mechanisms of tissue-dependent enzyme deficiency. Am. J. Hum. Genet. 1991, 48, 799–808. [Google Scholar] [PubMed]
- Galeeva, N.; Voevoda, M.; Spiridonova, M.; Stepanov, V.; Polyakov, A. Population frequency and age of c. 806 C> T mutation in CYB5R3 gene as cause of recessive congenital methemoglobinemia in Yakutia. Russ. J. Genet. 2013, 49, 457–463. [Google Scholar] [CrossRef]
- Lorenzo, F.R.; Phillips, J.D.; Nussenzveig, R.; Lingam, B.; Koul, P.A.; Schrier, S.L.; Prchal, J.T. Molecular basis of two novel mutations found in type I methemoglobinemia. Blood Cells Mol. Dis. 2011, 46, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.J.; Aslan, D. NADH-cytochrome b5 reductase in a Turkish family with recessive congenital methaemoglobinaemia type I. J. Clin. Pathol. 2008, 61, 1122. [Google Scholar] [CrossRef] [PubMed]
- Higasa, K.; Manabe, J.I.; Yubisui, T.; Sumimoto, H.; Pung-Amritt, P.; Tanphaichitr, V.S.; Fukumaki, Y. Molecular basis of hereditary methaemoglobinaemia, types I and II: Two novel mutations in the NADH-cytochrome b5 reductase gene. Br. J. Haematol. 1998, 103, 922–930. [Google Scholar] [CrossRef]
- Bando, S.; Takano, T.; Yubisui, T.; Shirabe, K.; Takeshita, M.; Nakagawa, A. Structure of human erythrocyte NADH-cytochrome b5 reductase. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1929–1934. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Parameter | Patient 1 (IV-I) | Patient 2 (IV-2) | Reference Value |
|---|---|---|---|
| Age | 16 | 17 | |
| Gender | F | M | |
| Consanguinity | Yes | Yes | |
| Hematological and biochemical analysis | |||
| Cyanosis | Yes + | + | |
| Dyspnea | + | + | |
| Orthopnea | + | − | |
| Hemoptysis | + | − | |
| Blood pressure | 120/70 | 110/90 | 120/80 |
| Heart rate (min) | 125 | 115 | 120/min |
| R.R (min) | 15 | 13 | 12–16/min |
| Arterial PO2 | 32 | 44 | 75–100 mmHg |
| PH, CO2, HCO3− | N | N | |
| Hemoglobin (g/dL) | 15.8 | 17 | 13.2–16.6 g/dL (men), 11.6–15 g/dL (women) |
| Leukocytes (g/dL) | 8.57 × 109 | 10.1 × 109 | 4.5–11.0 × 109 g/dL |
| Thrombocytes/L | 241 × 109 | 241 × 109 | 150–400 × 109/L |
| Reticulocyte Count (%) | 3.80% | 3.40% | 0.5–2.5% |
| Red cell distribution width (fL) | 47.1 fL | 49.3 fL | 39–46 fL |
| Blood cell count | N | N | |
| Liver function test | N | N | |
| Renal function test | N | N | |
| Serum electrolytes | N | N | |
| Pulmonary tuberculosis | − | − | |
| Hb investigations | |||
| HbA | − | − | |
| HBA2 | 3.30% | 3.10% | 2.0–3.5% |
| MetHb | 49% | 50.50% | 1–2% |
| HbF | 1.20% | 1.50% | 0.8–2% |
| Recessive congenital methemoglobinemia | RCM Type I | RCM Type I | |
| Genetic analysis | CYB5R3; p.(Ile224Phe) | CYB5R3; p.(Ile224Phe) |
| Protein | Interacting Residues | Interaction Type | Distance | Energy (Kcal/mol) | Docking Score |
|---|---|---|---|---|---|
| Native | CYS 273 | H-donor | 3.10 | −0.3 | −8.34 |
| THR 181 | H-donor | 2.99 | −2.5 | ||
| CYS 273 | H-donor | 3.75 | −0.2 | ||
| CYS 273 | H-acceptor | 3.10 | −0.6 | ||
| HIS 117 | H-pi | 4.35 | −0.6 | ||
| PHE 120 | H-pi | 4.84 | −0.1 | ||
| TYR 112 | pi-pi | 3.96 | −0.0 | ||
| Mutant | PRO 275 | H-acceptor | 3.70 | −0.1 | −7.89 |
| PRO 276 | H-acceptor | 3.66 | −0.2 | ||
| TYR 112 | H-acceptor | 3.36 | −0.1 | ||
| TYR 112 | H-pi | 4.50 | −0.1 | ||
| PRO 275 | pi-H | 4.72 | −0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, A.; Shah, A.A.; Syed, F.; Mahmood, A.; Ur Rehman, H.; Khurshid, B.; Samad, A.; Ahmad, W.; Basit, S. Molecular Dynamic Simulation Analysis of a Novel Missense Variant in CYB5R3 Gene in Patients with Methemoglobinemia. Medicina 2023, 59, 379. https://doi.org/10.3390/medicina59020379
Ullah A, Shah AA, Syed F, Mahmood A, Ur Rehman H, Khurshid B, Samad A, Ahmad W, Basit S. Molecular Dynamic Simulation Analysis of a Novel Missense Variant in CYB5R3 Gene in Patients with Methemoglobinemia. Medicina. 2023; 59(2):379. https://doi.org/10.3390/medicina59020379
Chicago/Turabian StyleUllah, Asmat, Abid Ali Shah, Fibhaa Syed, Arif Mahmood, Hassan Ur Rehman, Beenish Khurshid, Abdus Samad, Wasim Ahmad, and Sulman Basit. 2023. "Molecular Dynamic Simulation Analysis of a Novel Missense Variant in CYB5R3 Gene in Patients with Methemoglobinemia" Medicina 59, no. 2: 379. https://doi.org/10.3390/medicina59020379