
Assembly and Comparative Analyses of the Chloroplast Genomes of the Threatened Plant Rosa anemoniflora

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection and Preservation of Experimental Material
2.2. Chloroplast Genome Assembly and Annotation
2.3. Chloroplast Genome Comparative Analysis
2.4. Molecular Marker Discovery
2.5. Phylogenetic Analysis
2.6. Molecular Dating Analysis
2.7. Biogeographical Analysis
3. Results
3.1. General Features and Comparative Analyses of Rosa anemoniflora Chloroplast Genomes
3.2. Comparative Analysis of Gene Number and Selection Pressure
3.3. Comparative Analysis of Chloroplast Genome Structure
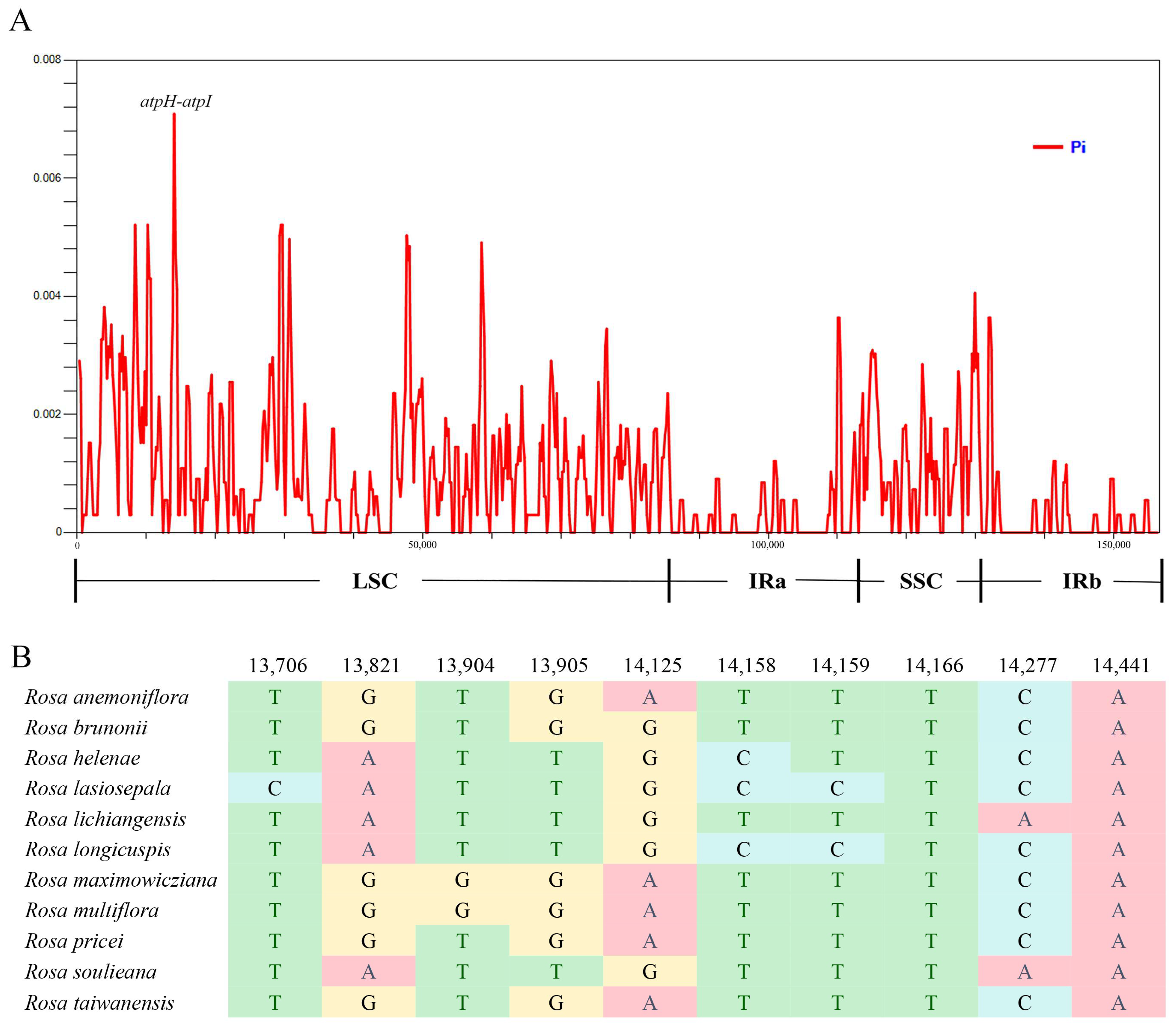
3.4. Highly Divergent Regions and Molecular Marker Development among Rosa Section Synstylae Plants
3.5. Phylogenetic Position, Species Divergence Time, and Ancestral Distribution Reconstruction of Rosa anemoniflora
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chinese Academy of Sciences, Editorial Committee of Flora of China. Flora of China; Science Press: Beijing, China, 1985; Volume 37. [Google Scholar]
- Ku, T.; Robertson, K. Rosa linnaeus . In Flora of China; Wu, C., Raven, P., Eds.; Science Press: Beijing, China, 2003; Volume 9, pp. 339–381. [Google Scholar]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef]
- Irisarri, I.; Strassert, J.F.H.; Burki, F. Phylogenomic insights into the origin of primary plastids. Syst. Biol. 2022, 71, 105–120. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, A.; Li, X.; Lu, C. The role of chloroplast gene expression in plant responses to environmental stress. Int. J. Mol. Sci. 2020, 21, 6082. [Google Scholar] [CrossRef]
- Mulo, P.; Sakurai, I.; Aro, E.M. Strategies for psbA gene expression in cyanobacteria, green algae and higher plants: From transcription to PSII repair. Biochim. Biophys. Acta 2012, 1817, 247–257. [Google Scholar] [CrossRef]
- Gao, Y.; Thiele, W.; Saleh, O.; Scossa, F.; Arabi, F.; Zhang, H.; Sampathkumar, A.; Kühn, K.; Fernie, A.; Bock, R.; et al. Chloroplast translational regulation uncovers nonessential photosynthesis genes as key players in plant cold acclimation. Plant Cell 2022, 34, 2056–2079. [Google Scholar] [CrossRef]
- Hu, S.; Ding, Y.; Zhu, C. Sensitivity and responses of chloroplasts to heat stress in plants. Front. Plant Sci. 2020, 11, 375. [Google Scholar] [CrossRef]
- Zhou, T.; Zhu, H.; Wang, J.; Xu, Y.; Xu, F.; Wang, X. Complete chloroplast genome sequence determination of Rheum species and comparative chloroplast genomics for the members of Rumiceae. Plant Cell Rep. 2020, 39, 811–824. [Google Scholar] [CrossRef]
- Yang, L.; Deng, S.; Zhu, Y.; Da, Q. Comparative chloroplast genomics of 34 species in subtribe Swertiinae (Gentianaceae) with implications for its phylogeny. BMC Plant Biol. 2023, 23, 164. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Liao, M.; Cheng, Y.-H.; Feng, Y.; Ju, W.-B.; Deng, H.-N.; Li, X.; Plenković-Moraj, A.; Xu, B. Comparative chloroplast genomics of seven edangered Cypripedium species and phylogenetic relationships of orchidaceae. Front. Plant Sci. 2022, 13, 911702. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Mount, D.W. Using the basic local alignment search tool (blast). Cold Spring Harb. 2007, 2007, pdb.top17. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile hmm searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. Aragorn, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Yang, Z.H. Paml: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. Dnasp 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. Misa-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. Modelfinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. Raxml-ng: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. Phylosuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using beast 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Zhu, Z.m.; Gao, X.f.; Fougère-Danezan, M. Phylogeny of Rosa sections Chinenses and Synstylae (Rosaceae) based on chloroplast and nuclear markers. Mol. Phylogen. Evol. 2015, 87, 50–64. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Yu, Y.; Blair, C.; He, X. Rasp 4: Ancestral state reconstruction tool for multiple genes and characters. Mol. Biol. Evol. 2019, 37, 604–606. [Google Scholar] [CrossRef]
- Shen, W.; Dong, Z.; Zhao, W.; Ma, L.; Wang, F.; Li, W.; Xin, P. Complete chloroplast genome sequence of Rosa lucieae and its characteristics. Horticulturae 2022, 8, 788. [Google Scholar] [CrossRef]
- Gao, C.; Li, T.; Zhao, X.; Wu, C.; Zhang, Q.; Zhao, X.; Wu, M.; Lian, Y.; Li, Z. Comparative analysis of the chloroplast genomes of Rosa species and rna editing analysis. BMC Plant Biol. 2023, 23, 318. [Google Scholar] [CrossRef]
- Ding, M.; Liao, M.; Liu, P.; Tan, G.; Chen, Y.; Shi, S. The complete chloroplast genome of Rosa cymosa (Rosaceae), a traditional medicinal plant in south China. Mitochondrial DNA Part B 2020, 5, 2571–2572. [Google Scholar] [CrossRef]
- Zhu, A.; Guo, W.; Gupta, S.; Fan, W.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef]
- Lu, Q.; Ye, W.; Lu, R.; Xu, W.; Qiu, Y. Phylogenomic and comparative analyses of complete plastomes of Croomia and Stemona (Stemonaceae). Int. J. Mol. Sci. 2018, 19, 2383. [Google Scholar] [CrossRef]
- Bell, G.; Collins, S. Adaptation, extinction and global change. Evol. Appl. 2008, 1, 3–16. [Google Scholar] [CrossRef]
- Huda, M.K.; Wilcock, C.C. Impact of floral traits on the reproductive success of epiphytic and terrestrial tropical orchids. Oecologia 2008, 154, 731–741. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Landis, J.B.; Shen, J.; Zhang, H.; Kuang, T.; Sun, W.; Sun, J.; Tiamiyu, B.B.; Deng, T.; et al. Transcriptomes of Saussurea (Asteraceae) provide insights into high-altitude adaptation. Plants 2021, 10, 1715. [Google Scholar] [CrossRef]
- Lahti, D.C.; Johnson, N.A.; Ajie, B.C.; Otto, S.P.; Hendry, A.P.; Blumstein, D.T.; Coss, R.G.; Donohue, K.; Foster, S.A. Relaxed selection in the wild. Trends Ecol. Evol. 2009, 24, 487–496. [Google Scholar] [CrossRef]
- Wissemann, V. Conventional taxonomy of wild roses. In Encyclopedia of Rose; Roberts, A., Debener, T., Gudin, S., Eds.; Science Press: London, UK, 2003; pp. 111–117. [Google Scholar]
- Bruneau, A.; Starr, J.R.; Joly, S. Phylogenetic relationships in the genus Rosa: New evidence from chloroplast DNA sequences and an appraisal of current knowledge. Syst. Bot. 2007, 32, 366–378. [Google Scholar] [CrossRef]
- Joly, S.; Starr, J.R.; Lewis, W.H.; Bruneau, A. Polyploid and hybrid evolution in roses east of the Rocky mountains. Am. J. Bot. 2006, 93, 412–425. [Google Scholar] [CrossRef]
- Matthews, J. Hybridism and classification in the genus Rosa. New Phytol. 1920, 19, 153–171. [Google Scholar] [CrossRef]
- Borsch, T.; Quandt, D. Mutational dynamics and phylogenetic utility of noncoding chloroplast DNA. Plant Syst. Evol. 2009, 282, 169–199. [Google Scholar] [CrossRef]
- Jian, H.; Zhang, Y.; Yan, H.; Qiu, X.; Wang, Q.; Li, S.; Zhang, S. The complete chloroplast genome of a key ancestor of modern roses, Rosa chinensis var. spontanea, and a comparison with congeneric species. Molecules 2018, 23, 389. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Liao, B.; Guo, S.; Liang, C.; Pei, J.; Xu, J.; Chen, S. The chloroplasts genomic analyses of Rosa laevigata, R. rugosa and R. canina. Chin. Med. 2020, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, X.; Wu, T.; Lu, H.; Cai, Y. Genetic differences between wild and artificial populations of Metasequoia glyptostroboides: Implications for species recovery. Conserv. Biol. 2005, 19, 224–231. [Google Scholar] [CrossRef]
- Edmands, S. Between a rock and a hard place: Evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol. Ecol. 2007, 16, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.Y.; Che, W.J. A revision of Rosa transmorrisonensis hayata and allied species in Taiwan. Taiwania 2022, 67, 484–496. [Google Scholar] [CrossRef]
- Chang, K. The neolithic Taiwan strait. Kaogu 1989, 6, 541–569. [Google Scholar]
- Revell, L.J. Comparing the rates of speciation and extinction between phylogenetic trees. Ecol. Evol. 2018, 8, 5303–5312. [Google Scholar] [CrossRef]
- Mace, G.M.; Gittleman, J.L.; Purvis, A. Preserving the tree of life. Science 2003, 300, 1707–1709. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | Rosa anemoniflora | Rosa brunonii | Rosa maximowicziana | Rosa multiflora | Rosa lucidissima | Rosa chinensis | Rosa odorata |
|---|---|---|---|---|---|---|---|
| Genome size (bp) | 156,578 | 156,544 | 156,405 | 156,519 | 156,588 | 156,591 | 156,580 |
| LSC size (bp) | 85,722 | 85,705 | 85,529 | 85,643 | 85,713 | 85,737 | 85,704 |
| SSC size (bp) | 18,740 | 18,743 | 18,760 | 18,760 | 18,779 | 18,766 | 18,780 |
| IR size (bp) | 26,058 | 26,048 | 26,058 | 26,058 | 26,048 | 26,044 | 26,048 |
| Total GC content | 37.23 | 37.24 | 37.25 | 37.24 | 37.25 | 37.24 | 37.25 |
| GC content in LSC | 35.18 | 35.2 | 31.32 | 35.2 | 35.2 | 35.2 | 35.21 |
| GC content in SSC | 31.34 | 31.3 | 35.21 | 31.32 | 31.35 | 31.32 | 31.35 |
| GC content in IR | 42.73 | 42.73 | 42.73 | 42.73 | 42.73 | 42.71 | 42.73 |
| Number of genes | 130 | 130 | 134 | 134 | 129 | 130 | 130 |
| Protein-coding genes | 84 | 85 | 88 | 88 | 83 | 85 | 85 |
| tRNA genes | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| rRNA genes | 37 | 37 | 37 | 37 | 37 | 37 | 37 |
| Pseudo genes | 1 | 0 | 1 | 1 | 1 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, W.; Zhou, X.; Yu, Q.; Lin, G.; Fu, C.; Kang, T.; Zeng, H. Assembly and Comparative Analyses of the Chloroplast Genomes of the Threatened Plant Rosa anemoniflora. Forests 2024, 15, 940. https://doi.org/10.3390/f15060940
Gao W, Zhou X, Yu Q, Lin G, Fu C, Kang T, Zeng H. Assembly and Comparative Analyses of the Chloroplast Genomes of the Threatened Plant Rosa anemoniflora. Forests. 2024; 15(6):940. https://doi.org/10.3390/f15060940
Chicago/Turabian StyleGao, Wei, Xianzhen Zhou, Qun Yu, Guojiang Lin, Chengjie Fu, Tianqi Kang, and Huahao Zeng. 2024. "Assembly and Comparative Analyses of the Chloroplast Genomes of the Threatened Plant Rosa anemoniflora" Forests 15, no. 6: 940. https://doi.org/10.3390/f15060940
APA StyleGao, W., Zhou, X., Yu, Q., Lin, G., Fu, C., Kang, T., & Zeng, H. (2024). Assembly and Comparative Analyses of the Chloroplast Genomes of the Threatened Plant Rosa anemoniflora. Forests, 15(6), 940. https://doi.org/10.3390/f15060940