Immunogenicity of Non-Mutated Ovarian Cancer-Specific Antigens

 ,
,  , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of the Most Prevalent TSAs
2.1.1. Selection of Highly Prevalent TSAs
2.1.2. Evaluation of Cancer Specificity and Sharing of TSA Source Transcripts
2.2. Peripheral Blood Mononuclear Cell Isolation
2.3. Generation of Monocyte-Derived Dendritic Cells
2.4. Peptide Pulsing and Electroporation of moDCs
2.5. MS Experiments
2.5.1. Cell Sample Preparation
2.5.2. Immunoprecipitation of HLA Class I Molecules
2.5.3. Mass Spectrometry Analyses
2.5.4. Bioinformatic Analysis
2.6. Immunogenicity Testing
2.6.1. Coculture of Naïve CD8 T Cells with moDCs
2.6.2. Tetramer Staining
2.6.3. Functional Expansion of Antigen-Specific T Cells Followed by TCR Vβ CDR3 Sequencing (FEST Assay)
2.6.4. PRIME 2.0 Score for Prediction of T-Cell Recognition
3. Results
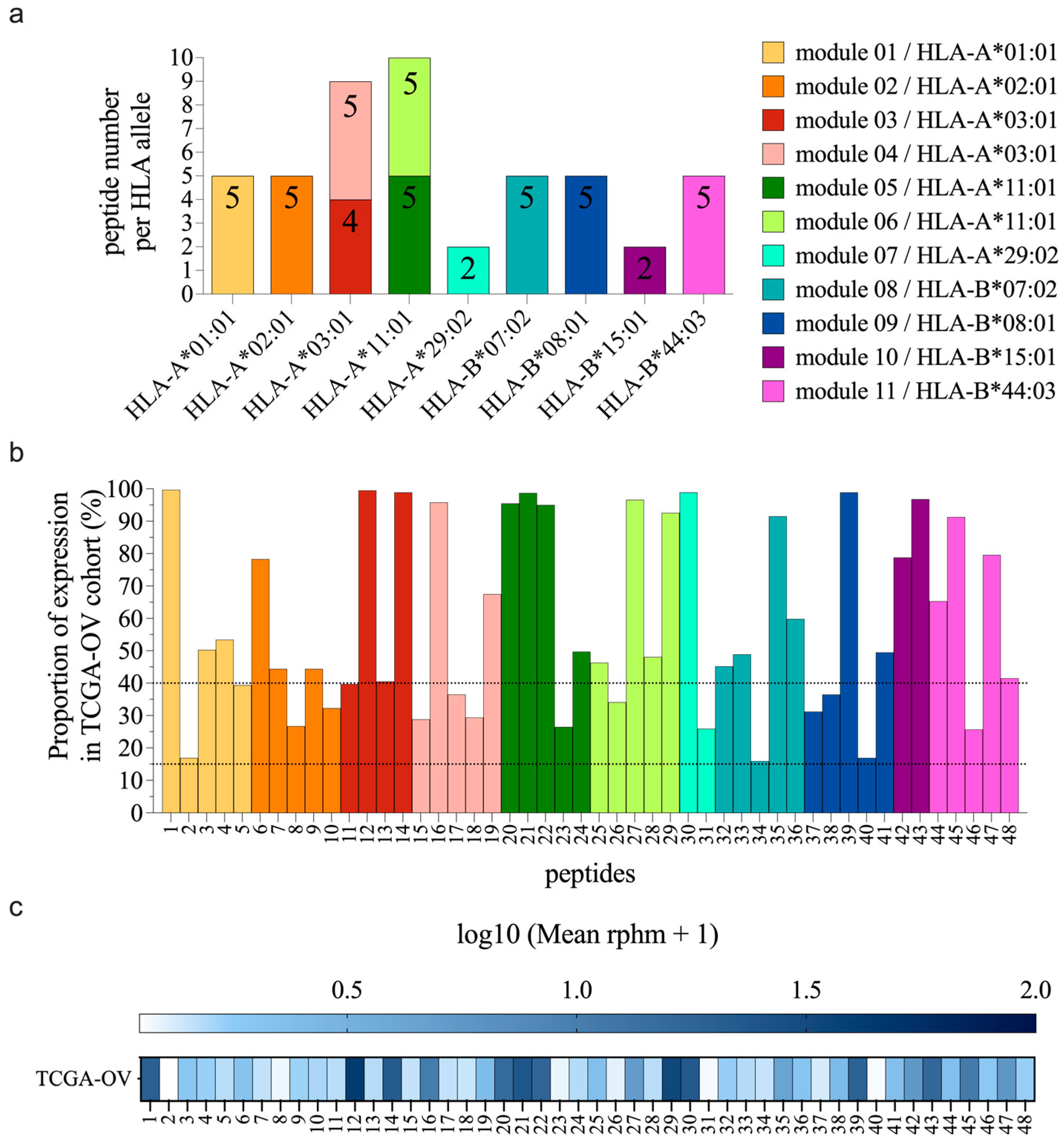
3.1. Selection of the Best TSA Candidates
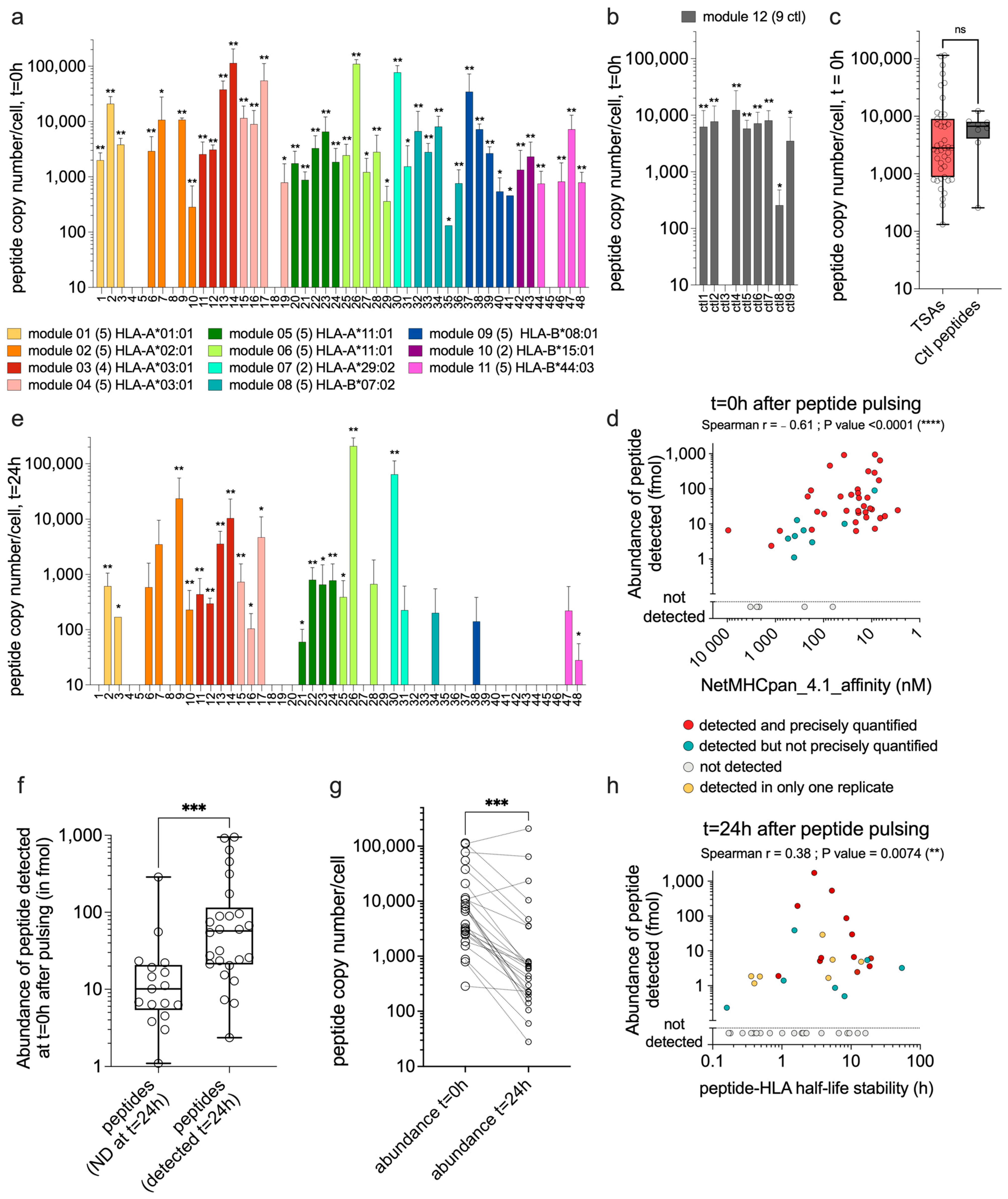
3.2. TSAs Are Well Presented by moDCs after Peptide Pulsing
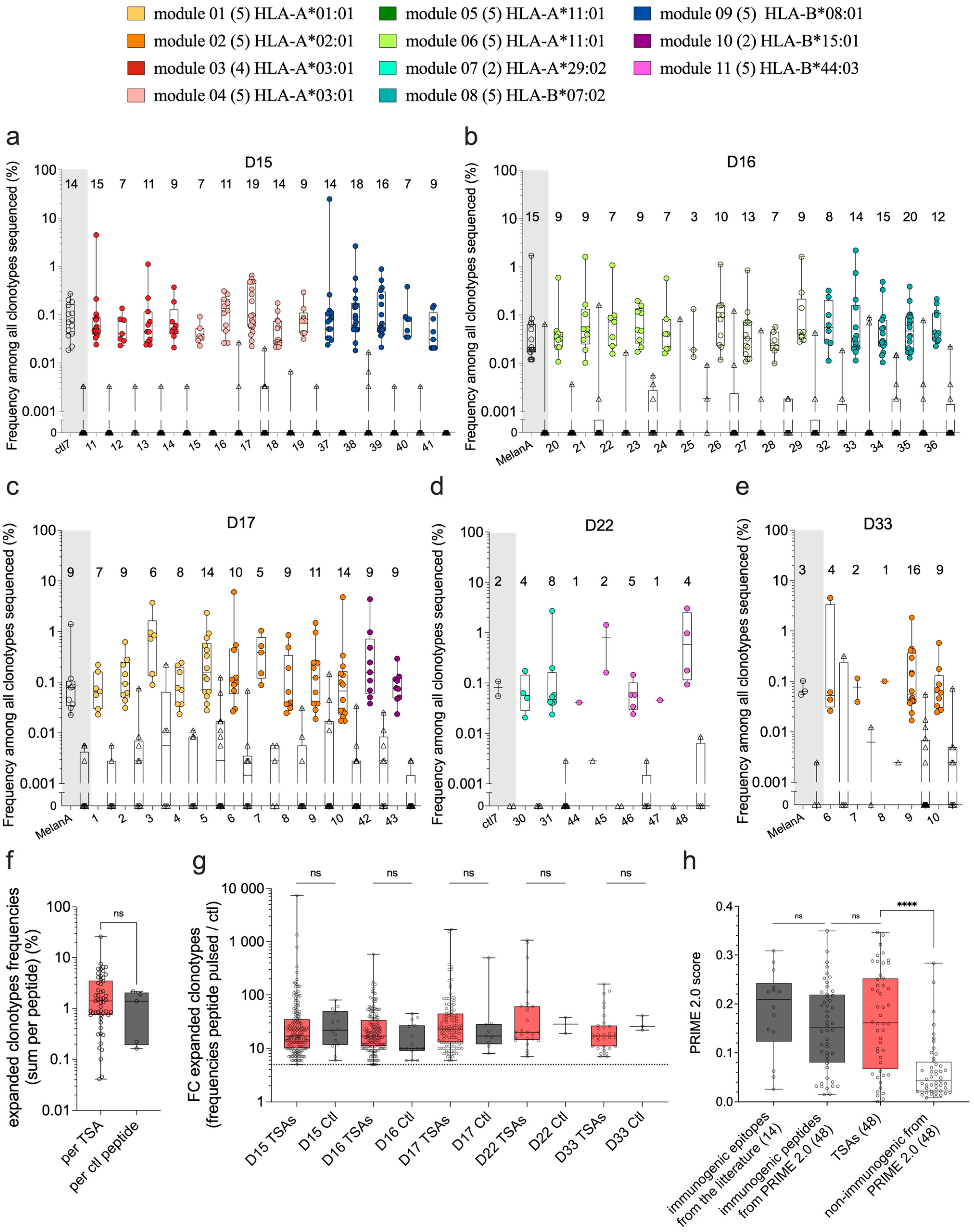
3.3. TCR-Vβ CDR3 Sequencing after Functional Expansion
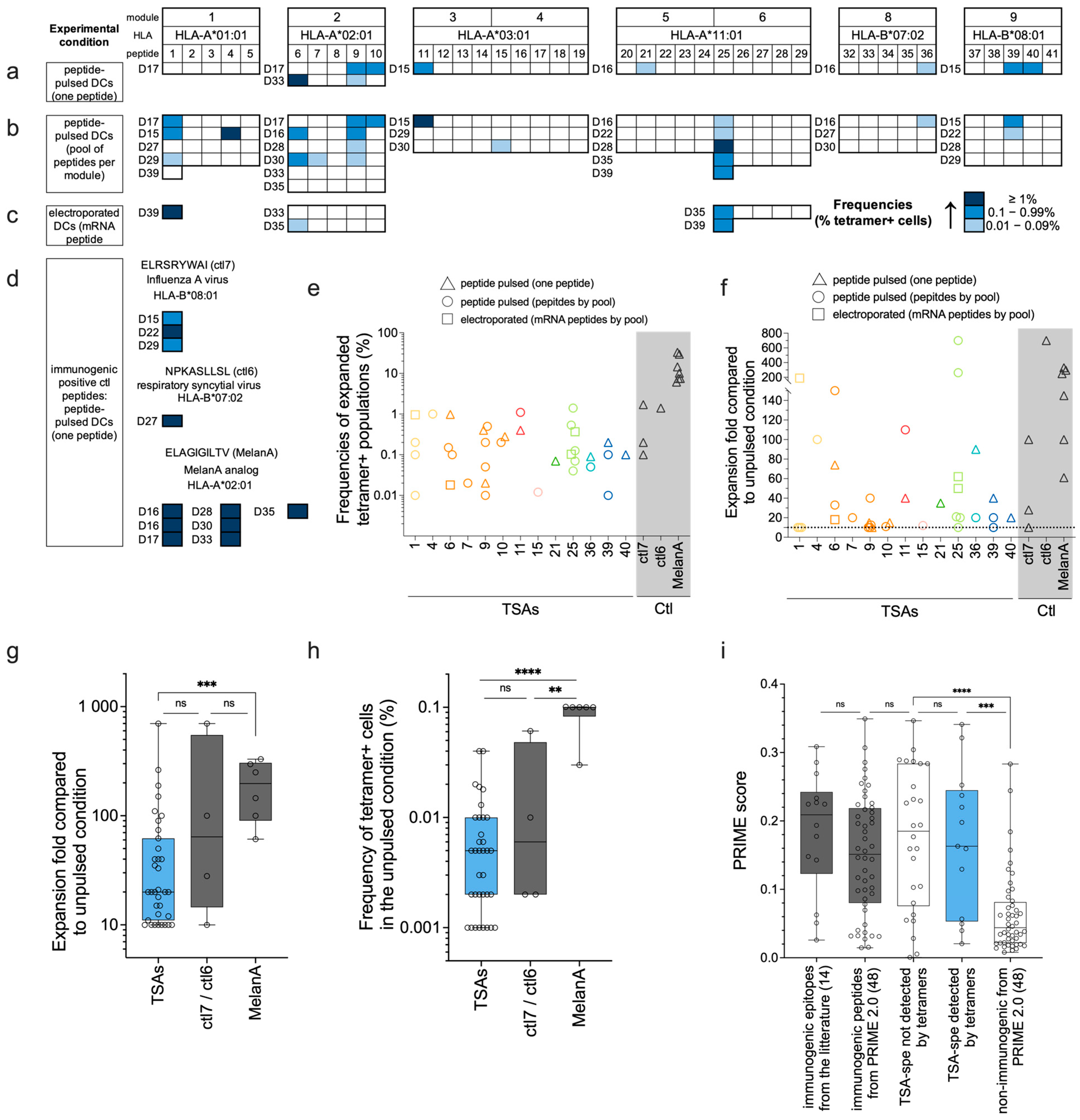
3.4. Tetramer Staining after Functional Expansion
4. Discussion
5. Patents
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, D.R.; Milne, K.; Nelson, B.H. Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Odunsi, K.; Coukos, G. Immunotherapy in Ovarian Cancer: Are We There Yet? J. Clin. Oncol. 2019, 37, 2460–2471. [Google Scholar] [CrossRef]
- Le Page, C.; Chung, J.; Rahimi, K.; Köbel, M.; Provencher, D.; Mes-Masson, A.M. Exploring the Clinical Impact of Predictive Biomarkers in Serous Ovarian Carcinomas. Curr. Drug Targets 2020, 21, 974–995. [Google Scholar] [CrossRef] [PubMed]
- Clouthier, D.L.; Lien, S.C.; Yang, S.Y.C.; Nguyen, L.T.; Manem, V.S.K.; Gray, D.; Ryczko, M.; Razak, A.R.A.; Lewin, J.; Lheureux, S.; et al. An interim report on the investigator-initiated phase 2 study of pembrolizumab immunological response evaluation (INSPIRE). J. Immunother. Cancer 2019, 7, 72. [Google Scholar] [CrossRef]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.J.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019, 25, 89–94. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, É.; Bonneil, É.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Côté, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef]
- Laumont, C.M.; Perreault, C. Exploiting non-canonical translation to identify new targets for T cell-based cancer immunotherapy. Cell. Mol. Life Sci. 2018, 75, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Frankiw, L.; Baltimore, D.; Li, G. Alternative mRNA splicing in cancer immunotherapy. Nat. Rev. Immunol. 2019, 19, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Ehx, G.; Larouche, J.D.; Durette, C.; Laverdure, J.P.; Hesnard, L.; Vincent, K.; Hardy, M.P.; Thériault, C.; Rulleau, C.; Lanoix, J.; et al. Atypical acute myeloid leukemia-specific transcripts generate shared and immunogenic MHC class-I-associated epitopes. Immunity 2021, 54, 737–752.e710. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [PubMed]
- Löffler, M.W.; Mohr, C.; Bichmann, L.; Freudenmann, L.K.; Walzer, M.; Schroeder, C.M.; Trautwein, N.; Hilke, F.J.; Zinser, R.S.; Mühlenbruch, L.; et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med. 2019, 11, 28. [Google Scholar] [CrossRef]
- Newey, A.; Griffiths, B.; Michaux, J.; Pak, H.S.; Stevenson, B.J.; Woolston, A.; Semiannikova, M.; Spain, G.; Barber, L.J.; Matthews, N.; et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J. Immunother. Cancer 2019, 7, 309. [Google Scholar] [CrossRef]
- Kraemer, A.I.; Chong, C.; Huber, F.; Pak, H.; Stevenson, B.J.; Müller, M.; Michaux, J.; Altimiras, E.R.; Rusakiewicz, S.; Simó-Riudalbas, L.; et al. The immunopeptidome landscape associated with T cell infiltration, inflammation and immune editing in lung cancer. Nat. Cancer 2023, 4, 608–628. [Google Scholar] [CrossRef] [PubMed]
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; van de Haar, J.; Engin, H.B.; de Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell 2017, 171, 1272–1283.e1215. [Google Scholar] [CrossRef] [PubMed]
- Nature Biotechnology. The problem with neoantigen prediction. Nat. Biotechnol. 2017, 35, 97. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Laverdure, J.P.; Lanoix, J.; Durette, C.; Côté, C.; Bonneil, É.; Laumont, C.M.; Gendron, P.; Vincent, K.; Courcelles, M.; et al. Proteogenomics Uncovers a Vast Repertoire of Shared Tumor-Specific Antigens in Ovarian Cancer. Cancer Immunol. Res. 2020, 8, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Kina, E.; Laverdure, J.P.; Durette, C.; Lanoix, J.; Courcelles, M.; Zhao, Q.; Apavaloaei, A.; Larouche, J.D.; Hardy, M.P.; Vincent, K.; et al. Breast cancer immunopeptidomes contain numerous shared tumor antigens. J. Clin. Investig. 2024, 134, e166740. [Google Scholar] [CrossRef] [PubMed]
- Ehx, G.; Perreault, C. Discovery and characterization of actionable tumor antigens. Genome Med. 2019, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Hardy, M.P.; Vincent, K.; Perreault, C. The Genomic Landscape of Antigenic Targets for T Cell-Based Leukemia Immunotherapy. Front. Immunol. 2019, 10, 2934. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Jørgensen, K.W.; Rasmussen, M.; Buus, S.; Nielsen, M. NetMHCstab—Predicting stability of peptide-MHC-I complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology 2014, 141, 18–26. [Google Scholar] [CrossRef]
- Maiers, M.; Gragert, L.; Klitz, W. High-resolution HLA alleles and haplotypes in the United States population. Hum. Immunol. 2007, 68, 779–788. [Google Scholar] [CrossRef]
- Cuevas, M.V.R.; Hardy, M.-P.; Larouche, J.-D.; Apavaloaei, A.; Kina, E.; Vincent, K.; Gendron, P.; Laverdure, J.-P.; Durette, C.; Thibault, P.; et al. BamQuery: A proteogenomic tool to explore the immunopeptidome and prioritize actionable tumor antigens. Genome Biol. 2023, 24, 188. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Larouche, J.D.; Trofimov, A.; Hesnard, L.; Ehx, G.; Zhao, Q.; Vincent, K.; Durette, C.; Gendron, P.; Laverdure, J.P.; Bonneil, É.; et al. Widespread and tissue-specific expression of endogenous retroelements in human somatic tissues. Genome Med. 2020, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Fergusson, J.R.; Morgan, M.D.; Bruchard, M.; Huitema, L.; Heesters, B.A.; van Unen, V.; van Hamburg, J.P.; van der Wel, N.N.; Picavet, D.; Koning, F.; et al. Maturing Human CD127+ CCR7+ PDL1+ Dendritic Cells Express AIRE in the Absence of Tissue Restricted Antigens. Front. Immunol. 2018, 9, 2902. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Foldvari, Z.; Giannakopoulou, E.; Böschen, M.L.; Strønen, E.; Yang, W.; Toebes, M.; Schubert, B.; Kohlbacher, O.; Schumacher, T.N.; et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019, 14, 1926–1943. [Google Scholar] [CrossRef] [PubMed]
- Sirois, I.; Isabelle, M.; Duquette, J.D.; Saab, F.; Caron, E. Immunopeptidomics: Isolation of Mouse and Human MHC Class I- and II-Associated Peptides for Mass Spectrometry Analysis. J. Vis. Exp. 2021, 176, e63052. [Google Scholar] [CrossRef] [PubMed]
- Lanoix, J.; Durette, C.; Courcelles, M.; Cossette, É.; Comtois-Marotte, S.; Hardy, M.P.; Côté, C.; Perreault, C.; Thibault, P. Comparison of the MHC I Immunopeptidome Repertoire of B-Cell Lymphoblasts Using Two Isolation Methods. Proteomics 2018, 18, e1700251. [Google Scholar] [CrossRef] [PubMed]
- Danilova, L.; Anagnostou, V.; Caushi, J.X.; Sidhom, J.W.; Guo, H.; Chan, H.Y.; Suri, P.; Tam, A.; Zhang, J.; Asmar, M.E.; et al. The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Gfeller, D.; Schmidt, J.; Croce, G.; Guillaume, P.; Bobisse, S.; Genolet, R.; Queiroz, L.; Cesbron, J.; Racle, J.; Harari, A. Improved predictions of antigen presentation and TCR recognition with MixMHCpred2.2 and PRIME2.0 reveal potent SARS-CoV-2 CD8+ T-cell epitopes. Cell Syst. 2023, 14, 72–83.e75. [Google Scholar] [CrossRef]
- Snary, D.; Barnstable, C.J.; Bodmer, W.F.; Crumpton, M.J. Molecular structure of human histocompatibility antigens: The HLA-C series. Eur. J. Immunol. 1977, 7, 580–585. [Google Scholar] [CrossRef]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Nik, H.; Michaux, J.; Corwin, W.L.; Keller, G.L.; Shcheglova, T.; Pak, H.; Coukos, G.; Baker, B.M.; Mandoiu, I.I.; Bassani-Sternberg, M.; et al. Mass spectrometry driven exploration reveals nuances of neoepitope-driven tumor rejection. JCI Insight 2019, 4, e129152. [Google Scholar] [CrossRef] [PubMed]
- Campillo-Davo, D.; Flumens, D.; Lion, E. The Quest for the Best: How TCR Affinity, Avidity, and Functional Avidity Affect TCR-Engineered T-Cell Antitumor Responses. Cells 2020, 9, 1720. [Google Scholar] [CrossRef] [PubMed]
- Burrows, S.R.; Kienzle, N.; Winterhalter, A.; Bharadwaj, M.; Altman, J.D.; Brooks, A. Peptide-MHC class I tetrameric complexes display exquisite ligand specificity. J. Immunol. 2000, 165, 6229–6234. [Google Scholar] [CrossRef] [PubMed]
- Goulder, P.J.R.; Lechner, F.; Klenerman, P.; McIntosh, K.; Walker, B.D. Characterization of a Novel Respiratory Syncytial Virus-Specific Human Cytotoxic T-Lymphocyte Epitope. J. Virol. 2000, 74, 7694–7697. [Google Scholar] [CrossRef] [PubMed]
- Pittet, M.J.; Valmori, D.; Dunbar, P.R.; Speiser, D.E.; Liénard, D.; Lejeune, F.; Fleischhauer, K.; Cerundolo, V.; Cerottini, J.C.; Romero, P. High frequencies of naive Melan-A/MART-1-specific CD8+ T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J. Exp. Med. 1999, 190, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Heidema, J.; de Bree, G.J.; de Graaff, P.M.A.; van Maren, W.W.C.; Hoogerhout, P.; Out, T.A.; Kimpen, J.L.L.; van Bleek, G.M. Human CD8+ T cell responses against five newly identified respiratory syncytial virus-derived epitopes. J. Gen. Virol. 2004, 85 Pt 8, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Alanio, C.; Lemaitre, F.; Law, H.K.; Hasan, M.; Albert, M.L. Enumeration of human antigen-specific naive CD8+ T cells reveals conserved precursor frequencies. Blood 2010, 115, 3718–3725. [Google Scholar] [CrossRef] [PubMed]
- Legoux, F.; Debeaupuis, E.; Echasserieau, K.; De La Salle, H.; Saulquin, X.; Bonneville, M. Impact of TCR reactivity and HLA phenotype on naive CD8 T cell frequency in humans. J. Immunol. 2010, 184, 6731–6738. [Google Scholar] [CrossRef]
- Romero, P.; Valmori, D.; Pittet, M.J.; Zippelius, A.; Rimoldi, D.; Lévy, F.; Dutoit, V.; Ayyoub, M.; Rubio-Godoy, V.; Michielin, O.; et al. Antigenicity and immunogenicity of Melan-A/MART-1 derived peptides as targets for tumor reactive CTL in human melanoma. Immunol. Rev. 2002, 188, 81–96. [Google Scholar] [CrossRef]
- Neller, M.A.; Ladell, K.; McLaren, J.E.; Matthews, K.K.; Gostick, E.; Pentier, J.M.; Dolton, G.; Schauenburg, A.J.; Koning, D.; Fontaine Costa, A.I.; et al. Naive CD8⁺ T-cell precursors display structured TCR repertoires and composite antigen-driven selection dynamics. Immunol. Cell Biol. 2015, 93, 625–633. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Tan, A.C.; Xiang, S.D.; Goubier, A.; Harland, K.L.; Clemens, E.B.; Plebanski, M.; Kedzierska, K. Understanding CD8+ T-cell responses toward the native and alternate HLA-A*02:01-restricted WT1 epitope. Clin. Transl. Immunol. 2017, 6, e134. [Google Scholar] [CrossRef] [PubMed]
- Tauber, C.; Schultheiss, M.; Luca, R.; Buettner, N.; Llewellyn-Lacey, S.; Emmerich, F.; Zehe, S.; Price, D.A.; Neumann-Haefelin, C.; Schmitt-Graeff, A.; et al. Inefficient induction of circulating TAA-specific CD8+ T-cell responses in hepatocellular carcinoma. Oncotarget 2019, 10, 5194–5206. [Google Scholar] [CrossRef] [PubMed]
- Azoury, M.E.; Samassa, F.; Buitinga, M.; Nigi, L.; Brusco, N.; Callebaut, A.; Giraud, M.; Irla, M.; Lalanne, A.I.; Carré, A.; et al. CD8+ T Cells Variably Recognize Native Versus Citrullinated GRP78 Epitopes in Type 1 Diabetes. Diabetes 2021, 70, 2879–2891. [Google Scholar] [CrossRef] [PubMed]
- Apavaloaei, A.; Hesnard, L.; Hardy, M.P.; Benabdallah, B.; Ehx, G.; Thériault, C.; Laverdure, J.P.; Durette, C.; Lanoix, J.; Courcelles, M.; et al. Induced pluripotent stem cells display a distinct set of MHC I-associated peptides shared by human cancers. Cell Rep. 2022, 40, 111241. [Google Scholar] [CrossRef] [PubMed]
- Schuster, H.; Peper, J.K.; Bösmüller, H.C.; Röhle, K.; Backert, L.; Bilich, T.; Ney, B.; Löffler, M.W.; Kowalewski, D.J.; Trautwein, N.; et al. The immunopeptidomic landscape of ovarian carcinomas. Proc. Natl. Acad. Sci. USA 2017, 114, e9942–e9951. [Google Scholar] [CrossRef] [PubMed]
- Nelde, A.; Schuster, H.; Heitmann, J.S.; Bauer, J.; Maringer, Y.; Zwick, M.; Volkmer, J.P.; Chen, J.Y.; Stanger, A.M.P.; Lehmann, A.; et al. Immune Surveillance of Acute Myeloid Leukemia Is Mediated by HLA-Presented Antigens on Leukemia Progenitor Cells. Blood Cancer Discov. 2023, 4, 468–489. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Huber, F.; Arnaud, M.; Kraemer, A.I.; Altimiras, E.R.; Michaux, J.; Taillandier-Coindard, M.; Chiffelle, J.; Murgues, B.; Gehret, T.; et al. Machine learning methods and harmonized datasets improve immunogenic neoantigen prediction. Immunity 2023, 56, 2650–2663.e2656. [Google Scholar] [CrossRef] [PubMed]
- Henrickson, S.E.; Mempel, T.R.; Mazo, I.B.; Liu, B.; Artyomov, M.N.; Zheng, H.; Peixoto, A.; Flynn, M.P.; Senman, B.; Junt, T.; et al. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat. Immunol. 2008, 9, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Prlic, M.; Hernandez-Hoyos, G.; Bevan, M.J. Duration of the initial TCR stimulus controls the magnitude but not functionality of the CD8+ T cell response. J. Exp. Med. 2006, 203, 2135–2143. [Google Scholar] [CrossRef]
- Obst, R.; van Santen, H.M.; Mathis, D.; Benoist, C. Antigen persistence is required throughout the expansion phase of a CD4+ T cell response. J. Exp. Med. 2005, 201, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Itano, A.A.; McSorley, S.J.; Reinhardt, R.L.; Ehst, B.D.; Ingulli, E.; Rudensky, A.Y.; Jenkins, M.K. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity 2003, 19, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Fonteneau, J.F.; Larsson, M.; Somersan, S.; Sanders, C.; Münz, C.; Kwok, W.W.; Bhardwaj, N.; Jotereau, F. Generation of high quantities of viral and tumor-specific human CD4+ and CD8+ T-cell clones using peptide pulsed mature dendritic cells. J. Immunol. Methods 2001, 258, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Leloup, C.; Reche, S.; Plumas, J. pDCs efficiently process synthetic long peptides to induce functional virus- and tumour-specific T-cell responses. Eur. J. Immunol. 2014, 44, 2880–2892. [Google Scholar] [CrossRef] [PubMed]
- Hoppes, R.; Oostvogels, R.; Luimstra, J.J.; Wals, K.; Toebes, M.; Bies, L.; Ekkebus, R.; Rijal, P.; Celie, P.H.; Huang, J.H.; et al. Altered peptide ligands revisited: Vaccine design through chemically modified HLA-A2-restricted T cell epitopes. J. Immunol. 2014, 193, 4803–4813. [Google Scholar] [CrossRef] [PubMed]
- Carretero-Iglesia, L.; Couturaud, B.; Baumgaertner, P.; Schmidt, J.; Maby-El Hajjami, H.; Speiser, D.E.; Hebeisen, M.; Rufer, N. High Peptide Dose Vaccination Promotes the Early Selection of Tumor Antigen-Specific CD8 T-Cells of Enhanced Functional Competence. Front. Immunol. 2019, 10, 3016. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, J.; Yoshida, T.; Isser, A.; Laino, A.S.; Vassallo, M.; Woods, D.; Kim, S.; Oelke, M.; Jones, K.; Schneck, J.P.; et al. Rapid Expansion of Highly Functional Antigen-Specific T Cells from Patients with Melanoma by Nanoscale Artificial Antigen-Presenting Cells. Clin. Cancer Res. 2020, 26, 3384–3396. [Google Scholar] [CrossRef] [PubMed]
- Stolk, D.A.; de Haas, A.; Vree, J.; Duinkerken, S.; Lübbers, J.; van de Ven, R.; Ambrosini, M.; Kalay, H.; Bruijns, S.; van der Vliet, H.J.; et al. Lipo-Based Vaccines as an Approach to Target Dendritic Cells for Induction of T- and iNKT Cell Responses. Front. Immunol. 2020, 11, 990. [Google Scholar] [CrossRef]
- Sykulev, Y.; Joo, M.; Vturina, I.; Tsomides, T.J.; Eisen, H.N. Evidence that a Single Peptide–MHC Complex on a Target Cell Can Elicit a Cytolytic T Cell Response. Immunity 1996, 4, 565–571. [Google Scholar] [CrossRef]
- Stutzmann, C.; Peng, J.; Wu, Z.; Savoie, C.; Sirois, I.; Thibault, P.; Wheeler, A.R.; Caron, E. Unlocking the potential of microfluidics in mass spectrometry-based immunopeptidomics for tumor antigen discovery. Cell Rep. Methods 2023, 3, 100511. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass Spectrometry of Human Leukocyte Antigen Class I Peptidomes Reveals Strong Effects of Protein Abundance and Turnover on Antigen Presentation*[S]. Mol. Cell. Proteom. 2015, 14, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, I.; Tsai, V.; Southwood, S.; Takesako, K.; Celis, E.; Sette, A. Identification of gp100-derived, melanoma-specific cytotoxic T-lymphocyte epitopes restricted by HLA-A3 supertype molecules by primary in vitro immunization with peptide-pulsed dendritic cells. Int. J. Cancer 1998, 78, 518–524. [Google Scholar] [CrossRef]
- Walter, S.; Weinschenk, T.; Stenzl, A.; Zdrojowy, R.; Pluzanska, A.; Szczylik, C.; Staehler, M.; Brugger, W.; Dietrich, P.Y.; Mendrzyk, R.; et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat. Med. 2012, 18, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Slingluff, C.L., Jr.; Petroni, G.R.; Chianese-Bullock, K.A.; Smolkin, M.E.; Hibbitts, S.; Murphy, C.; Johansen, N.; Grosh, W.W.; Yamshchikov, G.V.; Neese, P.Y.; et al. Immunologic and clinical outcomes of a randomized phase II trial of two multipeptide vaccines for melanoma in the adjuvant setting. Clin. Cancer Res. 2007, 13, 6386–6395. [Google Scholar] [CrossRef] [PubMed]
- Aurisicchio, L.; Fridman, A.; Bagchi, A.; Scarselli, E.; La Monica, N.; Ciliberto, G. A novel minigene scaffold for therapeutic cancer vaccines. Oncoimmunology 2014, 3, e27529. [Google Scholar] [CrossRef]
- Kuznetsov, A.; Voronina, A.; Govorun, V.; Arapidi, G. Critical Review of Existing MHC I Immunopeptidome Isolation Methods. Molecules 2020, 25, 5409. [Google Scholar] [CrossRef]
- Sturm, T.; Sautter, B.; Wörner, T.P.; Stevanović, S.; Rammensee, H.G.; Planz, O.; Heck, A.J.R.; Aebersold, R. Mild Acid Elution and MHC Immunoaffinity Chromatography Reveal Similar Albeit Not Identical Profiles of the HLA Class I Immunopeptidome. J. Proteome Res. 2021, 20, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Laugel, B.; van den Berg, H.A.; Gostick, E.; Cole, D.K.; Wooldridge, L.; Boulter, J.; Milicic, A.; Price, D.A.; Sewell, A.K. Different T cell receptor affinity thresholds and CD8 coreceptor dependence govern cytotoxic T lymphocyte activation and tetramer binding properties. J. Biol. Chem. 2007, 282, 23799–23810. [Google Scholar] [CrossRef]
- Lissina, A.; Ladell, K.; Skowera, A.; Clement, M.; Edwards, E.; Seggewiss, R.; van den Berg, H.A.; Gostick, E.; Gallagher, K.; Jones, E.; et al. Protein kinase inhibitors substantially improve the physical detection of T-cells with peptide-MHC tetramers. J. Immunol. Methods 2009, 340, 11–24. [Google Scholar] [CrossRef]
- Dolton, G.; Lissina, A.; Skowera, A.; Ladell, K.; Tungatt, K.; Jones, E.; Kronenberg-Versteeg, D.; Akpovwa, H.; Pentier, J.M.; Holland, C.J.; et al. Comparison of peptide-major histocompatibility complex tetramers and dextramers for the identification of antigen-specific T cells. Clin. Exp. Immunol. 2014, 177, 47–63. [Google Scholar] [CrossRef]
- Tungatt, K.; Bianchi, V.; Crowther, M.D.; Powell, W.E.; Schauenburg, A.J.; Trimby, A.; Donia, M.; Miles, J.J.; Holland, C.J.; Cole, D.K.; et al. Antibody stabilization of peptide-MHC multimers reveals functional T cells bearing extremely low-affinity TCRs. J. Immunol. 2015, 194, 463–474. [Google Scholar] [CrossRef]
- Lozano-Rabella, M.; Garcia-Garijo, A.; Palomero, J.; Yuste-Estevanez, A.; Erhard, F.; Farriol-Duran, R.; Martín-Liberal, J.; Ochoa-de-Olza, M.; Matos, I.; Gartner, J.J.; et al. Exploring the Immunogenicity of Noncanonical HLA-I Tumor Ligands Identified through Proteogenomics. Clin. Cancer Res. 2023, 29, 2250–2265. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.A.; Urba, W.J.; Jensen, S.M.; Page, D.B.; Curti, B.D.; Sanborn, R.E.; Leidner, R.S. Cancer’s Dark Matter: Lighting the Abyss Unveils Universe of New Therapies. Clin. Cancer Res. 2023, 29, 2173–2175. [Google Scholar] [CrossRef] [PubMed]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Rasmussen, M.; Fenoy, E.; Harndahl, M.; Kristensen, A.B.; Nielsen, I.K.; Nielsen, M.; Buus, S. Pan-Specific Prediction of Peptide-MHC Class I Complex Stability, a Correlate of T Cell Immunogenicity. J. Immunol. 2016, 197, 1517–1524. [Google Scholar] [CrossRef]
- Trofimov, A.; Brouillard, P.; Larouche, J.D.; Séguin, J.; Laverdure, J.P.; Brasey, A.; Ehx, G.; Roy, D.C.; Busque, L.; Lachance, S.; et al. Two types of human TCR differentially regulate reactivity to self and non-self antigens. iScience 2022, 25, 104968. [Google Scholar] [CrossRef]
- Kvedaraite, E.; Ginhoux, F. Human dendritic cells in cancer. Sci. Immunol. 2022, 7, eabm9409. [Google Scholar] [CrossRef] [PubMed]
- Laoui, D.; Keirsse, J.; Morias, Y.; Van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat. Commun. 2016, 7, 13720. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Bevers, S.; Kooijmans, S.A.A.; Van de Velde, E.; Evers, M.J.W.; Seghers, S.; Gitz-Francois, J.; van Kronenburg, N.C.H.; Fens, M.; Mastrobattista, E.; Hassler, L.; et al. mRNA-LNP vaccines tuned for systemic immunization induce strong antitumor immunity by engaging splenic immune cells. Mol. Ther. 2022, 30, 3078–3094. [Google Scholar] [CrossRef]
- Gebre, M.S.; Rauch, S.; Roth, N.; Yu, J.; Chandrashekar, A.; Mercado, N.B.; He, X.; Liu, J.; McMahan, K.; Martinot, A.; et al. Optimization of non-coding regions for a non-modified mRNA COVID-19 vaccine. Nature 2022, 601, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Huff, A.L.; Jaffee, E.M.; Zaidi, N. Messenger RNA vaccines for cancer immunotherapy: Progress promotes promise. J. Clin. Investig. 2022, 132, e156211. [Google Scholar] [CrossRef]
- Lutz, J.; Meister, M.; Habbeddine, M.; Fiedler, K.; Kowalczyk, A.; Heidenreich, R. Local immunotherapy with the RNA-based immune stimulator CV8102 induces substantial anti-tumor responses and enhances checkpoint inhibitor activity. Cancer Immunol. Immunother. 2023, 72, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Ramos da Silva, J.; Bitencourt Rodrigues, K.; Formoso Pelegrin, G.; Silva Sales, N.; Muramatsu, H.; de Oliveira Silva, M.; Porchia, B.; Moreno, A.C.R.; Aps, L.; Venceslau-Carvalho, A.A.; et al. Single immunizations of self-amplifying or non-replicating mRNA-LNP vaccines control HPV-associated tumors in mice. Sci. Transl. Med. 2023, 15, eabn3464. [Google Scholar] [CrossRef] [PubMed]
- Sittplangkoon, C.; Alameh, M.G.; Weissman, D.; Lin, P.J.C.; Tam, Y.K.; Prompetchara, E.; Palaga, T. mRNA vaccine with unmodified uridine induces robust type I interferon-dependent anti-tumor immunity in a melanoma model. Front. Immunol. 2022, 13, 983000. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence (This Study) | Module ID | Peptide ID |
|---|---|---|
| TSDRLFLGY | 01 | 1 |
| ESDEQTLNY | 01 | 2 |
| YLDTAQKNLY | 01 | 3 |
| TPSSHLGLLSY | 01 | 4 |
| LSGCCSLY | 01 | 5 |
| LLLDKLYFL | 02 | 6 |
| SLLNVIGLSV | 02 | 7 |
| SLIPTALSL | 02 | 8 |
| LLSSKLLLM | 02 | 9 |
| IIHSSSLLL | 02 | 10 |
| RTATPLTMKK | 03 | 11 |
| VLAGTVLFK | 03 | 12 |
| RTATPLTMK | 03 | 13 |
| SVYMATTLK | 03 | 14 |
| RLATAPSEK | 04 | 15 |
| RLVTEPSGPK | 04 | 16 |
| RMKTFMMSH | 04 | 17 |
| RLLLPLQSR | 04 | 18 |
| ATWQSVLAR | 04 | 19 |
| STQMTITTQK | 05 | 20 |
| TTLKYLWKK | 05 | 21 |
| GTSPPSVEK | 05 | 22 |
| GTAQVGITK | 05 | 23 |
| SSALLAVALK | 05 | 24 |
| ATLQAAILYEK | 06 | 25 |
| LVFNIILHR | 06 | 26 |
| ASNPVIKKK | 06 | 27 |
| SSSALLAVALK | 06 | 28 |
| RTHQMNTFQR | 06 | 29 |
| VYMATTLKY | 07 | 30 |
| TLVVSIIIY | 07 | 31 |
| SPQTQTHTL | 08 | 32 |
| RPGAGPPGIL | 08 | 33 |
| RVKSTISSL | 08 | 34 |
| PSPLRPSL | 08 | 35 |
| NPSEGSGIRL | 08 | 36 |
| YLATKFMPI | 09 | 37 |
| TPKLRETSV | 09 | 38 |
| YGLPRVVAV | 09 | 39 |
| DILGKSLTL | 09 | 40 |
| YLKWAQIL | 09 | 41 |
| IMKKIRESY | 10 | 42 |
| SQGFSHSQM | 10 | 43 |
| AEHQEGTGTW | 11 | 44 |
| TEISNSQAA | 11 | 45 |
| KEVDPASNTY | 11 | 46 |
| AEEEIMKKI | 11 | 47 |
| AETGVKKPQ | 11 | 48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hesnard, L.; Thériault, C.; Cahuzac, M.; Durette, C.; Vincent, K.; Hardy, M.-P.; Lanoix, J.; Lavallée, G.O.; Humeau, J.; Thibault, P.; et al. Immunogenicity of Non-Mutated Ovarian Cancer-Specific Antigens. Curr. Oncol. 2024, 31, 3099-3121. https://doi.org/10.3390/curroncol31060236
Hesnard L, Thériault C, Cahuzac M, Durette C, Vincent K, Hardy M-P, Lanoix J, Lavallée GO, Humeau J, Thibault P, et al. Immunogenicity of Non-Mutated Ovarian Cancer-Specific Antigens. Current Oncology. 2024; 31(6):3099-3121. https://doi.org/10.3390/curroncol31060236
Chicago/Turabian StyleHesnard, Leslie, Catherine Thériault, Maxime Cahuzac, Chantal Durette, Krystel Vincent, Marie-Pierre Hardy, Joël Lanoix, Gabriel Ouellet Lavallée, Juliette Humeau, Pierre Thibault, and et al. 2024. "Immunogenicity of Non-Mutated Ovarian Cancer-Specific Antigens" Current Oncology 31, no. 6: 3099-3121. https://doi.org/10.3390/curroncol31060236