
Energetic Materials Based on N-substituted 4(5)-nitro-1,2,3-triazoles

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
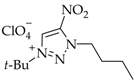
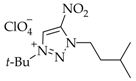
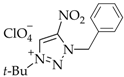


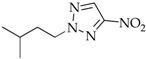

3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steppeler, F.; Kłopotowska, D.; Wietrzyk, J.; Wojaczyńska, E. Synthesis and Antiproliferative Activity of Triazoles Based on 2-Azabicycloalkanes. Materials 2021, 14, 2039. [Google Scholar] [CrossRef] [PubMed]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Medic. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef] [PubMed]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click chemistry: 1,2,3-triazoles as pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Zhang, W. New lead structures in antifungal drug discovery. Curr Med. Chem. 2011, 18, 733–766. [Google Scholar] [CrossRef]
- Neu, H.C.; Fu, K.P. Cefatrizine activity compared with that of other cephalosporins. Antimicrob. Agents Chemother. 1979, 15, 209–212. [Google Scholar] [CrossRef] [Green Version]
- Soltis, M.J.; Yeh, H.J.; Cole, K.A.; Whittaker, N.; Wersto, R.P.; Kohn, E.C. Identification and characterization of human metabolites of CAI [5-amino-1-1(4’-chlorobenzoyl-3,5-dichlorobenzyl)-1,2,3-triazole-4-carboxamide). Drug Metab. Dispos. 1996, 24, 799–806. [Google Scholar]
- Higashitani, F.; Hyodo, A.; Ishida, N.; Inoue, M.; Mitsuhashi, S. Inhibition of betalactamases by tazobactam and in-vitro antibacterial activity of tazobactam combined with piperacillin. J. Antimicrob. Chemother. 1990, 25, 567–574. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, Z.; Gao, C. Triazole derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 138, 501–513. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Parrish, D.A.; Shreeve, J.M. Derivatives of 5-nitro-1,2,3-2H-triazole—High performance energetic materials. J. Mat. Chem. A. 2013, 1, 585–593. [Google Scholar] [CrossRef]
- Cao, W.; Qin, J.; Zhang, J.; Sinditskii, V.P. 4,5-Dicyano-1,2,3-Triazole—A Promising Precursor for a New Family of Energetic Compounds and Its Nitrogen-Rich Derivatives: Synthesis and Crystal Structures. Molecules 2021, 26, 6735. [Google Scholar] [CrossRef]
- Gu, H.; Xiong, H.; Yang, H.; Cheng, G. Tricyclic nitrogen-rich cation salts based on 1,2,3-triazole: Chemically stable and insensitive candidates for novel gas generant. Chem. Eng. J. 2021, 408, 128021. [Google Scholar] [CrossRef]
- Feng, S.; Yin, P.; He, C.; Pang, S.P.; Shreeve, J.M. Tunable Dimroth rearrangement of versatile 1,2,3-triazoles towards highperformance energetic materials. J. Mat. Chem. A. 2021, 9, 12291–12298. [Google Scholar] [CrossRef]
- Lai, Q.; Fei, T.; Yin, P.; Shreeve, J.M. 1,2,3-Triazole with linear and branched catenated nitrogen chains—The role of regiochemistry in Energetic Materials. Chem. Eng. J. 2020, 410, 128148. [Google Scholar] [CrossRef]
- Yao, W.; Xue, Y.; Qian, L.; Yang, H.; Cheng, G. Combination of 1,2,3-triazole and 1,2,4-triazole frameworks for new high-energy and low-sensitivity compounds. Energ. Mat. Front. 2021, 2, 131–138. [Google Scholar] [CrossRef]
- Cao, Y.; Huang, H.; Pang, A.; Lin, X.; Yang, J.; Gong, X.; Fan, G. Synthesis of a bi-heterocyclic skeleton with high HOF and corresponding energetic salts with high heat of detonation. Chem. Eng. J. 2020, 393, 124683. [Google Scholar] [CrossRef]
- Zhao, X.X.; Li, S.H.; Wang, Y.; Li, Y.C.; Zhao, F.Q.; Pang, S.P. Design and Synthesis of Energetic Materials Towards High Density and Positive Oxygen Balance by N-Dinitromethyl Functionalization of Nirtoazoles. J. Mater. Chem. A. 2016, 4, 5495–5504. [Google Scholar] [CrossRef]
- Zhang, Q.; Shreeve, J.M. Energetic Ion Liquids as Explosives and Propellant Fuels: A New Journey of Liquid Chemistry. Chem. Rev. 2014, 114, 10527–10574. [Google Scholar] [CrossRef] [PubMed]
- Larina, L.; Lopyrev, V. Nitroazoles: Synthesis, Structure and Applications. Topics in Applied Chemistry; Springer Science + Business Media: Dordrecht, The Netherlands, 2009; pp. 1–441. ISBN 978-1-4614-2445-1. [Google Scholar]
- Krivopalov, V.P.; Shkurko, O.P. 1,2,3-Triazole and its derivatives. Development of methods for the formation of the triazole ring. Russ. Chem. Rev. 2005, 74, 339–379. [Google Scholar] [CrossRef]
- Vereshchagin, L.I.; Kuznetsova, N.I.; Kirillova, L.P.; Shcherbakov, V.V.; Sukhanov, G.T.; Gareev, G.A. Reactions of 4-nitro-1,2,3-triazole with alkylating agents and compounds with activated multiple bonds. Chem. Heterocycl. Compd. 1986, 22, 745–748. [Google Scholar] [CrossRef]
- Tsutomu, K.; Motonobu, M.; Nakahara, Y.; Ryoji, K.; Tsuneo, T.; Ryochi, O.; Sakano, K. A radiation sensitizer. EP Patent 0212558A2, 4 March 1987. [Google Scholar]
- Vereschagin, L.I.; Pokatilov, F.A.; Kizhnyaev, V.N. Synthesis and properties of nitro-1,2,3-triazoles (Review). Chem. Heterocycl. Compd. 2008, 44, 1–19. [Google Scholar] [CrossRef]
- Licht, H.H.; Ritter, H.; Bircher, H.R.; Bigler, P. Tautomerism in nitrotriazoles: Structure investigation by combined 1H, 13C and 15N NMR spectroscopy. Magn. Reson. Chem. 1998, 36, 343–350. [Google Scholar] [CrossRef]
- Verkhozina, O.N.; Kizhnyaev, V.N.; Vereshchagin, L.I.; Rokhin, A.V.; Smirnov, A.I. Synthesis of PolynuclearNonfused Azoles. Russ. J. Org. Chem. 2003, 39, 1792–1796. [Google Scholar] [CrossRef]
- Pryde, D.C.; Maw, G.N.; Planken, S.; Platts, M.Y.; Sanderson, V.; Corless, M.; Stobie, A.; Barber, C.G.; Russell, R.; Foster, L.; et al. Novel Selective Inhibitors of Neutral Endopeptidase for the Treatment of Female Sexual Arousal Disorder. Synthesis and Activity of Functionalized Glutaramides. J. Med. Chem. 2006, 49, 4409–4424. [Google Scholar] [CrossRef] [PubMed]
- Shangbiao, F.; Fengsheng, L.; Xinyuan, Z.; Yadong, Q.; Teng, F.; Ping, Y.; Siping, P. Comparative study on 1,2,3-triazole based azo- and triazene-bridged high-nitrogen energetic materials. Energetic Mater. Front. 2021, 2, 125–130. [Google Scholar] [CrossRef]
- Licht, H.H.; Ritter, H. Synthesis and explosive properties of dinitrobitriazole. Propellants Explos. Pyrotech. 1997, 22, 333–336. [Google Scholar] [CrossRef]
- Hirai, R.; Watanabe, T.; Ono, T. Design of Clickable Ionic Liquid Monomers to Enhance Ionic Conductivity for Main-Chain 1,2,3-Triazolium-Based Poly(Ionic Liquid)s. ACS Omega 2021, 6, 10030–10038. [Google Scholar] [CrossRef]
- González-Perdomo, P.; González, J.; Martínez-Otero, D.; Unnamatla, B.; García-Eleno, M.A.; Corona-Becerril, D.; Cuevas-Yañez, E. Synthesis of 3-alkyl-1,2,3-triazol-1-ium hydrogen sulphate derivatives. J. Chem Res. 2021, 45, 322–325. [Google Scholar] [CrossRef]
- Yacob, Z.; Liebscher, J. Chemistry of 1,2,3-Triazolium Salts. In Chemistry of 1,2,3-triazoles. Topics in Heterocyclic Chemistry; Dehaen, W., Bakulev, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 40, pp. 1–384. [Google Scholar] [CrossRef]
- Kizhnyaev, V.N.; Golobokova, T.V.; Pokatilov, F.A.; Vereshchagin, L.I.; Estrin, Y.I. Synthesis of energetic triazole- and tetrazole-containing oligomers and polymers. Chem. Heterocycl. Compd. 2017, 53, 682–692. [Google Scholar] [CrossRef]
- Voitekhovich, S.V.; Gaponik, P.N.; Lyakhov, A.S.; Filipova, J.V.; Sukhanova, A.G.; Sukhanov, G.T.; Ivashkevich, O.A. N-Alkylation of 4-nitro-1,2,3-triazole revisited. Detection and characterization of the N3-ethylation product, 1-ethyl-5-nitro-1,2,3-triazole. Tetrahedron Lett. 2009, 50, 2577–2579. [Google Scholar] [CrossRef]
- Voitekhovich, S.V.; Filippova, J.V.; Sukhanova, A.G.; Lyakhov, A.S.; Ivashkevich, L.S.; Sukhanov, G.T.; Grigoriev, Y.V. Selective complexation of 1-ethyl-5-nitro-1,2,3-triazole (entz) with copper(II) salts: Preparation and characterization of [Cu(entz)2Cl2] and [Cu(entz)4(H2O)2](ClO4)2. Inorg. Chem. Comm. 2012, 24, 77–80. [Google Scholar] [CrossRef]
- Matulis, V.E.; Ivashkevich, O.A.; Gaponik, P.N.; Elkind, P.D.; Sukhanov, G.T.; Bazyleva, A.B.; Zaitsau, D.H. Theoretical study of gas-phase formation enthalpies and isomerism for 4(5)-nitro-1,2,3-triazole and its N-alkyl derivatives and experimental determination of formation enthalpy for 2-methyl-4-nitro-1,2,3-triazole. J. Mol. Struct. 2008, 854, 18–25. [Google Scholar] [CrossRef]
- Ivashkevich, O.A.; Matulis, V.E.; Gaponik, P.N.; Sukhanov, G.T.; Filippova, J.V.; Sukhanova, A.G. Quantum-chemical investigation of certain physicochemical properties of C-nitro-1,2,3-triazole and N-alkyl-4(5)-nitro-1,2,3-triazoles. Chem. Heterocycl. Compd. 2008, 44, 1472–1482. [Google Scholar] [CrossRef]
- Sukhanov, G.T.; Sakovich, G.V.; Filippova, Y.V.; Bagryanskaya, I.Y.; Sukhanova, A.G. Regioselective quaternization of N-alkyl-4-nitro-1,2,3-triazoles in ButOH–HClO4 system. Mendeleev. Commun. 2014, 24, 280–282. [Google Scholar] [CrossRef]
- Baryshnikov, A.T.; Erashko, V.I.; Zubanova, N.I.; Ugrak, B.I.; Shevelev, S.A.; Fainzil’berg, A.A.; Laikhter, A.L.; Mel’nikova, L.G.; Semenov, V.V. Gem-dinitro compounds in organic synthesis. 3. Syntheses of 4-nitro-1,2,3-triazoles from gem-dinitro compounds. Russ. Chem. Bull. 1992, 41, 751–757. [Google Scholar] [CrossRef]
- Sakovich, G.V.; Sukhanov, G.T.; Filippova, Y.V.; Sukhanova, A.G.; Bosov, K.K. Alkylation of 4(5)-nitro-1,2,3-triazole with alcohols in strongly acidic media. Russ. Chem. Bull. Int. Ed. 2013, 62, 111–116. [Google Scholar] [CrossRef]
- Filippova, Y.V.; Sukhanova, A.G.; Voitekhovich, S.V.; Matulis, V.E.; Sukhanov, G.T.; Grigoriev, Y.V.; Ivashkevich, O.A. Acid Catalyzed tert-Butylation and Tritylation of 4-Nitro-1,2,3-triazole: Selective Synthesis of 1-Methyl-5-nitro-1,2,3-triazole via 1-tert-Butyl-4-nitro-1,2,3-triazole. J. Heterocycl. Chem. 2012, 49, 965–968. [Google Scholar] [CrossRef]
- Sukhanov, G.T.; Filippova, Y.V.; Gatilov, Y.V.; Sukhanova, A.G.; Bosov, K.K.; Krupnova, I.A.; Pivovarova, E.V. Acidic N-dealkylation in nitrotriazolium salts. Mendeleev Commun. 2022, 1, in press. [Google Scholar]
- Grigoriev, Y.V.; Voitekhovich, S.V.; Ivashkevich, O.A. Alkylation of 3-nitro-1,2,4-triazole with allyl bromide and cyclohexa-1,3-diene in acid medium. Russ. J. Org. Chem. 2012, 48, 610–612. [Google Scholar] [CrossRef]
- Gaponik, P.N.; Voitekhovich, S.V.; Klyaus, B.G. Formation of 2-(2-Cyclohexenyl)-5-R-tetrazoles in Acid-Catalyzed Alkylation of 5-Substituted Tetrazoles with 1,3-Cyclohexadiene. Russ. J. Org. Chem. 2004, 40, 598–600. [Google Scholar] [CrossRef]
- CCDC-2119451 Contains the Supplementary Crystallographic Data of 6 for This Paper. These Data Can be Obtained Free of Charge from The Cambridge Crystallographic Data Centre. Available online: www.ccdc.cam.ac.uk/data_request/cif (accessed on 1 November 2021).
- Li, T.; Xing, Z. Crystal structure of hexa-μ2-chlorido-μ4-oxido-tetrakis(1-vinyl-1H-imidazole-κN)tetracopper(II), C20H24Cu4Cl6N8O. Z.Kristallogr. New Cryst.Struct. 2019, 234, 363–365. [Google Scholar] [CrossRef]
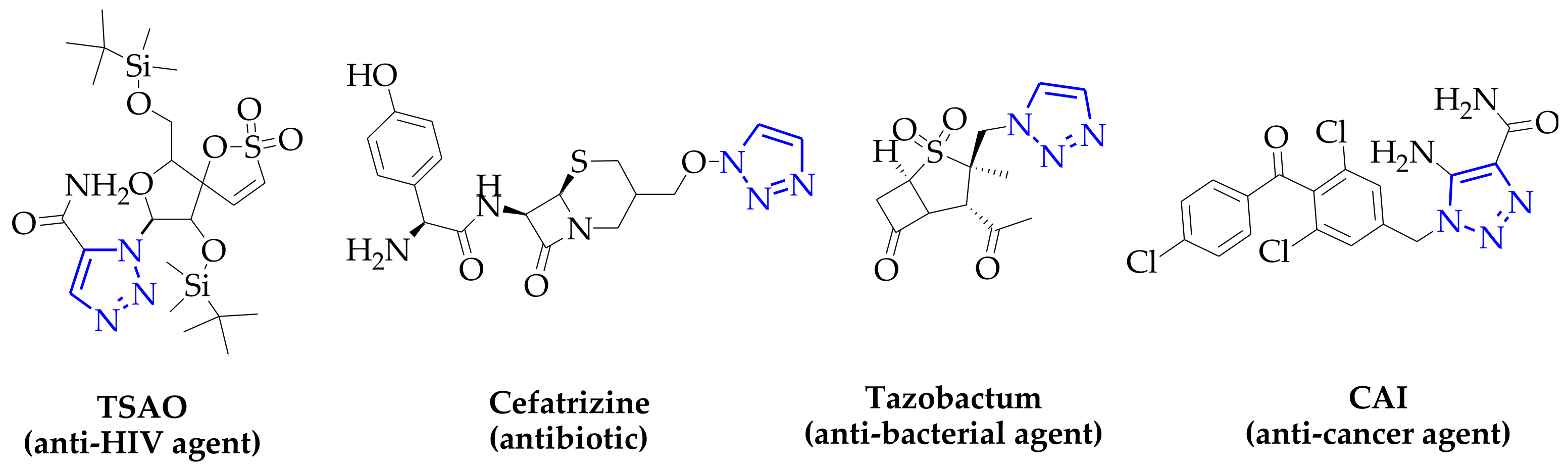
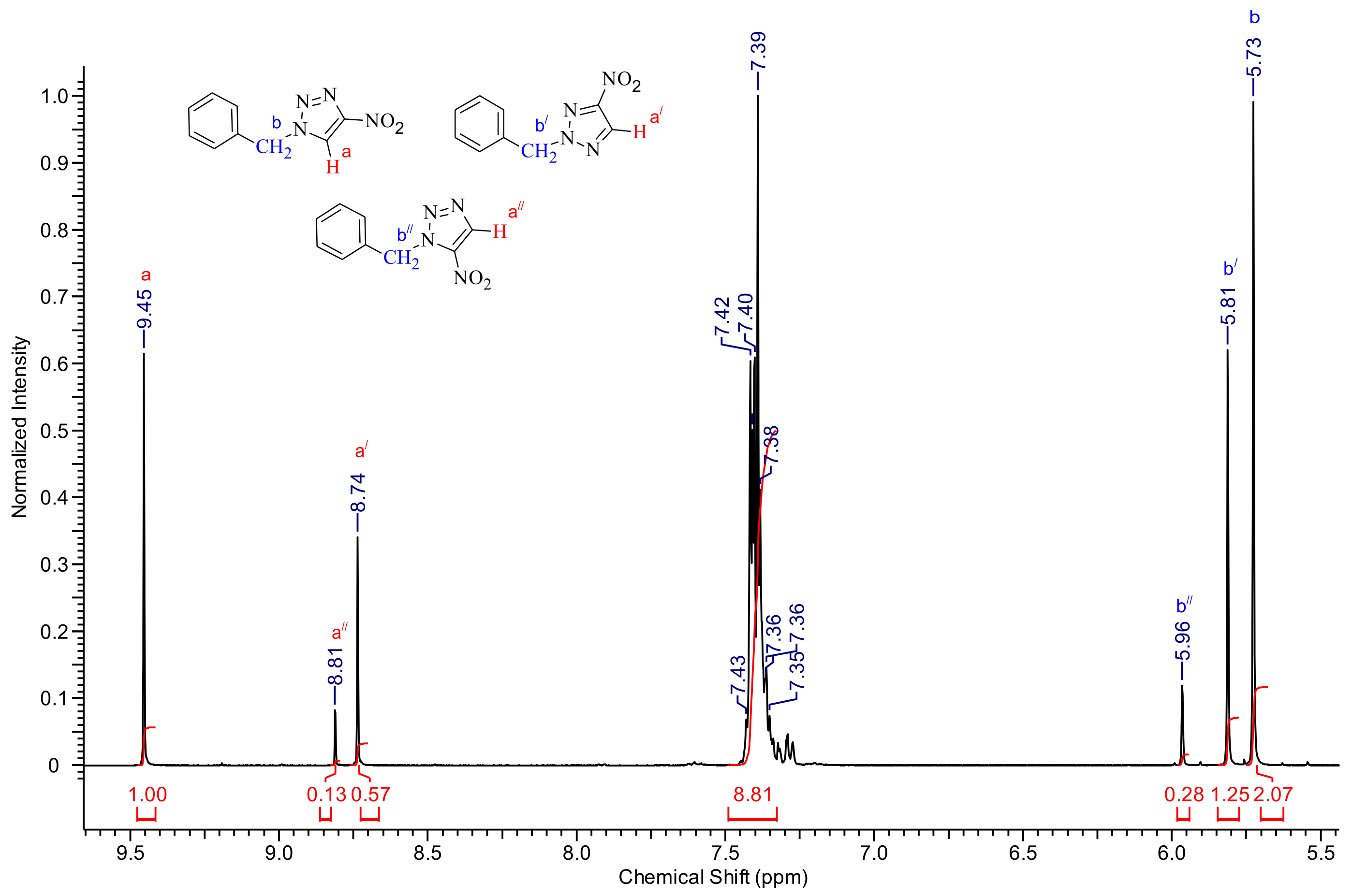
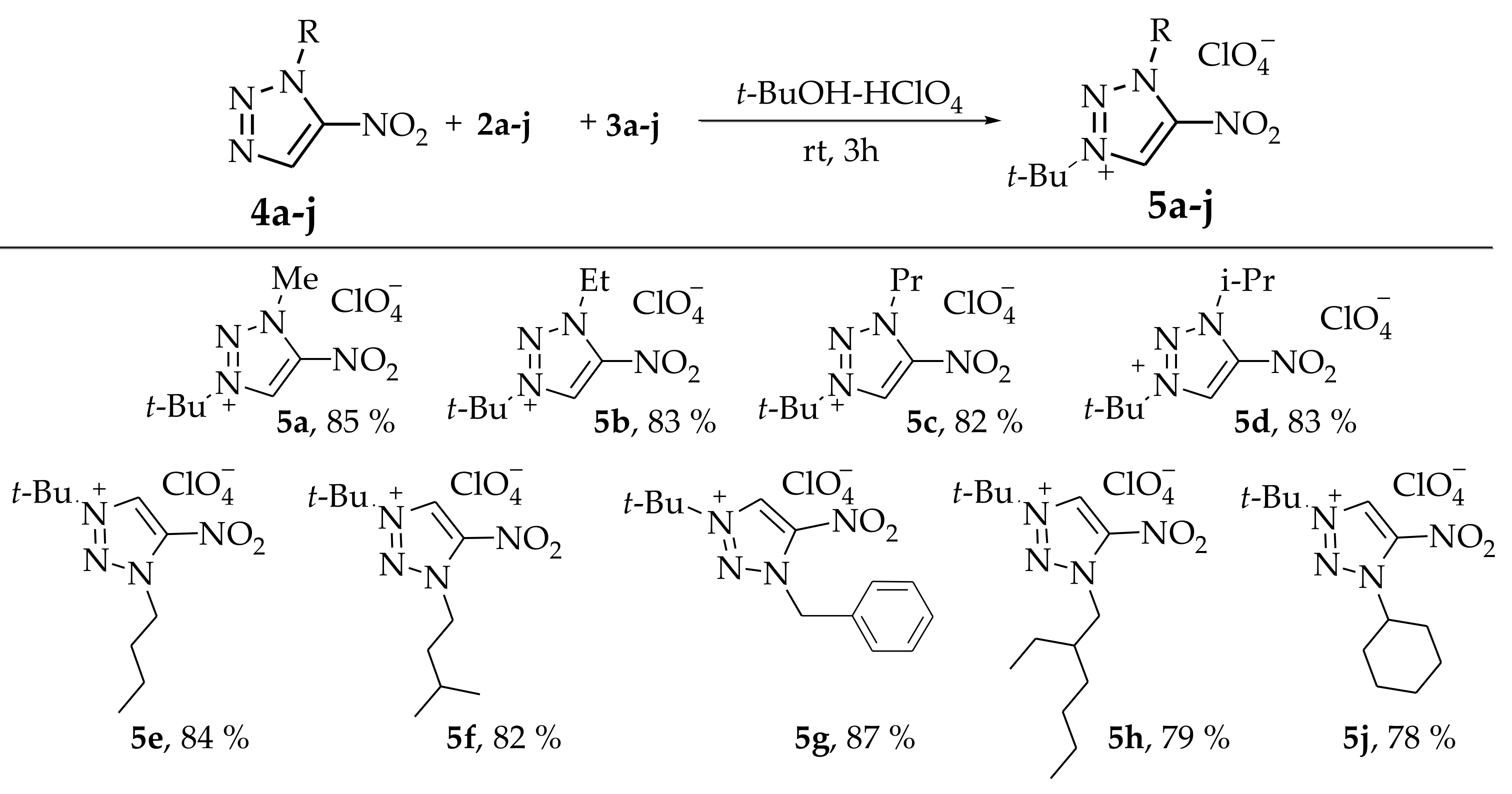
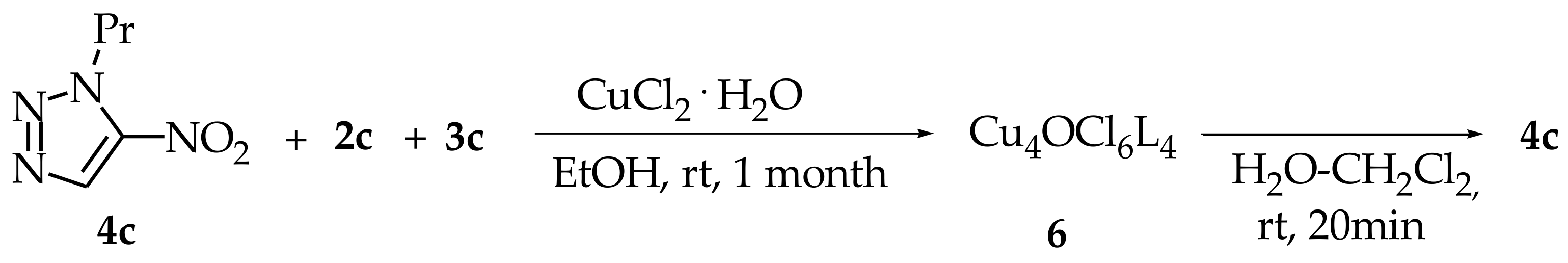


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
| Entry | R | X | 2−4 | Solvent | Temp, °C | Time, h | Yield, % | 2:3:4 Ratio c |
|---|---|---|---|---|---|---|---|---|
| 1 | Me | MeOSO3 | a | EtOH | 78 | 10 b | 83 | 36:53:11 |
| 2 | Me | MeOSO3 | a | H2O | 84 | 5 b | 81 | 45:43:12 |
| 3 | Et | EtOSO3 | b | EtOH | 78 | 10 b | 81 | 33:56:11 |
| 4 | Et | EtOSO3 | b | H2O | 84 | 5 b | 85 | 40:48:12 |
| 5 | Et | EtOSO3 | b | H2O | 90 | 10 | 82 | 42:47:11 |
| 6 | Et | EtOSO3 | b | H2O | 90 | 15 | 80 | 41:48:11 |
| 7 | Me | I | a | EtOH | 28 | 2 | 79 | 40:56:4 |
| 8 a | Et | Br | b | EtOH | 70 | 10 | 85 | 37:57:6 |
| 9 | Et | Br | b | H2O | 50 | 25 | 85 | 43:50:7 |
| 10 | n-Pr | Br | c | EtOH | 70 | 10 | 80 | 35:56:9 |
| 11 | i-Pr | Br | d | EtOH | 68 | 10 | 82 | 35:56:9 |
| 12 | n-Bu | Br | e | EtOH | 80 | 5 | 82 | 41:53:6 |
| 13 | i-amyl | Br | f | EtOH | 80 | 5 | 82 | 41:53:6 |
| 14 | Bn | Cl | g | EtOH | 80 | 5 | 88 | 64:30:6 |
| 15 | 2-ethylhexyl | ONO2 | h | EtOH | 78–100 | 10 | 78 | 53:45:2 |
| 16 | cyclohexyl | ONO2 | j | EtOH-H2O | 78–95 | 25 | 8 | 37:45:18 |
| N1-Isomer 2a | N2-Isomer 3a | N3-Isomer 4a | |
|---|---|---|---|
| ρ, g/cm3 | 1.525 | 1.537 | 1.566 |
| Δf, kJ/mol (calcd and exp) [34] | 468 calcd | 436 calcd 427 exp | 490 calcd. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukhanov, G.T.; Filippova, Y.V.; Gatilov, Y.V.; Sukhanova, A.G.; Krupnova, I.A.; Bosov, K.K.; Pivovarova, E.V.; Krasnov, V.I. Energetic Materials Based on N-substituted 4(5)-nitro-1,2,3-triazoles. Materials 2022, 15, 1119. https://doi.org/10.3390/ma15031119
Sukhanov GT, Filippova YV, Gatilov YV, Sukhanova AG, Krupnova IA, Bosov KK, Pivovarova EV, Krasnov VI. Energetic Materials Based on N-substituted 4(5)-nitro-1,2,3-triazoles. Materials. 2022; 15(3):1119. https://doi.org/10.3390/ma15031119
Chicago/Turabian StyleSukhanov, Gennady T., Yulia V. Filippova, Yuri V. Gatilov, Anna G. Sukhanova, Irina A. Krupnova, Konstantin K. Bosov, Ekaterina V. Pivovarova, and Vyacheslav I. Krasnov. 2022. "Energetic Materials Based on N-substituted 4(5)-nitro-1,2,3-triazoles" Materials 15, no. 3: 1119. https://doi.org/10.3390/ma15031119