Abstract
NO formation, which is one of the main disadvantages of ammonia combustion, was studied during the isochoric, adiabatic autoignition of ammonia/air mixtures using the algorithm of Computational Singular Perturbation (CSP). The chemical reactions supporting the action of the mode relating the most to NO were shown to be essentially the ones of the extended Zeldovich mechanism, thus indicating that NO formation is mainly thermal and not due to fuel-bound nitrogen. Because of this, addition of water vapor reduced NO formation, because of its action as a thermal buffer, but increased ignition delay, thus exacerbating the second important caveat of ammonia combustion, which is unrealistically long ignition delay. However, it was also shown that further addition of just 2% molar of H2O2 does not only reduce the ignition delay by a factor of 30, but also reverses the way water vapor affects ignition delay. Specifically, in the ternary mixture NH/HO/HO, addition of water vapor does not prolong but rather shortens ignition delay because it increases OH radicals. At the same time, the presence of HO does not affect the influence of HO in suppressing NO generation. In this manner, we were able to show that NH/HO/HO mixtures offer a way to use ammonia as carbon-less fuel with acceptable NO emissions and realistic ignition delay.
1. Introduction
In view of the strong societal interest in carbon neutrality, ammonia has been proposed as a hydrogen source that can be easily liquified, cost-effectively produced and stored at low pressures, as well as deployed through a reliable and very extensive distribution network [1]. Its production from natural gas through the Haber process, which is widely practiced in the industry, can be thus seen as a method of mass-production of a carbon-less hydrogen carrier, which can act as a fuel. Ammonia use as engine fuel was proposed as early as during World War II [2], but its combustion is severely caveated by two very serious handicaps, namely long ignition delay and increased NO emissions [3]. In a recent publication [4] we were able to show that ignition delay can be shortened drastically through HO addition to the mixture (even a 2% molar addition can decrease the ignition delay by a factor of 30). However, the issue of NO emissions remains to our knowledge unresolved and, in fact, in [4], we showed that HO addition causes a modest increase in NO.
A substantial amount of experimental data has been generated for ammonia combustion in engines and burners. Duynslaegher et al. [5] studied ammonia combustion characteristics in SI engines using a wide range of compression ratios ranging from 12 to 20 with different equivalence ratios ranging from lean to rich compositions. The results revealed that NO emission drops drastically with rich mixtures, with 50% decrease between equivalence ratio of 1.2 and 0.95. Moreover, with higher initial temperatures, the laminar flame velocity, adiabatic flame temperature and thermal NO formation increased, while the increase of pressure exhibited a minimum decrease in the adiabatic flame temperature and no effect on the laminar flame velocity although a decrease in NO mole fractions was observed. This indicates that pressure affects the chemical kinetics of the reactions producing NO [5]. Westlye et al. [6] conducted several experiments using a 1:4 volumetric fuel mixture of hydrogen to ammonia at various compression ratios in SI engines. The results revealed that near stoichiometric conditions lower NO emissions are achieved with ammonia combustion compared to hydrocarbons. Somarathne et al. [7] studied the combustion of turbulent non-premixed ammonia-air in a gas turbine combustion chamber under high pressure and different thermal boundary conditions. The findings revealed that NO emissions are related to the local OH mass fraction. Cai et al. [8] numerically investigated NO emissions associated with the combustion of a premixed ammonia-air mixture on a rectangular micro planer combustor. The results confirmed that NO emissions are highly dependent on the stoichiometric ratio and that it increases with lean combustion mainly due to the higher temperature and concentration of O radicals. Moreover, in line with [9], NO mass fraction was shown to decrease by preheating the fuel mixture which results in a narrow high-temperature region due to the increased flame velocity [8]. Li et al. [10] studied the effect of oxygen enrichment on the combustion characteristics of ammonia and it was shown that it led to an increase of both the laminar burning velocity and flame temperature. It was also shown that HNO, which is considered a significant predecessor for NO formation, also increased with oxygen enrichment. Lee et al. [11] developed a combustion strategy for internal combustion engines using NH as a fuel and showed that NO production was divided into four regimes: pre-combustion, combustion, reduction, and thermal.
The studies of the underlying chemical dynamics that would explain these experimental findings have been relatively few. Hayakawa et al. [12] investigated the characteristics of NO emissions at various stoichiometric ratios and pressures. The results revealed that the NO mass fraction decreased in fuel-rich mixtures and increased pressures. Using flow reactor analysis, the authors identified the reactions in the mechanism by Tian et al. [13] which either formed or consumed NO, as shown in Table 1.

Table 1.
NO forming and reducing reactions [12].
Overall, the rate of NO reduction was faster than that of NO formation, which explained the deNO action of ammonia. The reactions of the same mechanism that depended on pressure were identified. The third body equation OH+H+M ↔ HO+M had the highest rate increase with increased pressure. Through this reaction, H and OH radicals get depleted at higher pressures thus decreasing thermal NO formation through the Zeldovich mechanism [14,15].
Some recent studies were devoted to identifying additives that would assist ammonia in reaching the optimal combustion behavior for its application in combustion engines [4,16,17,18]. In an earlier paper, we showed that hydrogen peroxide reduced the ignition delay of ammonia-air mixtures drastically with a slight increase in NO emissions [4]. An addition of 2%-molar HO, the ignition delay was reduced by a factor of 30 with a cost of 2% increase in NO emission, as shown in Figure 1. This effect was the result of both thermal and chemical action; i.e., due to an early temperature rise due to two-stage autoignition and to the enhanced action of reaction HO(+M)→ OH+OH(+M). It was also shown that the NO emission increased with the amount of HO in the mixture. HO had the same effect on NO and NO, although the final mass fraction of these species was much smaller than that of NO.
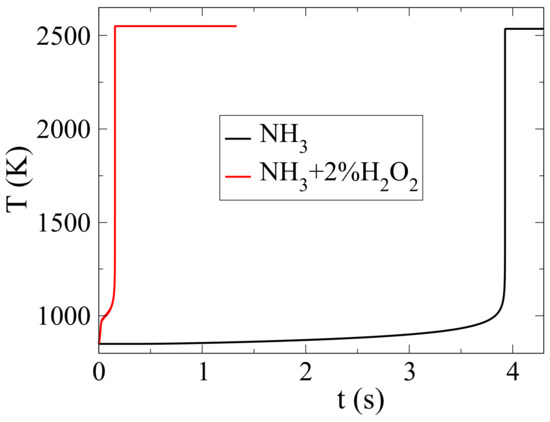
Figure 1.
Temperature evolution during autoignition of (i) pure NH-air and (ii) NH-air with 2%HO mixtures [4].
The drastic reduction of ignition delay with HO was an important finding because it showed that HO addition offered a means to overcome a significant caveat of NO combustion, i.e., unrealistically long ignition delays. However, the issue of NO emissions remained and was actually slightly exacerbated. In [4], mass fractions of NO on the order of 1% were reported, whereas current regulatory limits are on the order of 1000 ppm molar and decreasing. To address this issue, we have studied the dynamics of NO formation during the autoignition of NH-air mixtures using the method of Computational Singular Perturbation (CSP). We particularly focused on the slow chemical dynamics that relate to NO formation, by identifying the reactions that significantly contributed to the time scale that characterized NO emissions as well as on the identification of an additive that would reduce these emissions, possibly without canceling the positive effect that HO had with respect to the ignition delay.
2. CSP Methodology
The autoignition of an isochoric adiabatic homogeneous mixture of ammonia/air was examined using CSP, which provides algorithmic tools for the analysis of multi-scale systems [19]. A chemical kinetics mechanism that consists of N = 34 species, E = 5 elements (O, H, N, He and Ar), and K = 211 reversible elementary reactions was used in this study. This mechanism was generated by removing all carbon chemistry from the mechanism by Glarborg et al. in [20]. The evolution of the modeling of nitrogen chemistry in combustion is discussed thoroughly in [20], where it is shown how the mechanism originally proposed in [13] has evolved to a high-fidelity kinetic model of nitrogen chemistry, validated against a substantial body of experimental results. With the K elementary reactions considered to be unidirectional, the species and energy governing equations can be described as:
where y is the N-dimension mass fraction state column vector of the species, R and S are the reaction rate and stoichiometric vector respectively of the kth unidirectional reaction, is the mixture density, W is a N × N diagonal matrix with the species molecular weights, c is the heat capacity, h is the N-dim. vector of the species absolute enthalpies, T is the temperature, R is the universal gas constant, and U = [1, 1, …, 1] [21,22]. The governing equations, Equations (1) and (2), can be cast in CSP form as:
where is (N+1) dimension state column vector defined as , g (z) is the (N+1) dimension column vector field, and is the generalized stoichiometric vector [19,23]. Resolving Equation (3) along the CSP basis vectors, yields:
where and are the column and row CSP basis vector of the nth mode, respectively () and is the related amplitude. The vector field is decomposed into N-E+1 modes [24,25,26]. The amplitudes to are by definition zero as per the law of atom conservation, since they represent the conservation of elements E. Ultimately, Equation (3) can be written as:
Each CSP mode is characterized by (i) its time scale () that defines the duration of its action, (ii) its amplitude () that measures its contribution to the system evolution and (iii) the variables associated the most with that mode [23,27,28,29,30,31,32,33]. The time scale is approximated by , with being the nth non-zero eigenvalue of the Jacobian of the vector field g. The time scale related to a positive (negative) is explosive (dissipative) in nature and tends to drive the system away from (towards) equilibrium. The eigenvalue can be expressed in terms of the 2K elementary reactions and the th right column and left row eigenvectors of , and respectively, as:
Assuming that the M fastest time scales in the system dynamics are dissipative in nature and substantially faster than the remaining time scales, the M fastest modes can be considered exhausted, so that the system can be reduced as:
The M algebraic relations define a low-dimensional surface in phase space, on which the solution is constrained to evolve, known as Slow Invariant Manifold (SIM) [34,35]. The system of ordinary differential equations in equation Equation (7) defines the flow along the SIM and its dynamics is characterized by the fastest of the remaining slow time scales.
To identify the contribution of the th reaction to the value of the th timescale, the Time Scale Participation Index (TPI) is defined from Equation (6) as:
where varies from 1 to N-E+1, varies from 1 to 2K, and . Positive values of the TPI relate to the reactions that promote the explosive character of the th mode, while negative values of the TPI promote its dissipative character.
Since the 2K unidirectional reactions contribute by different amounts to the amplitude of each CSP mode, the amplitude participation index (API) is used to assess the relative contribution of each reaction to the mode amplitude.
where varies from 1 to N-E+1, varies from 1 to 2K and [4]. In an exhausted mode, the amplitude participation index will identify the reactions contributing to the cancelations in the algebraic relations . In the explosive mode, the API would reflect the contribution of the th reaction to the amplitude of the CSP explosive mode.
The CSP pointer is a tool that identifies the variables related the most to each mode; i.e., the variables that will respond the most to a perturbation along the direction of each mode. It is defined as:
where [23,25,36].
The CSP methodology employed was introduced in 1989 [24] and it has since been studied, employed, and verified extensively in various fields; among them combustion, chemical kinetics, mathematics, biology and pharmacokinetics. In particular, we have been able to verify the methodology for the study of oxidation and autoignition problems in [23,25,27,29,32,33,36,37,38].
3. NO Diagnostics in Ammonia Autoignition
Ammonia combustion is caveated by two significant disadvantages, namely prolonged ignition delays and elevated NO emissions. In a previous work [4] we examined the chemical dynamics of autoignition and showed that addition of HO could drastically decrease the otherwise unacceptably long ignition delay of ammonia. There, we were able to investigate the autoignition dynamics of NH with and without HO addition and determine the chemical and thermal processes that drove NH(/HO) towards ignition. We are now focusing on the slow dynamics that determine the formation of NO. NO emission remains a substantial challenge for ammonia combustion that hinders its application as a fuel due to environmental regulations. Because ammonia has a relatively low flame temperature, it is expected that thermal NO formation will be diminished and that NO will mainly form from fuel-bound nitrogen [39]. This is an intuitive assumption that has never been checked quantitatively. NO accounts for most of the NO emissions of ammonia combustion [11], which indicates the criticality of adequately understanding the chemical dynamics of the NO formation in order to control and reduce it.
Considering an ammonia/air mixture of equivalence ratio at initial temperature and pressure of 850 K and 20 bar, respectively, it is shown in Figure 2 that during the isochoric and adiabatic autoignition of an ammonia-air mixture, substantial NO formation occurs very late in the process; i.e., at high temperature values.
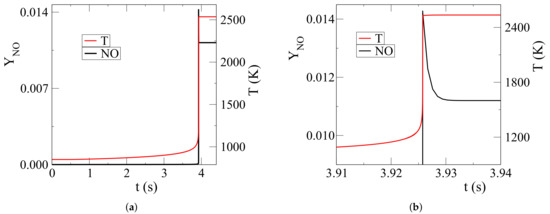
Figure 2.
NO mass fraction and temperature evolution associated with pure NH/air autoignition: (a) full explosive stage, (b) final part of the explosive stage.
In Figure 3, we are focusing on the time that NO initially overshoots, before ultimately reaching its equilibrium value. Three indicative points were selected after ignition, when the NO-producing mechanism was activated and before it reached equilibrium; P was taken at ignition when the temperature reached its maximum value, P midway to equilibrium and P right before equilibrium was reached. CSP diagnostics, computed at these locations, will be studied in order to determine the reactions responsible the most for the observed NO formation. For that purpose, at each location, the CSP mode related the most to NO was identified, using the CSP Pointer in Equation (10). The reactions with the highest contribution to the time scale and to the amplitude of that specific mode were identified, using the TPI in Equation (8) and the API in in Equation (9). These reactions are listed in Table 2.
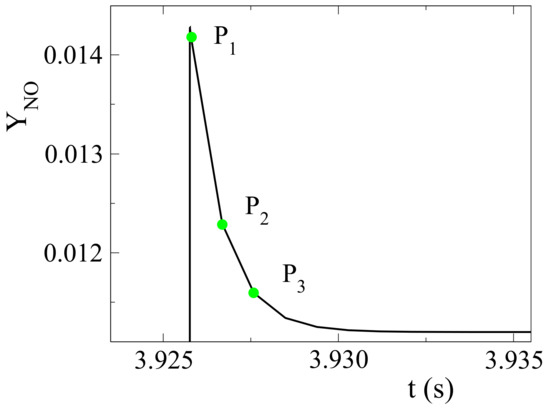
Figure 3.
Diagnostic points identified on NO mass fraction profile; autoignition of pure NH/air.
Table 2.
The reactions most influencing the slow chemical dynamics related to NO formation.
With the exception of the stable species H, O, HO, HO, N, NH, NO, and NO, all the other species discussed in the paper are radicals. Because of this and for facility of presentation, we have not used the occasionally used convention of indicating radicals with a dot.
Mixtures with = 0.75, T(0) = 850 K and p(0) = 20 bar have been considered, in order to account for conditions that are typical of gas turbines and homogeneous combustion compression ignition (HCCI) reciprocating engines. The related CSP diagnostics are shown in Table 3; the symbols “f” and “b” in the number of the reactions denote forward and backward direction, respectively.
Table 3.
The largest TPI, API and Po values for the slow mode that relates to NO at the points P to P shown in Figure 3; Only contributions larger than 5% are shown. In italics are contributions to TPI and API by pairs of forward and backward directions of the same reaction canceling each other.
In the period of interest (P to P) the mode that exerts the most significant influence on the evolution of the process is the one related to NO, say a f. This influence is established by the fact that the amplitude f of this mode is the largest. In this period, most of the chemical reactions have equilibrated. This is manifested in Table 3 by the large number of contributions to TPI and API by pairs of forward and backward directions of the same reaction canceling each other. Neglecting the reactions that provide very small net contributions to TPI and API, leaves reactions 69f (N + NO → N + O), 67b (N + OH ← NO + H) and to a smaller extend 68b (N + O← NO + O) as those that contribute the most to the characteristic time scale and amplitude of the af mode. It is noted that although very frequent in hydrocarbon combustion, thermal NO formation does not strictly require he presence of C/H radicals, but rather formation through the set of reactions 67–69, which constitute the extended Zeldovich mechanism [40]. Especially equation 69b shows that the mechanism for formation of N, which is a necessary prerequisite for the formation of NO, is the attack of O to the N molecule of the air, i.e., it does not depend on the presence of ammonia. This challenges the intuitive expectation that NO formation during ammonia ignition would be due to fuel-bound nitrogen [39], but it is in agreement with the findings of [4], which established that in NH autoignition, the chemical runaway is suspended very early during the process and the largest part of the ignition delay comprises a thermal runaway that leads to a Semenov-type explosion.
NO can be reduced either by increasing its consumption or reducing its production. Since the TPIs reported in Table 3 indicate that the NO-consuming reactions 69f, 67b and 68b are the ones that mainly determine the time frame and the impact of the NO evolution, it follows that their reactants, atomic N, H and O, must be increased in order to reduce NO. Since reactions 69f, 67b and 68b constitute the Zeldovich mechanism and since there is no realistic chemical way to suppress N, H or O, reducing the ignition temperature is the only practical way to reduce NO, possibly with the identification of an appropriate additive, as in [37].
In practice, temperature reduction is achieved by dilution with water vapor [41,42]. Tingas et al. [38] evaluated the effect of HO dilution on the ignition delay of CH/air mixture and on the subsequent NO formation. That study concluded that HO dilution exerted:
- a chemical effect that led to a shortening in the ignition delay, mostly through the increase of OH production by the chain-initiating reaction 22f,
- a different chemical effect that led to the depletion of the O-radical after ignition, thus limiting the NO production and
- a thermal effect in reducing the temperature, acting as a thermal buffer.
On the other hand, the results in [4] indicated that the explosive mode throughout the ignition delay of NH/air mixture was also promoted by OH-producing reaction 22f. This action of 22f was reinforced by smaller contributions from the ammonia dissociation reactions NH + HNO → NH + OH and NH + HN → NH + OH that assisted in building up the pool of OH radicals required for explosion. Since the reactions that relate to OH production play a substantial role also with NH/air combustion, it is worth investigating the effect HO dilution has on both NO emission and ignition delay.
4. The Effect of HO Dilution on the Combustion of the NH/Air Mixture
Using the same mixture and initial conditions defined earlier ( = 0.75, T(0) = 850 K and p(0) = 20 bar), HO was added to NH/air mixture in various molar percentages (5%,10%, 15%, 20%, 30%, 40%, and 50%). Similarly positioned indicative points in the post-ignition regime, as those defined earlier in Figure 3, were used to compute the CSP diagnostics. As in the case of pure ammonia, the mode exhibiting the largest NO CSP pointer was identified and the reactions with the largest contributions to the time scale and the amplitude of that specific mode are listed in Table 4 for the case with 10% HO. The same table also lists the variables that relate to the NO-mode the most. The diagnostics are almost identical to those of Table 3. This development leads to the conclusion that the effect of HO dilution in the NO generation is mainly thermal.
Table 4.
The largest TPI, API and Po for the slow mode that points to NO in the mixture of NH/air with 10%HO for the points shown in Figure 3; Contributions larger than 5% are shown.
The changes in ignition delay, final temperature, and NO mass fraction, for various degrees of HO dilution, are listed in Table 5. It can be seen that HO dilution reduces the final equilibrium temperature and the NO mass fraction; additional reduction is achieved with higher percentages of HO. On the other hand, the ignition delay increases with the amount of dilution. The results also indicate that the drop in final temperature is relatively small compared to the reduction in NO emissions.
Table 5.
Change in ignition delay, final temperature and NO mass fraction with various mole fraction of HO in the initial mixture.
To understand the effect that HO dilution has on ignition delay, we present in Figure 4 the evolution of selected species prior to and post-ignition; O, OH, HO, N, and H mass fractions along with the temperature are presented as a function of scaled time (time divided by the ignition delay). It is shown that in all three cases of HO dilution considered, there is a short initial period (up to about 5 to 15% of t) in which radicals are generated at a very high rate. This rate diminishes considerably after this period and picks-up again close to t. These features suggest that the chemical runaway regime is very short in this case and that the thermal one is relatively long, occupying the largest portion of ignition delay. Figure 4 shows that increasing HO dilution accelerates the action in the chemical runaway regime, the most notable effect there is the increased OH generation. In contrast, during the thermal runaway regime increasing HO dilution does not introduce significant additional changes. This indicates that the chemical activity of water vapor is significant only during the chemical runaway. A similar behavior was reported in [38], where it was shown that the presence of water vapor resulted in decreasing the ignition delay of CH/air autoignition, mainly via the action of reaction 22f: HO(+M) → OH+OH(+M). The absence of significant chemical activity during the long thermal runaway regime, displayed in Figure 4 and the increasing t with increasing HO dilution suggest that vapor acts mainly as a thermal buffer there, in accordance to what is reported in [42].
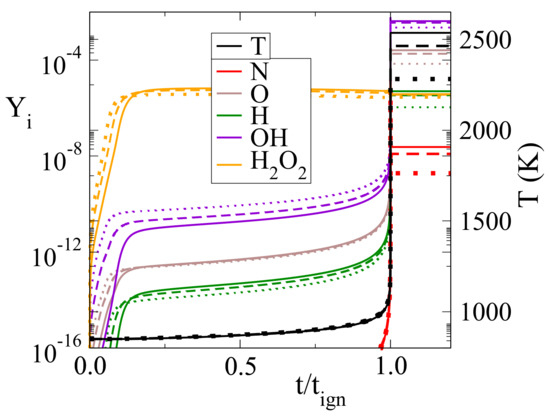
Figure 4.
Evolution of species mass fraction against scaled time (t/t) with various HO percentages (0%: solid lines, 20%: dashed lines and 50%: dotted lines).
Although it was established that the main influence of HO dilution is based on its action as a thermal buffer, it also exerts a chemical influence. This action can be established by investigating the dynamics of reaction 69f, which was shown by the results displayed in Table 3 and Table 5 to influence the most the CSP mode related to NO. Figure 4 shows that in the post-ignition region O-radical mass fraction decreases with increasing HO diluent. Since O is one of the reactants in reaction 69b and a primary agent for NO formation, this feature leads to a reduced NO formation. To understand the chemical effect related to the reduction of O, the CSP mode related to O was investigated at points P to P, shown in Figure 3. The reactions with the largest contribution to the time scale and the amplitude of that specific mode were identified. The results computed at P are displayed in Table 6; those at P and P being qualitatively similar.
Table 6.
The largest TPI, API and Po for the mode that points to O at P; Only contributions larger than 5% are shown.
The mode related to O, say f, is a fast exhausted mode (f). As Table 6 shows, this mode expresses the equilibration of reaction 5: OH + OH ↔ O + HO (large APIs of its two directions, canceling each other), which is mainly driven by its backward direction (involves the most pointed species and exhibits the largest TPI). Clearly, insertion of HO into the mixture shifts this equilibrium to lower mass fractions of O, which will lead to decreased NO generation via reaction 69b.
5. The Effect of HO Dilution on the Dynamics NH/Air and 2%HO Mixture
In [4], we were able to show that mixing NH with HO tackled a first important issue of ammonia combustion, namely it shortened the ignition delay. In particular, we were able to show that inserting just 2% molar HO in the reactant mixture reduced the ignition delay by a factor of approximately 30, as seen in Figure 1. It is noted that as explained in detail in [4], HO is not only a component of the initial mixture but an intermediate product of oxidation with a pivotal role in chemical kinetics. As with what has been shown in [43,44,45], its formation is preceded by the formation of HO through the reaction of the H radical, which comes from H-abstraction from the fuel molecule, with O2 according to the reaction H + O2(+M) → HO2(+M) [4]. The formation of H2O2 follows as a result of HO chemistry. Insertion of HO into the initial mixture causes a drastic increase of OH production through reaction 22f and, therefore a drastic decrease of ignition delay.
However, the ignition delay decrease came with a modest increase in NO emissions. On the other hand, in the previous sections of this paper, we have shown that insertion of water vapor into the mixture can decrease NO, but at the expense of an increase in ignition delay (Table 5). This brings up the need for the investigation of the combustion of ternary NH/HO/HO mixtures with the aim to perhaps identify an optimum composition. To achieve this, HO was added to a mixture of NH-2% HO-air in various mole fractions of HO (5%,10%, 15%, 20%, 30%, 40%, and 50%). Similar to the previous case in Section 3, the change in ignition delay, final temperature, and NO mass fraction of three-part mixture are listed in Table 7.
Table 7.
Change in ignition delay, final temperature and NO mass fraction of NH-2%HO-air mixture, with various percentages of HO.
It can be seen from Table 7 that when HO is present, HO dilution reduces the ignition delay of the mixture significantly and that the reduction increases with the increase of HO content. Also, the decrease of the final temperature and of the final NO mass fraction is very similar to the one with the same HO dilution, as reported in Table 5, but without HO. This implies that the effect of HO dilution on NO, as a thermal buffer, remains essentially unaffected by the presence of HO.
Figure 5 shows the effect of HO and/or HO dilution on ignition delay. In the absence of HO, a 2% HO addition decreases considerably ignition delay. On the other hand, a 10% HO dilution of pure NH increases slightly ignition delay, while a similar dilution of the NH-2%HO mixture causes a slight decrease.
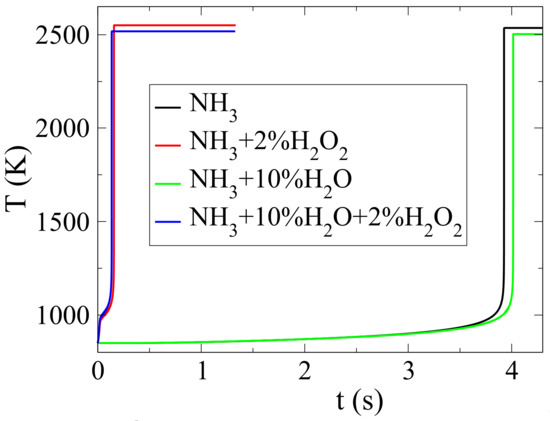
Figure 5.
Temperature evolution of pure NH-air, NH-air with 2%HO, NH-air with 10%HO and NH-air with 2%HO10%HO mixtures.
This response of the NH autoignition in the presence of HO and HO addition can be explained on the basis of the results displayed in Figure 6. In particular, Figure 6a shows that the addition of HO increases considerably the OH levels during the early stage of the ignition delay (chemical runaway regime). This is in agreement with the findings of [4], where it was shown that this feature was due to the enhanced action of reaction 22f: HO(+M)→ OH+OH(+M). Figure 6a also shows that the addition of HO leads to increased levels of OH in both the absence and the presence of HO. Evidently, the rise of OH with the addition of HO is higher in the presence of HO in both the relative and absolute sense. As a result, the increase of the OH contribution towards autoignition caused by HO is higher in the presence of HO. Figure 6b shows that in the presence of HO two-stage ignition occurs, with an early increase in temperature, as a result of the early generation of OH via reaction 22f. In closure, the decrease of the ignition delay with the addition of HO is due to the enhanced chemical action in the early stage of the process, while the addition of HO results in a small increase in the absence of HO (due to the thermal buffer effect) and in a small decrease in its presence (due to the enhancement of the HO chemical action).
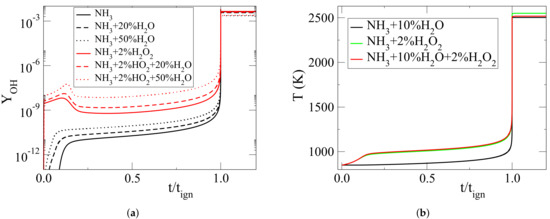
Figure 6.
(a) Evolution of OH mass fraction against scaled time (t/t), when considering NH-2%HO mixture with various HO molar percentages (0% denoted by solid lines, 20% denoted by dashed lines and 50% denoted by dotted lines), (b) Temperature evolution against scaled time (t/t) for NH-10%HO, NH-2%HO and NH-10%HO-2%HO mixtures.
6. Conclusions
The conclusions of this work can be summarized as follows:
- NO formation during isochoric, adiabatic autoignition of ammonia/air mixtures is a slow process that develops after temperature has reached its maximum value and the rest of autoignition chemistry has equilibrated.
- NO formation occurs through the reactions of the extended Zeldovich mechanism, which implies that NO formation is thermal and not due to fuel-bound nitrogen.
- As such, it can be suppressed by addition of water vapor to the fuel mixture, but this causes an increase of the already unacceptably long ignition delay of ammonia.
- Addition of just 2% molar of hydrogen peroxide to the mixture does not only decrease the ignition delay by a factor of 30, but also changes the way water vapor acts. Specifically, in the ternary mixture NH/HO/HO, addition of water vapor does not prolong, but rather it shortens the ignition delay, because it facilitates the increased—due to the HO presence—production of OH radicals.
In closure, it was shown that NH/HO/HO mixtures can be used to overcome two very important disadvantages of ammonia combustion, namely NO formation (through the action of HO) and long ignition delay (through the action of HO). The great advantage of this formulation is that the action of each additive does not counter the action of the other. This can potentially lead to the establishment of a carbon-less fuel and hydrogen carrier, for the storage and transportation of which extensive knowledge and infrastructure already exist.
Author Contributions
Conceptualization, D.A.G. and D.C.K.; methodology, D.A.G.; software, D.A.G., D.M.M.; validation, D.M.M.; formal analysis, D.A.G.; investigation, A.T.K. and D.M.M.; data curation, D.M.M.; writing—original draft preparation, D.M.M. and A.T.K.; writing—review and editing, D.C.K. and D.M.M. and D.A.G.; visualization, D.M.M. and A.T.K.; supervision, D.C.K. and D.A.G.; project administration, D.C.K. and D.A.G.; funding acquisition, D.C.K. and D.A.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
All data reported in this manuscript are available from the corresponding author upon request.
Acknowledgments
We acknowledge support for this work from Khalifa University of Science and Technology under projects RC2-2019-007 and CIRA-2019-033.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Philibert, C. Producing Ammonia and Fertilizers: New Opportunities from Renewables; International Energy Agency: Paris, France, 2017. [Google Scholar]
- Koch, E. Ammonia–A fuel for motor buses. J. Inst. Pet 1945, 31, 213. [Google Scholar]
- Cox, L. Nitrogen Oxides (NOx) Why and How They Are Controlled; Diane Publishing: Collingdon, PA, USA, 1999. [Google Scholar]
- Khalil, A.T.; Manias, D.M.; Tingas, E.A.; Kyritsis, D.C.; Goussis, D.A. Algorithmic Analysis of Chemical Dynamics of the Autoignition of NH3–H2O2/Air Mixtures. Energies 2019, 12, 4422. [Google Scholar] [CrossRef]
- Duynslaegher, C.; Jeanmart, H.; Vandooren, J. Ammonia combustion at elevated pressure and temperature conditions. Fuel 2010, 89, 3540–3545. [Google Scholar] [CrossRef]
- Westlye, F.R.; Ivarsson, A.; Schramm, J. Experimental investigation of nitrogen based emissions from an ammonia fueled SI-engine. Fuel 2013, 111, 239–247. [Google Scholar] [CrossRef]
- Somarathne, K.D.K.A.; Okafor, E.C.; Hayakawa, A.; Kudo, T.; Kurata, O.; Iki, N.; Kobayashi, H. Emission characteristics of turbulent non-premixed ammonia/air and methane/air swirl flames through a rich-lean combustor under various wall thermal boundary conditions at high pressure. Combust. Flame 2019, 210, 247–261. [Google Scholar] [CrossRef]
- Cai, T.; Zhao, D.; Wang, B.; Li, J.; Guan, Y. NOx emission and thermal performances studies on premixed ammonia-oxygen combustion in a CO2-free micro-planar combustor. Fuel 2020, 280, 118554. [Google Scholar] [CrossRef]
- Sorrentino, G.; Sabia, P.; Bozza, P.; Ragucci, R.; de Joannon, M. Low-NOx conversion of pure ammonia in a cyclonic burner under locally diluted and preheated conditions. Appl. Energy 2019, 254, 113676. [Google Scholar] [CrossRef]
- Li, J.; Huang, H.; Kobayashi, N.; He, Z.; Osaka, Y.; Zeng, T. Numerical study on effect of oxygen content in combustion air on ammonia combustion. Energy 2015, 93, 2053–2068. [Google Scholar] [CrossRef]
- Lee, D.; Song, H.H. Development of combustion strategy for the internal combustion engine fueled by ammonia and its operating characteristics. J. Mech. Sci. Technol. 2018, 32, 1905–1925. [Google Scholar] [CrossRef]
- Hayakawa, A.; Goto, T.; Mimoto, R.; Kudo, T.; Kobayashi, H. NO formation/reduction mechanisms of ammonia/air premixed flames at various equivalence ratios and pressures. Mech. Eng. J. 2015, 14-00402. [Google Scholar] [CrossRef]
- Tian, Z.; Li, Y.; Zhang, L.; Glarborg, P.; Qi, F. An experimental and kinetic modeling study of premixed NH3/CH4/O2/Ar flames at low pressure. Combust. Flame 2009, 156, 1413–1426. [Google Scholar] [CrossRef]
- Miller, J.A.; Smooke, M.D.; Green, R.M.; Kee, R.J. Kinetic modeling of the oxidation of ammonia in flames. Combust. Sci. Technol. 1983, 34, 149–176. [Google Scholar] [CrossRef]
- Miller, J.A.; Bowman, C.T. Mechanism and modeling of nitrogen chemistry in combustion. Prog. Energy Combust. Sci. 1989, 15, 287–338. [Google Scholar] [CrossRef]
- Reiter, A.J.; Kong, S.C. Combustion and emissions characteristics of compression-ignition engine using dual ammonia-diesel fuel. Fuel 2011, 90, 87–97. [Google Scholar] [CrossRef]
- Gill, S.; Chatha, G.; Tsolakis, A.; Golunski, S.E.; York, A. Assessing the effects of partially decarbonising a diesel engine by co-fuelling with dissociated ammonia. Int. J. Hydrogen Energy 2012, 37, 6074–6083. [Google Scholar] [CrossRef]
- Frigo, S.; Gentili, R. Analysis of the behaviour of a 4-stroke Si engine fuelled with ammonia and hydrogen. Int. J. Hydrogen Energy 2013, 38, 1607–1615. [Google Scholar] [CrossRef]
- Diamantis, D.J.; Mastorakos, E.; Goussis, D.A. H2/air autoignition: The nature and interaction of the developing explosive modes. Combust. Theory Model. 2015, 19, 382–433. [Google Scholar] [CrossRef]
- Glarborg, P.; Miller, J.A.; Ruscic, B.; Klippenstein, S.J. Modeling nitrogen chemistry in combustion. Prog. Energy Combust. Sci. 2018, 67, 31–68. [Google Scholar] [CrossRef]
- Williams, F.A. Combustion Theory; The Benjamin/Cummings Publ. Co.: Menlo Park, CA, USA, 1985. [Google Scholar]
- Law, C.K. Combustion Physics; Cambridge University Press: New York, NY, USA, 2006. [Google Scholar]
- Valorani, M.; Najm, H.N.; Goussis, D.A. CSP analysis of a transient flame-vortex interaction: Time scales and manifolds. Combust. Flame 2003, 134, 35–53. [Google Scholar] [CrossRef]
- Lam, S.H.; Goussis, D.A. Understanding complex chemical kinetics with computational singular perturbation. Proc. Combust. Inst. 1989, 22, 931–941. [Google Scholar] [CrossRef]
- Lam, S.H.; Goussis, D.A. The CSP method for simplifying kinetics. Int. J. Chem. Kinet. 1994, 26, 461–486. [Google Scholar] [CrossRef]
- Hadjinicolaou, M.; Goussis, D.A. Asymptotic solution of stiff PDEs with the CSP method: The reaction diffusion equation. SIAM J. Sci. Comput. 1998, 20, 781–810. [Google Scholar] [CrossRef]
- Najm, H.N.; Ponganis, D.; Prager, J. Analysis of NO structure in a methane–air edge flame. Proc. Combust. Inst. 2009, 32, 1117–1124. [Google Scholar] [CrossRef]
- Lu, T.; Yoo, C.S.; Chen, J.; Law, C.K. Three-dimensional direct numerical simulation of a turbulent lifted hydrogen jet flame in heated coflow: A chemical explosive mode analysis. J. Fluid Mech. 2010, 652, 45. [Google Scholar] [CrossRef]
- Goussis, D.A.; Valorani, M.; Creta, F.; Najm, H.N. Reactive and reactive-diffusive time scales in stiff reaction-diffusion systems. Prog. Comput. Fluid Dyn. Int. J. 2005, 5, 316–326. [Google Scholar] [CrossRef]
- Luo, Z.; Yoo, C.S.; Richardson, E.S.; Chen, J.H.; Law, C.K.; Lu, T. Chemical explosive mode analysis for a turbulent lifted ethylene jet flame in highly-heated coflow. Combust. Flame 2012, 159, 265–274. [Google Scholar] [CrossRef]
- Shan, R.; Yoo, C.S.; Chen, J.H.; Lu, T. Computational diagnostics for n-heptane flames with chemical explosive mode analysis. Combust. Flame 2012, 159, 3119–3127. [Google Scholar] [CrossRef]
- Kooshkbaghi, M.; Frouzakis, C.E.; Boulouchos, K.; Karlin, I.V. n-Heptane/air combustion in perfectly stirred reactors: Dynamics, bifurcations and dominant reactions at critical conditions. Combust. Flame 2015, 162, 3166–3179. [Google Scholar] [CrossRef]
- Tingas, E.A.; Kyritsis, D.C.; Goussis, D.A. Autoignition dynamics of DME/air and EtOH/air homogeneous mixtures. Combust. Flame 2015, 162, 3263–3276. [Google Scholar] [CrossRef]
- Goussis, D.A. The role of slow system dynamics in predicting the degeneracy of slow invariant manifolds: The case of vdP relaxation–oscillations. Phys. D Nonlinear Phenom. 2013, 248, 16–32. [Google Scholar] [CrossRef]
- Maris, D.T.; Goussis, D.A. The “hidden” dynamics of the Rössler attractor. Phys. D Nonlinear Phenom. 2015, 295, 66–90. [Google Scholar] [CrossRef]
- Goussis, D.A.; Lam, S.H. A study of homogeneous methanol oxidation kinetics using CSP. Symp. Combust. 1992, 24, 113–120. [Google Scholar] [CrossRef]
- Manias, D.M.; Tingas, E.A.; Frouzakis, C.E.; Boulouchos, K.; Goussis, D.A. The mechanism by which CH2O and H2O2 additives affect the autoignition of CH4/air mixtures. Combust. Flame 2016, 164, 111–125. [Google Scholar] [CrossRef]
- Tingas, E.A.; Kyritsis, D.C.; Goussis, D.A. Algorithmic determination of the mechanism through which H2O-dilution affects autoignition dynamics and NO formation in CH4/air mixtures. Fuel 2016, 183, 90–98. [Google Scholar] [CrossRef]
- Li, J.; Huang, H.; Kobayashi, N.; He, Z.; Nagai, Y. Study on using hydrogen and ammonia as fuels: Combustion characteristics and NOx formation. Int. J. Energy Res. 2014, 38, 1214–1223. [Google Scholar] [CrossRef]
- Glassman, I.; Yetter, R.A.; Glumac, N.G. Combustion; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Le Cong, T.; Dagaut, P. Experimental and detailed modeling study of the effect of water vapor on the kinetics of combustion of hydrogen and natural gas, impact on NOx. Energy Fuels 2009, 23, 725–734. [Google Scholar] [CrossRef]
- Donohoe, N.; Heufer, K.A.; Aul, C.J.; Petersen, E.L.; Bourque, G.; Gordon, R.; Curran, H.J. Influence of steam dilution on the ignition of hydrogen, syngas and natural gas blends at elevated pressures. Combust. Flame 2015, 162, 1126–1135. [Google Scholar] [CrossRef]
- Altarawneh, M.K.; Dlugogorski, B.Z.; Kennedy, E.M.; Mackie, J.C. Rate constants for reactions of ethylbenzene with hydroperoxyl radical. Combust. Flame 2013, 160, 9–16. [Google Scholar] [CrossRef]
- Rawadieh, S.E.; Altarawneh, I.S.; Batiha, M.A.; Al-Makhadmeh, L.A.; Almatarneh, M.H.; Altarawneh, M. Reaction of Hydroperoxy Radicals with Primary C1–5 Alcohols: A Profound Effect on Ignition Delay Times. Energy Fuels 2019, 33, 11781–11794. [Google Scholar] [CrossRef]
- Westbrook, C.K.; Dryer, F.L. Chemical kinetic modeling of hydrocarbon combustion. Prog. Energy Combust. Sci. 1984, 10, 1–57. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).