
Machine Learning-Accelerated First-Principles Study of Atomic Configuration and Ionic Diffusion in Li10GeP2S12 Solid Electrolyte


 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Enumerated Configurations and Electrostatic Energies
2.2. Machine Learning- and Active-Learning-Based LAsou Methods
2.3. First-Principles Calculations
2.4. AIMD Simulations
3. Results and Discussion
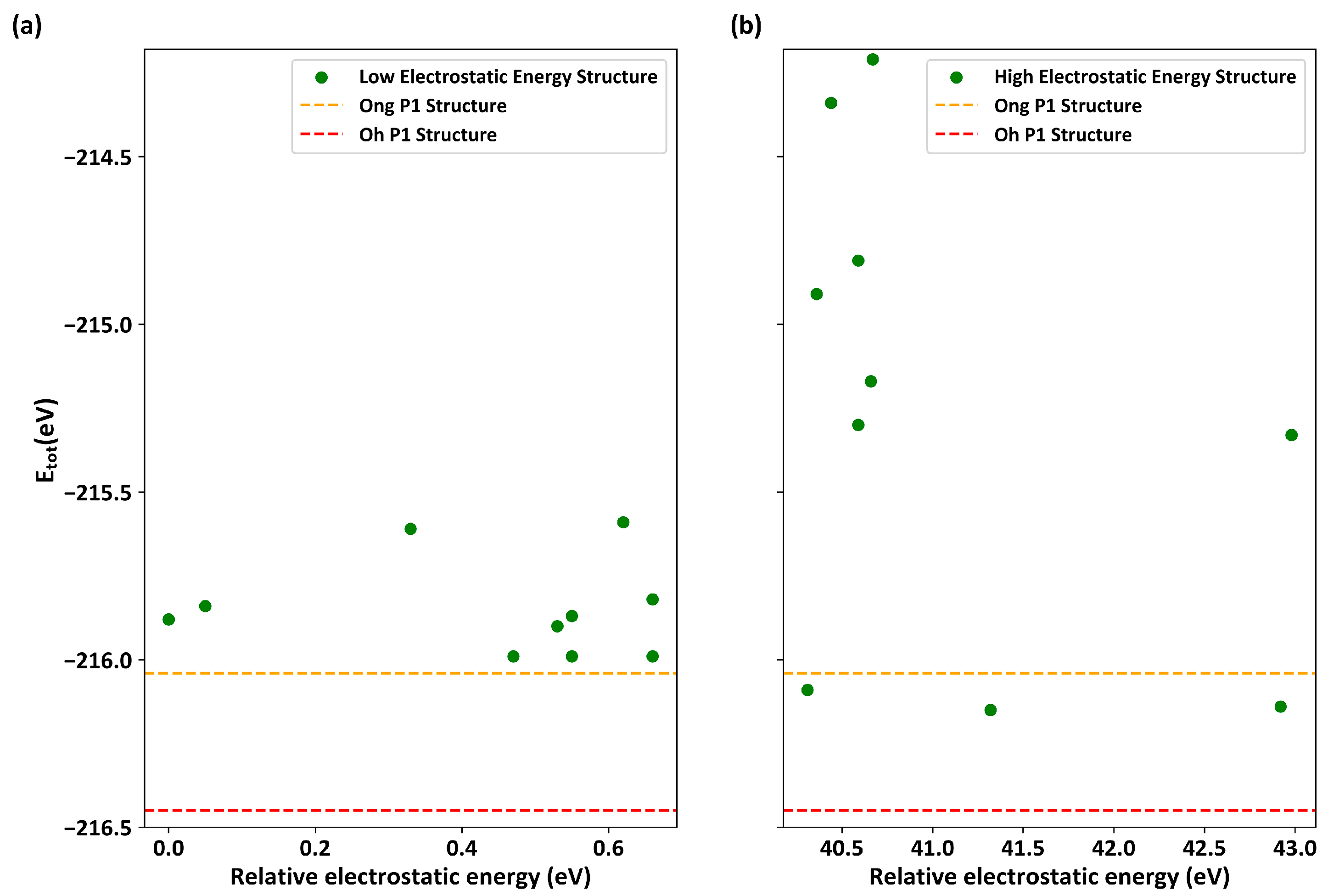
3.1. Evaluation of the Electrostatic Energy Criterion Method
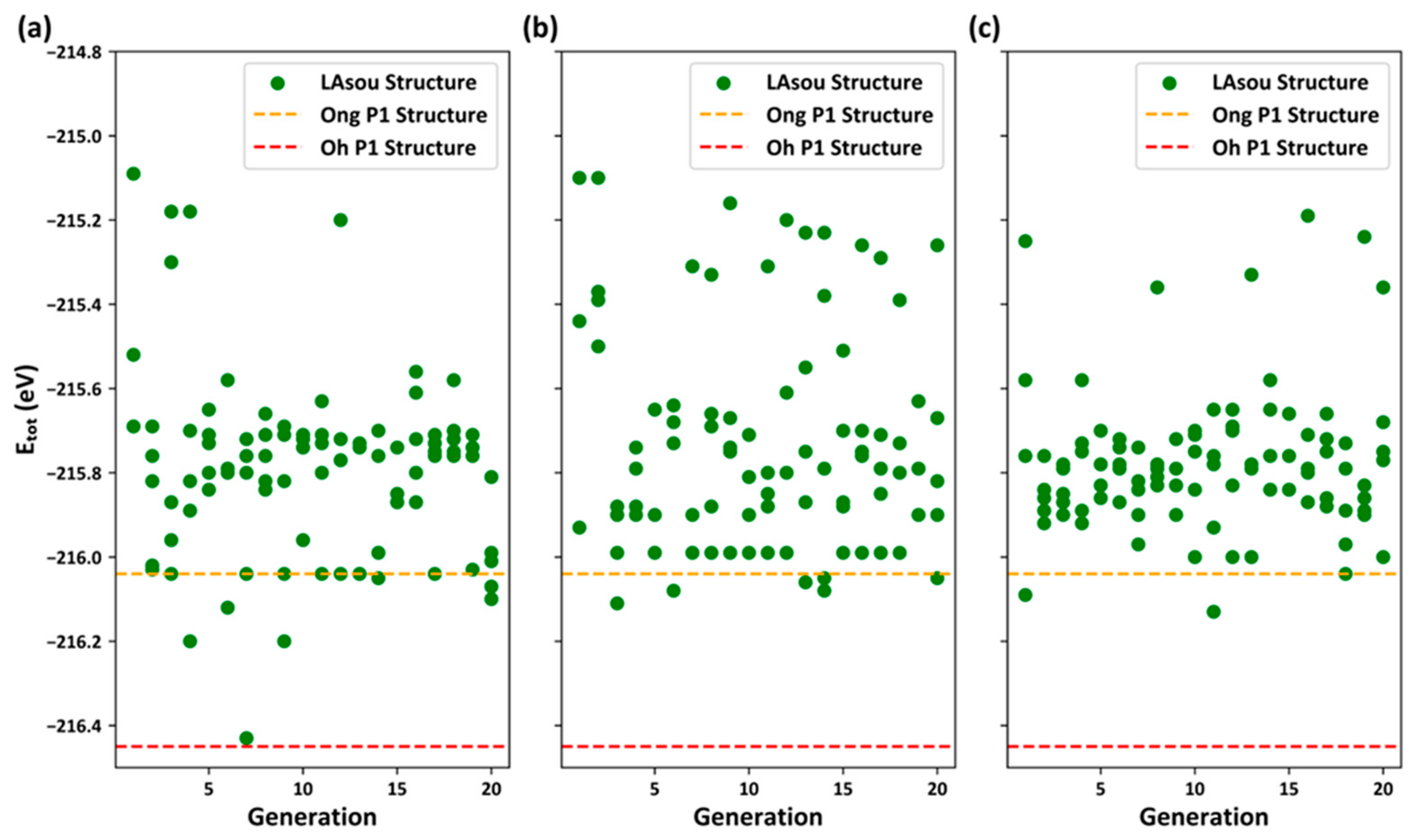
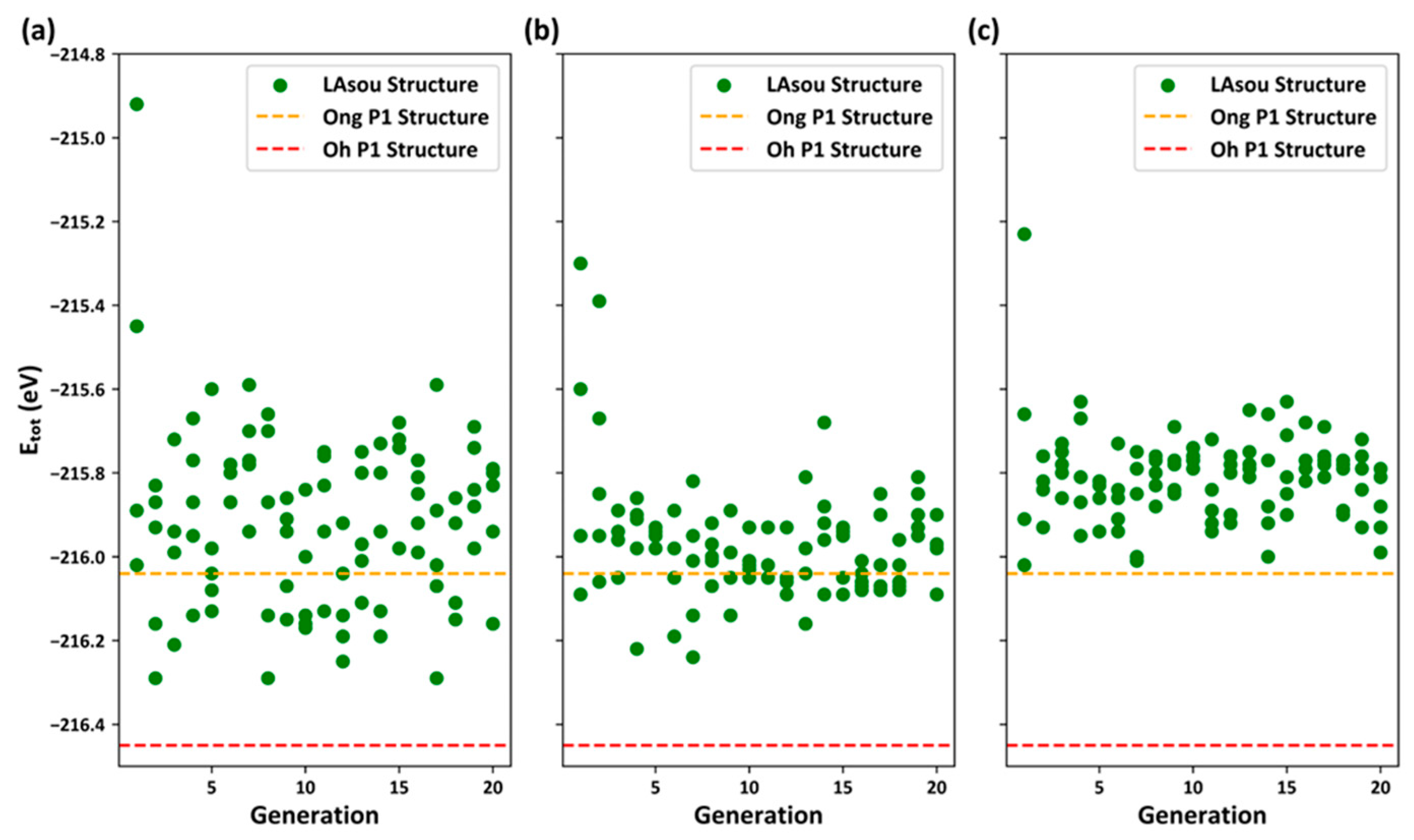
3.2. Stable Atomic Configurations Predicted using the LAsou Method
3.3. Influence of the Li Ion Distribution on Diffusion and Conductivity
4. Conclusions
- (1)
- Using the experimental crystal structures provided by Kamaya et al. and Kuhn et al., we utilized the Supercell package to generate 91,728 and 2,167,200 enumerated configurations, respectively. These configurations were categorized into Zig, Pa, and Pc skeletons based on the disordered arrangements of GeS4 and P1S4 tetrahedra. The presence of co-occupancy, defects, doping, and other fractional occupancies in crystal structures can result in an exceptionally large number of possible configurations in sampling (combinatorial) space, posing a big challenge for both experimental and theoretical research.
- (2)
- The electrostatic energy criterion, although very useful in screening a small subspace within a huge sampling space, has notable limitations. For example, configurations with low electrostatic energy often adhere to the lower Coulombic repulsion rule and tend to have a uniform distribution. Additionally, the small size of the subspace may result in the failure to obtain superior results in electrostatic regions with high energy.
- (3)
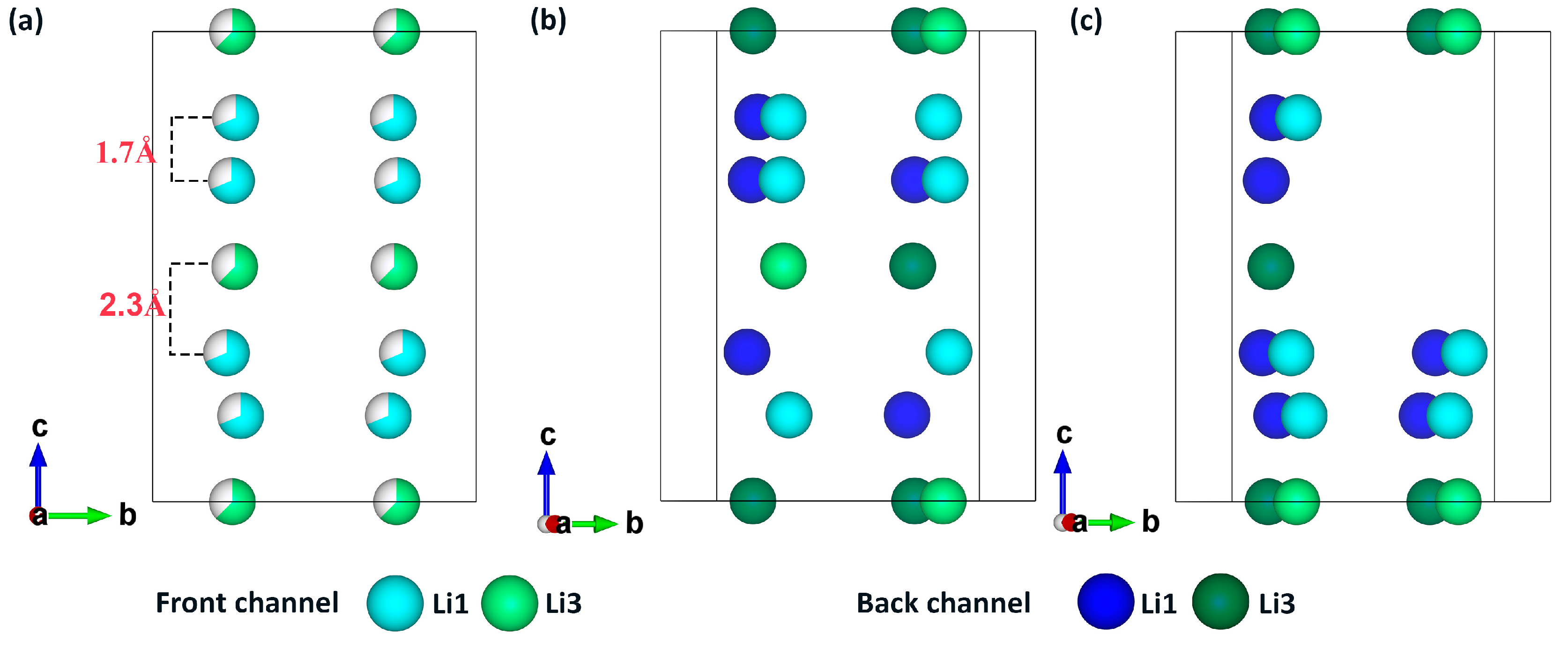
- In contrast to the electrostatic energy criterion, the LAsou method utilizes data-driven machine learning and active-learning algorithms. These algorithms facilitate iterative sampling and labeling in a large space, ultimately identifying stable configuration with minimal computational cost. For the Zig skeletons, only 35 configurations calculated using DFT were necessary to achieve similar theoretical results to those of Oh et al., who performed exhaustive DFT calculations on nearly 1000 configurations. Furthermore, the LAsou method successfully reproduced the new Li4 sites based on the low-resolution crystal structure reported by Kamaya et al. This suggests that the LAsou method can also contribute to the refinement of experimental crystal structures.
- (4)
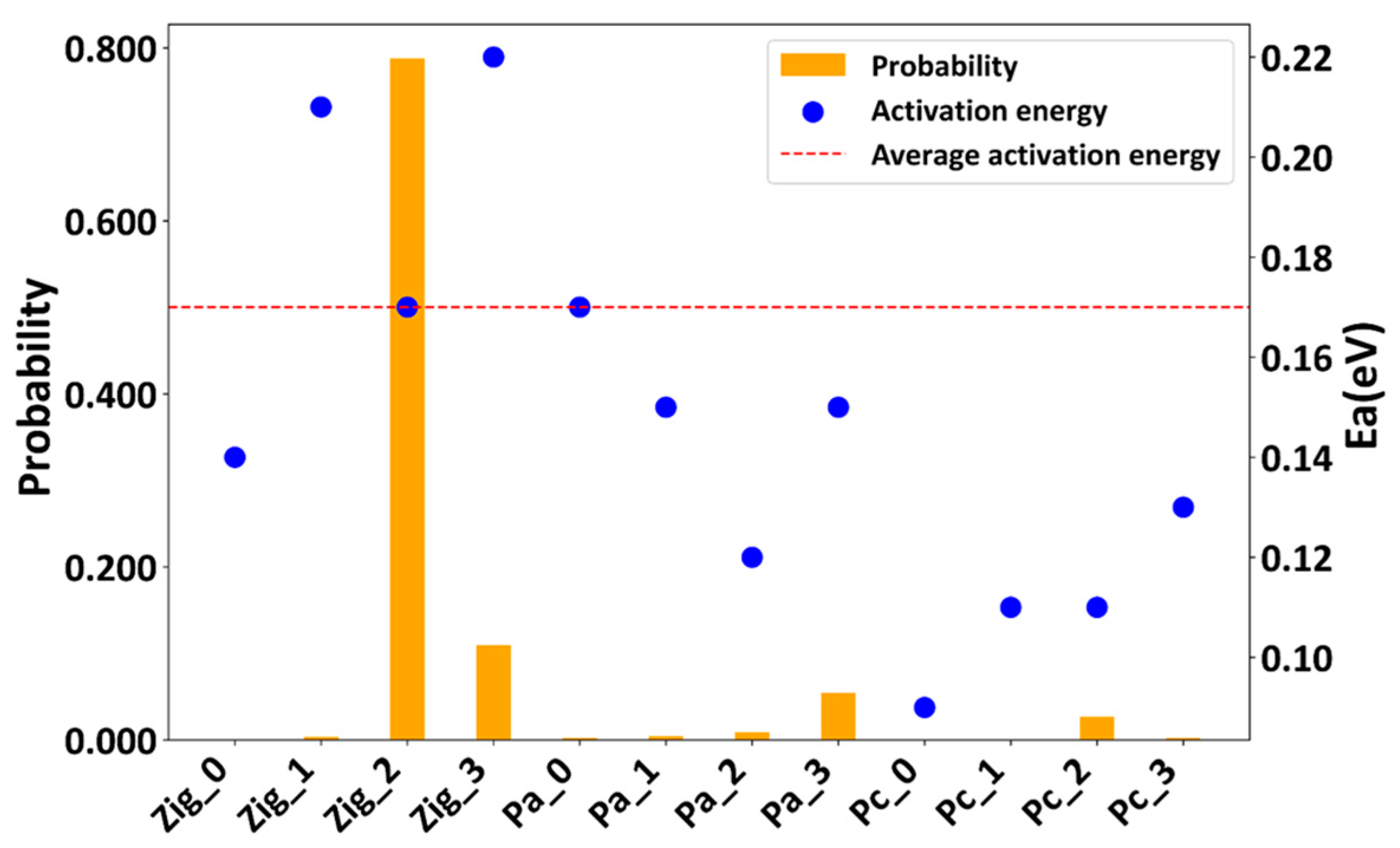
- Based on the predicted stable configurations of Zig_n, Pa_n, and Pc_n (n = 0, 1, 2, 3), we performed AIMD simulations to investigate Li ion diffusion at temperatures ranging from 500 to 900 K. The results highlighted the significant impact of the skeleton and Li ion distribution on diffusion activation energy, ionic conductivity, and electronic structure properties. Although various stable configurations may co-exist, the overall weighted average is closer to the experimental results, where the most stable configuration makes the greatest contributions.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Scrosati, B.; Garche, J. Lithium batteries: Status, prospects and future. J. Power Sources 2010, 195, 2419–2430. [Google Scholar] [CrossRef]
- Dunn, B.; Kamath, H.; Tarascon, J.-M. Electrical Energy Storage for the Grid: A Battery of Choices. Science 2011, 334, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Manthiram, A.; Yu, X.; Wang, S. Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2017, 2, 16103. [Google Scholar] [CrossRef]
- Fu, Z.-H.; Chen, X.; Yao, N.; Shen, X.; Ma, X.-X.; Feng, S.; Wang, S.; Zhang, R.; Zhang, L.; Zhang, Q. The chemical origin of temperature-dependent lithium-ion concerted diffusion in sulfide solid electrolyte Li10GeP2S12. J. Energy Chem. 2022, 70, 59–66. [Google Scholar] [CrossRef]
- Kamaya, N.; Homma, K.; Yamakawa, Y.; Hirayama, M.; Kanno, R.; Yonemura, M.; Kamiyama, T.; Kato, Y.; Hama, S.; Kawamoto, K.; et al. A lithium superionic conductor. Nat. Mater. 2011, 10, 682–686. [Google Scholar] [CrossRef]
- Adams, S.; Rao, R.P. Structural requirements for fast lithium ion migration in Li10GeP2S12. J. Mater. Chem. 2012, 22, 7687–7691. [Google Scholar] [CrossRef]
- Kuhn, A.; Kohler, J.; Lotsch, B.V. Single-crystal X-ray structure analysis of the superionic conductor Li10GeP2S12. Phys. Chem. Chem. Phys. 2013, 15, 11620–11622. [Google Scholar] [CrossRef]
- Kwon, O.; Hirayama, M.; Suzuki, K.; Kato, Y.; Saito, T.; Yonemura, M.; Kamiyama, T.; Kanno, R. Synthesis, structure, and conduction mechanism of the lithium superionic conductor Li10+δGe1+δP2−δS12. J. Mater. Chem. A 2015, 3, 438–446. [Google Scholar] [CrossRef]
- Weber, D.A.; Senyshyn, A.; Weldert, K.S.; Wenzel, S.; Zhang, W.; Kaiser, R.; Berendts, S.; Janek, J.; Zeier, W.G. Structural Insights and 3D Diffusion Pathways within the Lithium Superionic Conductor Li10GeP2S12. Chem. Mater. 2016, 28, 5905–5915. [Google Scholar] [CrossRef]
- Iwasaki, R.; Hori, S.; Kanno, R.; Yajima, T.; Hirai, D.; Kato, Y.; Hiroi, Z. Weak Anisotropic Lithium-Ion Conductivity in Single Crystals of Li10GeP2S12. Chem. Mater. 2019, 31, 3694–3699. [Google Scholar] [CrossRef]
- Yajima, T.; Hinuma, Y.; Hori, S.; Iwasaki, R.; Kanno, R.; Ohhara, T.; Nakao, A.; Munakata, K.; Hiroi, Z. Correlated Li-ion migration in the superionic conductor Li10GeP2S12. J. Mater. Chem. A 2021, 9, 11278–11284. [Google Scholar] [CrossRef]
- Kato, Y.; Hori, S.; Kanno, R. Li10GeP2S12-Type Superionic Conductors: Synthesis, Structure, and Ionic Transportation. Adv. Energy Mater. 2020, 10, 2002153. [Google Scholar] [CrossRef]
- Mo, Y.; Ong, S.P.; Ceder, G. First Principles Study of the Li10GeP2S12 Lithium Super Ionic Conductor Material. Chem. Mater. 2011, 24, 15–17. [Google Scholar] [CrossRef]
- Xu, M.; Ding, J.; Ma, E. One-dimensional stringlike cooperative migration of lithium ions in an ultrafast ionic conductor. Appl. Phys. Lett. 2012, 101, 031901. [Google Scholar] [CrossRef]
- Ong, S.P.; Mo, Y.; Richards, W.D.; Miara, L.; Lee, H.S.; Ceder, G. Phase stability, electrochemical stability and ionic conductivity of the Li10±1MP2X12(M = Ge, Si, Sn, Al or P, and X = O, S or Se) family of superionic conductors. Energy Environ. Sci. 2013, 6, 148–156. [Google Scholar] [CrossRef]
- Du, F.; Ren, X.; Yang, J.; Liu, J.; Zhang, W. Structures, Thermodynamics, and Li+Mobility of Li10GeP2S12: A First-Principles Analysis. J. Phys. Chem. C 2014, 118, 10590–10595. [Google Scholar] [CrossRef]
- Bhandari, A.; Bhattacharya, J. Origin of Fast Ion Conduction in Li10GeP2S12, a Superionic Conductor. J. Phys. Chem. C 2016, 120, 29002–29010. [Google Scholar] [CrossRef]
- Oh, K.; Chang, D.; Lee, B.; Kim, D.-H.; Yoon, G.; Park, I.; Kim, B.; Kang, K. Native Defects in Li10GeP2S12 and Their Effect on Lithium Diffusion. Chem. Mater. 2018, 30, 4995–5004. [Google Scholar] [CrossRef]
- Dobhal, G.; Walsh, T.R.; Tawfik, S.A. Blocking Directional Lithium Diffusion in Solid-State Electrolytes at the Interface: First-Principles Insights into the Impact of the Space Charge Layer. ACS Appl. Mater. Interfaces 2022, 14, 55471–55479. [Google Scholar] [CrossRef]
- Yin, Y.C.; Yang, J.T.; Luo, J.D.; Lu, G.X.; Huang, Z.; Wang, J.P.; Li, P.; Li, F.; Wu, Y.C.; Tian, T.; et al. A LaCl3-based lithium superionic conductor compatible with lithium metal. Nature 2023, 616, 77–83. [Google Scholar] [CrossRef]
- Gorai, P.; Long, H.; Jones, E.; Santhanagopalan, S.; Stevanović, V. Defect chemistry of disordered solid-state electrolyte Li10GeP2S12. J. Mater. Chem. A 2020, 8, 3851–3858. [Google Scholar] [CrossRef]
- Morita, K.; Hoshino, T. Ab initio molecular dynamics study of isotope effects in lithium-ion conductors. Solid State Ionics 2020, 355, 115434. [Google Scholar] [CrossRef]
- Jang, S.H.; Jalem, R.; Tateyama, Y. EwaldSolidSolution: A High-Throughput Application to Quickly Sample Stable Site Arrangements for Ionic Solid Solutions. J. Phys. Chem. A 2023, 127, 5734–5744. [Google Scholar] [CrossRef] [PubMed]
- Hautier, G.; Fischer, C.C.; Jain, A.; Mueller, T.; Ceder, G. Finding Nature’s Missing Ternary Oxide Compounds Using Machine Learning and Density Functional Theory. Chem. Mater. 2010, 22, 3762–3767. [Google Scholar]
- Yuan, X.; Zhou, Y.; Peng, Q.; Yang, Y.; Li, Y.; Wen, X. Active learning to overcome exponential-wall problem for effective structure prediction of chemical-disordered materials. NPJ Comput. Mater. 2023, 9, 12. [Google Scholar] [CrossRef]
- Okhotnikov, K.; Charpentier, T.; Cadars, S. Supercell program: A combinatorial structure-generation approach for the local-level modeling of atomic substitutions and partial occupancies in crystals. J. Cheminf. 2016, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Ewald, P.P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys. 1921, 369, 253–287. [Google Scholar] [CrossRef]
- Grau-Crespo, R.; Hamad, S.; Catlow, C.R.A.; de Leeuw, N.H. Symmetry-adapted configurational modelling of fractional site occupancy in solids. J. Phys. Condens. Matter 2007, 19, 256201. [Google Scholar] [CrossRef]
- Hart, G.L.W.; Forcade, R.W. Algorithm for generating derivative structures. Phys. Rev. B 2008, 77, 224115. [Google Scholar] [CrossRef]
- Hart, G.L.W.; Forcade, R.W. Generating derivative structures from multilattices: Algorithm and application to hcp alloys. Phys. Rev. B 2009, 80, 014120. [Google Scholar] [CrossRef]
- Hart, G.L.W.; Nelson, L.J.; Forcade, R.W. Generating derivative structures at a fixed concentration. Comput. Mater. Sci. 2012, 59, 101–107. [Google Scholar] [CrossRef]
- Lian, J.-C.; Wu, H.-Y.; Huang, W.-Q.; Hu, W.; Huang, G.-F. Algorithm for generating irreducible site-occupancy configurations. Phys. Rev. B 2020, 102, 134209. [Google Scholar] [CrossRef]
- Ong, S.P.; Richards, W.D.; Jain, A.; Hautier, G.; Kocher, M.; Cholia, S.; Gunter, D.; Chevrier, V.L.; Persson, K.A.; Ceder, G. Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis. Comput. Mater. Sci. 2013, 68, 314–319. [Google Scholar] [CrossRef]
- Peng, Q.; Zhao, S.; Yuan, X.; Chen, X.-J. Elasticity of Mg3Bi2-xSbx. Materials 2022, 15, 7161. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ma, H.; Jiao, Y.; Chen, Y.; Yuan, X.; Zhang, X.; Wang, F.; Xiao, D.; Wang, Y.; Hong, D.; et al. Water-Induced Structural Evolution of LaTMSi Ternary Intermetallic Electrides. Chem. Mater. 2023, 35, 1972–1979. [Google Scholar] [CrossRef]
- Oftelie, L.B.; Rajak, P.; Kalia, R.K.; Nakano, A.; Sha, F.; Sun, J.; Singh, D.J.; Aykol, M.; Huck, P.; Persson, K.; et al. Active learning for accelerated design of layered materials. NPJ Comput. Mater. 2018, 4, 74. [Google Scholar] [CrossRef]
- Lookman, T.; Balachandran, P.V.; Xue, D.; Yuan, R. Active learning in materials science with emphasis on adaptive sampling using uncertainties for targeted design. NPJ Comput. Mater. 2019, 5, 21. [Google Scholar] [CrossRef]
- Kusne, A.G.; Yu, H.; Wu, C.; Zhang, H.; Hattrick-Simpers, J.; DeCost, B.; Sarker, S.; Oses, C.; Toher, C.; Curtarolo, S.; et al. On-the-fly closed-loop materials discovery via Bayesian active learning. Nat. Commun. 2020, 11, 5966. [Google Scholar] [CrossRef]
- Min, K.; Cho, E. Accelerated discovery of potential ferroelectric perovskite via active learning. J. Mater. Chem. C 2020, 8, 7866–7872. [Google Scholar] [CrossRef]
- Jablonka, K.M.; Jothiappan, G.M.; Wang, S.; Smit, B.; Yoo, B. Bias free multiobjective active learning for materials design and discovery. Nat. Commun. 2021, 12, 2312. [Google Scholar] [CrossRef]
- Verduzco, J.C.; Marinero, E.E.; Strachan, A. An active learning approach for the design of doped LLZO ceramic garnets for battery applications. Integr. Mater. Manuf. Innov. 2021, 10, 299–310. [Google Scholar] [CrossRef]
- Szymanski, N.J.; Rendy, B.; Fei, Y.; Kumar, R.E.; He, T.; Milsted, D.; McDermott, M.J.; Gallant, M.; Cubuk, E.D.; Merchant, A.; et al. An autonomous laboratory for the accelerated synthesis of novel materials. Nature 2023, 624, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Kim, E.; Sun, J.; Kim, M.; Hong, E.; Min, K. Rapid discovery of promising materials via active learning with multi-objective optimization. Mater. Today Commun. 2023, 37, 107245. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Cussen, E.J. The structure of lithium garnets: Cation disorder and clustering in a new family of fast Li+ conductors. Chem. Commun. 2006, 412–413. [Google Scholar] [CrossRef] [PubMed]
- Urban, A.; Seo, D.-H.; Ceder, G. Computational understanding of Li-ion batteries. NPJ Comput. Mater. 2016, 2, 1–13. [Google Scholar] [CrossRef]
- He, X.; Zhu, Y.; Epstein, A.; Mo, Y. Statistical variances of diffusional properties from ab initio molecular dynamics simulations. NPJ Comput. Mater. 2018, 4, 18. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Park, K.S. The Li-ion rechargeable battery: A perspective. J. Am. Chem. Soc. 2013, 135, 1167–1176. [Google Scholar] [CrossRef]
- Materzanini, G.; Kahle, L.; Marcolongo, A.; Marzari, N. High Li-ion conductivity in tetragonal LGPO: A comparative first-principles study against known LISICON and LGPS phases. Phys. Rev. Mater. 2021, 5, 035408. [Google Scholar] [CrossRef]
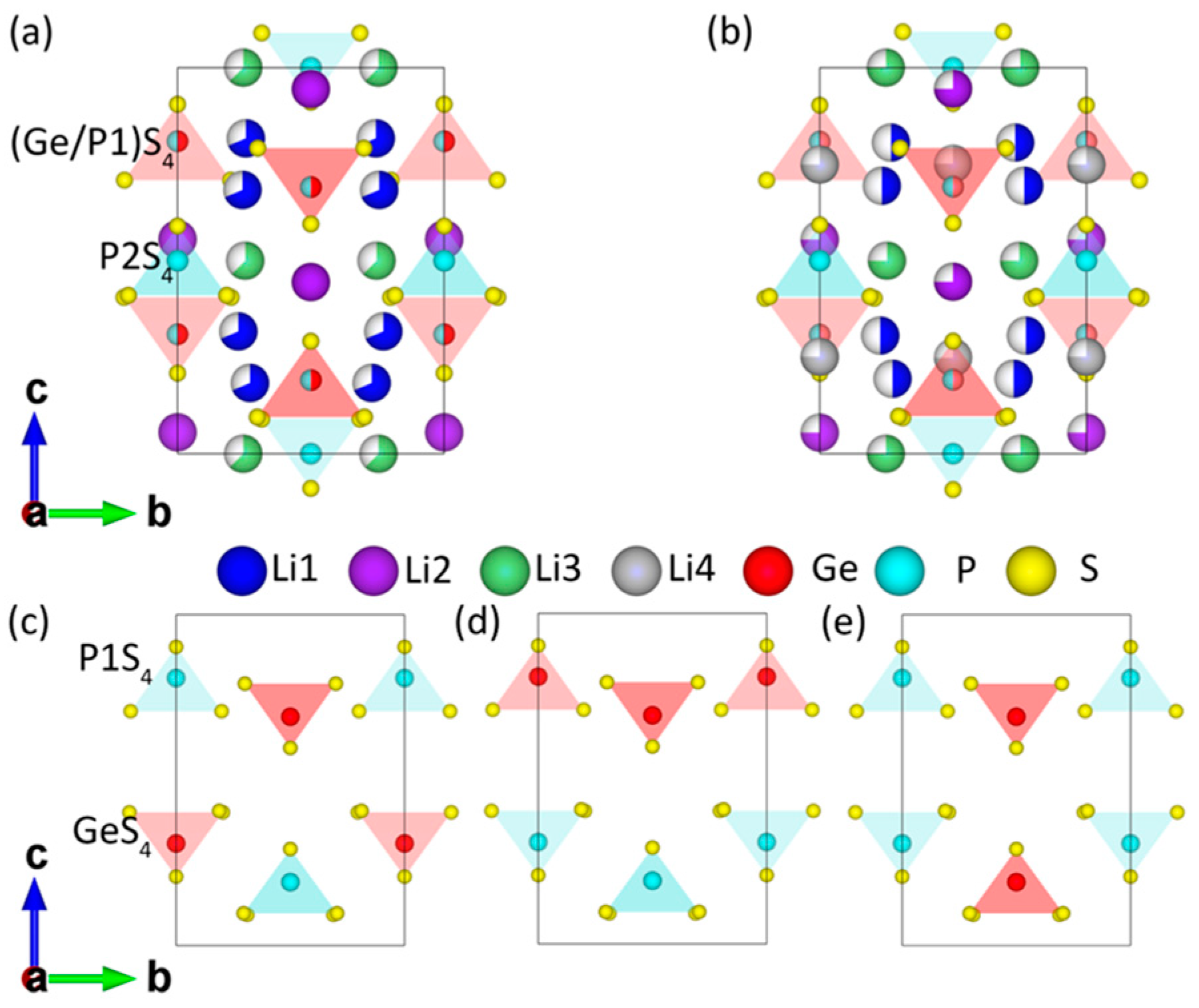

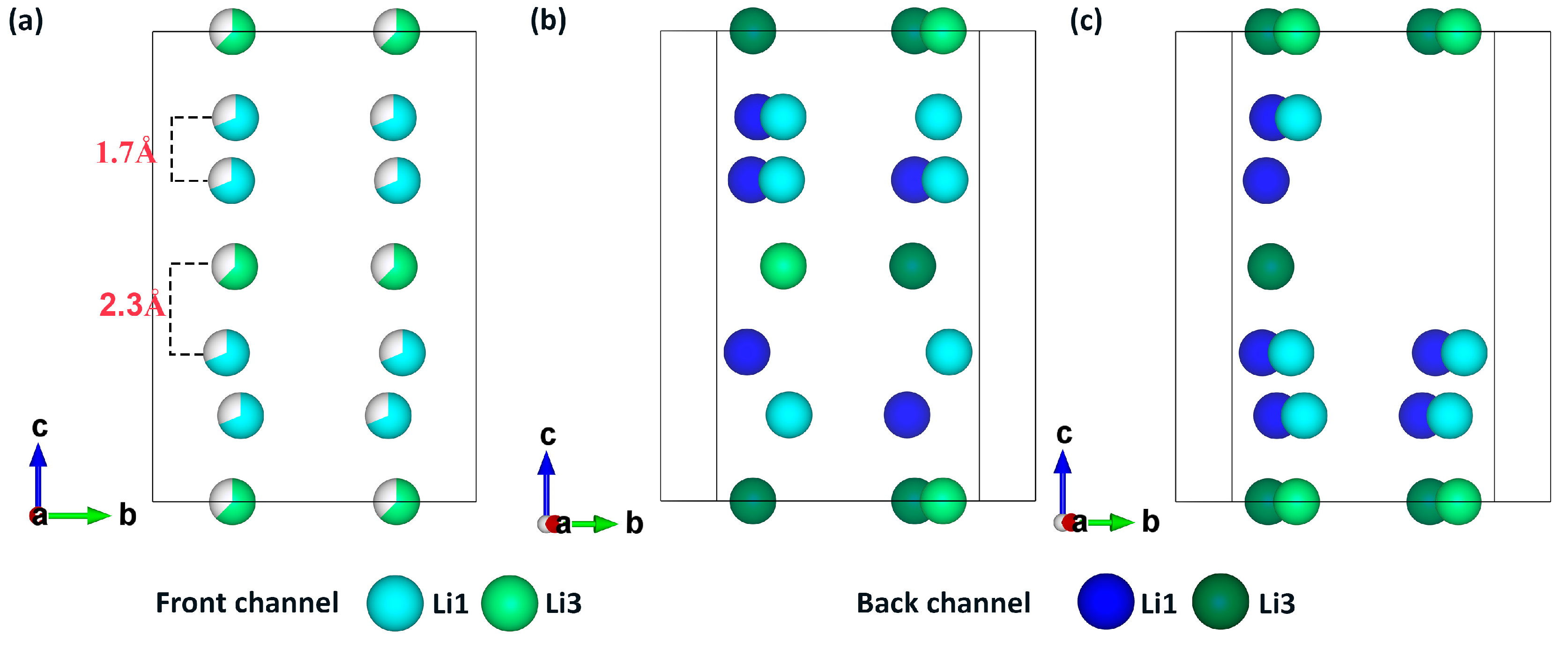

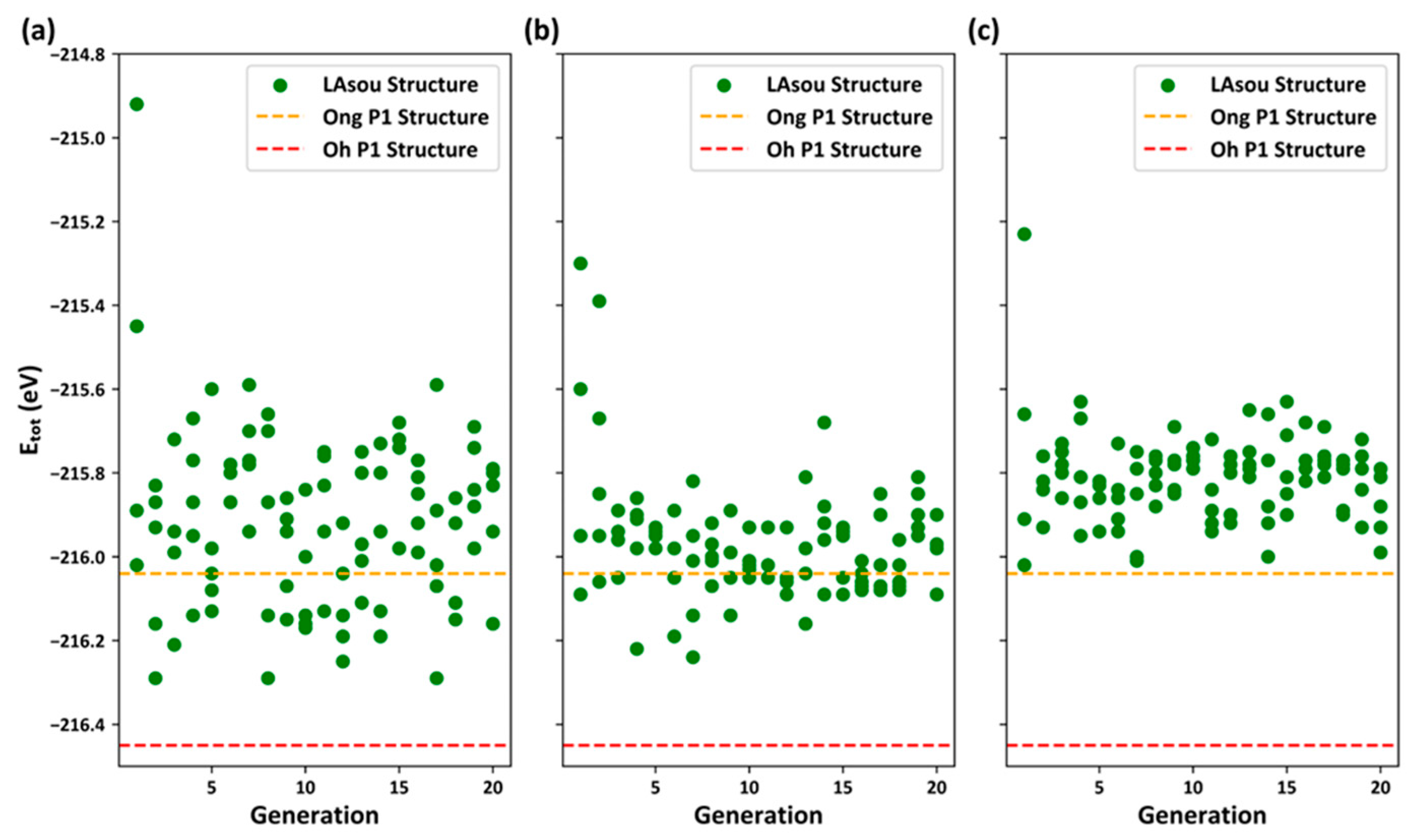


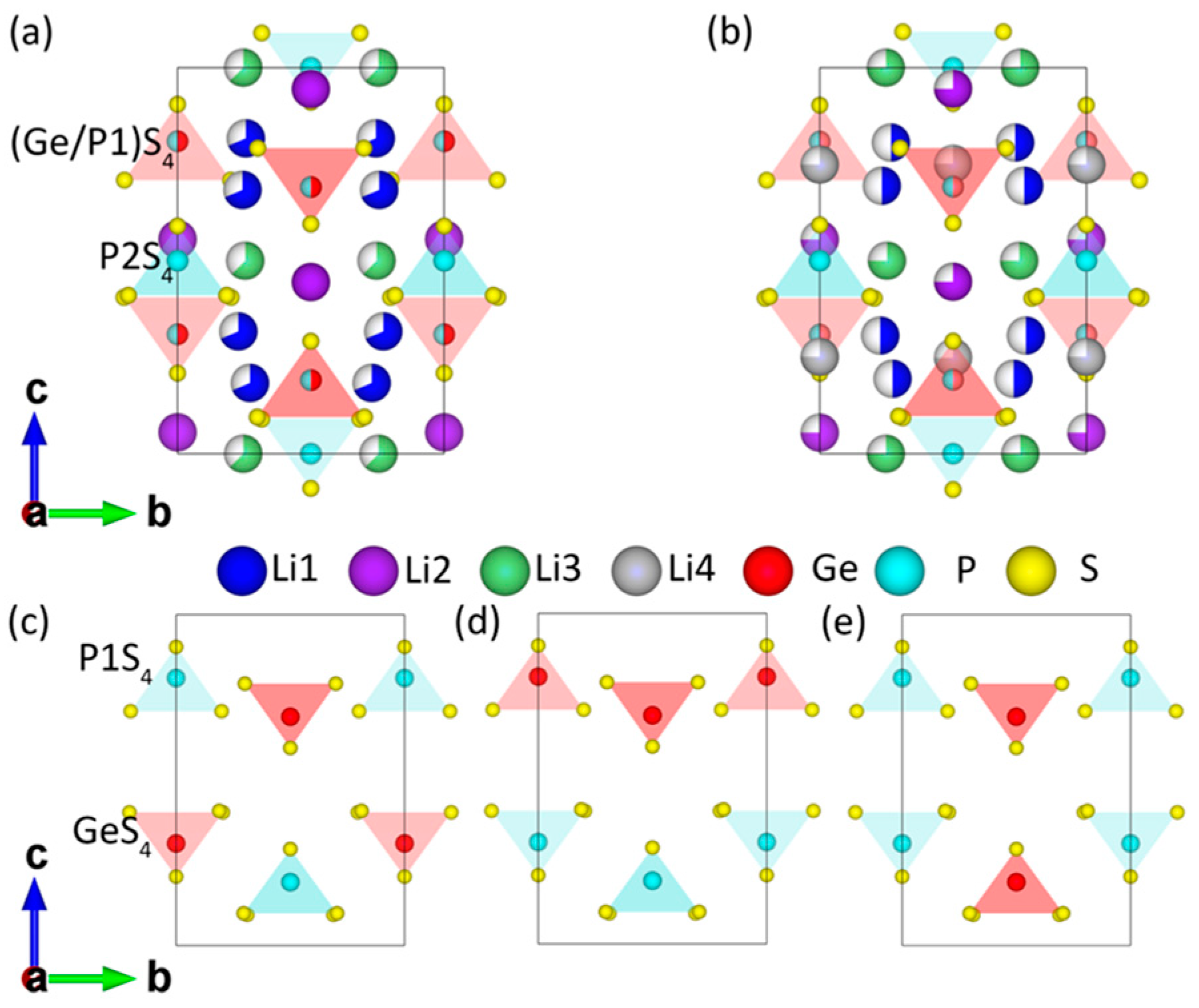{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Configuration | ΔEtot | σ | |||
|---|---|---|---|---|---|
| Zig_0 | 0.62 | 0.14 ± 0.023 | 0.12 ± 0.037 | 0.22 ± 0.022 | 95 |
| Zig_1 | 0.38 | 0.21 ± 0.028 | 0.19 ± 0.034 | 0.27 ± 0.015 | 23 |
| Zig_2 | 0 | 0.17 ± 0.008 | 0.15 ± 0.008 | 0.25 ± 0.026 | 43 |
| Zig_3 | 0.14 | 0.22 ± 0.019 | 0.20 ± 0.020 | 0.27 ± 0.005 | 15 |
| Pa_0 | 0.42 | 0.17 ± 0.009 | 0.15 ± 0.014 | 0.24 ± 0.011 | 45 |
| Pa_1 | 0.37 | 0.15 ± 0.027 | 0.13 ± 0.035 | 0.22 ± 0.015 | 80 |
| Pa_2 | 0.32 | 0.12 ± 0.019 | 0.09 ± 0.025 | 0.18 ± 0.035 | 172 |
| Pa_3 | 0.19 | 0.15 ± 0.023 | 0.12 ± 0.031 | 0.22 ± 0.022 | 84 |
| Pc_0 | 0.54 | 0.09 ± 0.016 | 0.07 ± 0.023 | 0.15 ± 0.013 | 332 |
| Pc_1 | 0.39 | 0.11 ± 0.031 | 0.09 ± 0.036 | 0.17 ± 0.023 | 243 |
| Pc_2 | 0.24 | 0.11 ± 0.014 | 0.08 ± 0.016 | 0.16 ± 0.014 | 207 |
| Pc_3 | 0.42 | 0.13 ± 0.017 | 0.11 ± 0.019 | 0.16 ± 0.02 | 148 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, C.; Zhou, Y.; Yuan, X.; Peng, Q.; Yang, Y.; Li, Y.; Wen, X. Machine Learning-Accelerated First-Principles Study of Atomic Configuration and Ionic Diffusion in Li10GeP2S12 Solid Electrolyte. Materials 2024, 17, 1810. https://doi.org/10.3390/ma17081810
Qi C, Zhou Y, Yuan X, Peng Q, Yang Y, Li Y, Wen X. Machine Learning-Accelerated First-Principles Study of Atomic Configuration and Ionic Diffusion in Li10GeP2S12 Solid Electrolyte. Materials. 2024; 17(8):1810. https://doi.org/10.3390/ma17081810
Chicago/Turabian StyleQi, Changlin, Yuwei Zhou, Xiaoze Yuan, Qing Peng, Yong Yang, Yongwang Li, and Xiaodong Wen. 2024. "Machine Learning-Accelerated First-Principles Study of Atomic Configuration and Ionic Diffusion in Li10GeP2S12 Solid Electrolyte" Materials 17, no. 8: 1810. https://doi.org/10.3390/ma17081810