
EBV-Associated Hub Genes as Potential Biomarkers for Predicting the Prognosis of Nasopharyngeal Carcinoma
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microarray Dataset Collection and Preprocessing
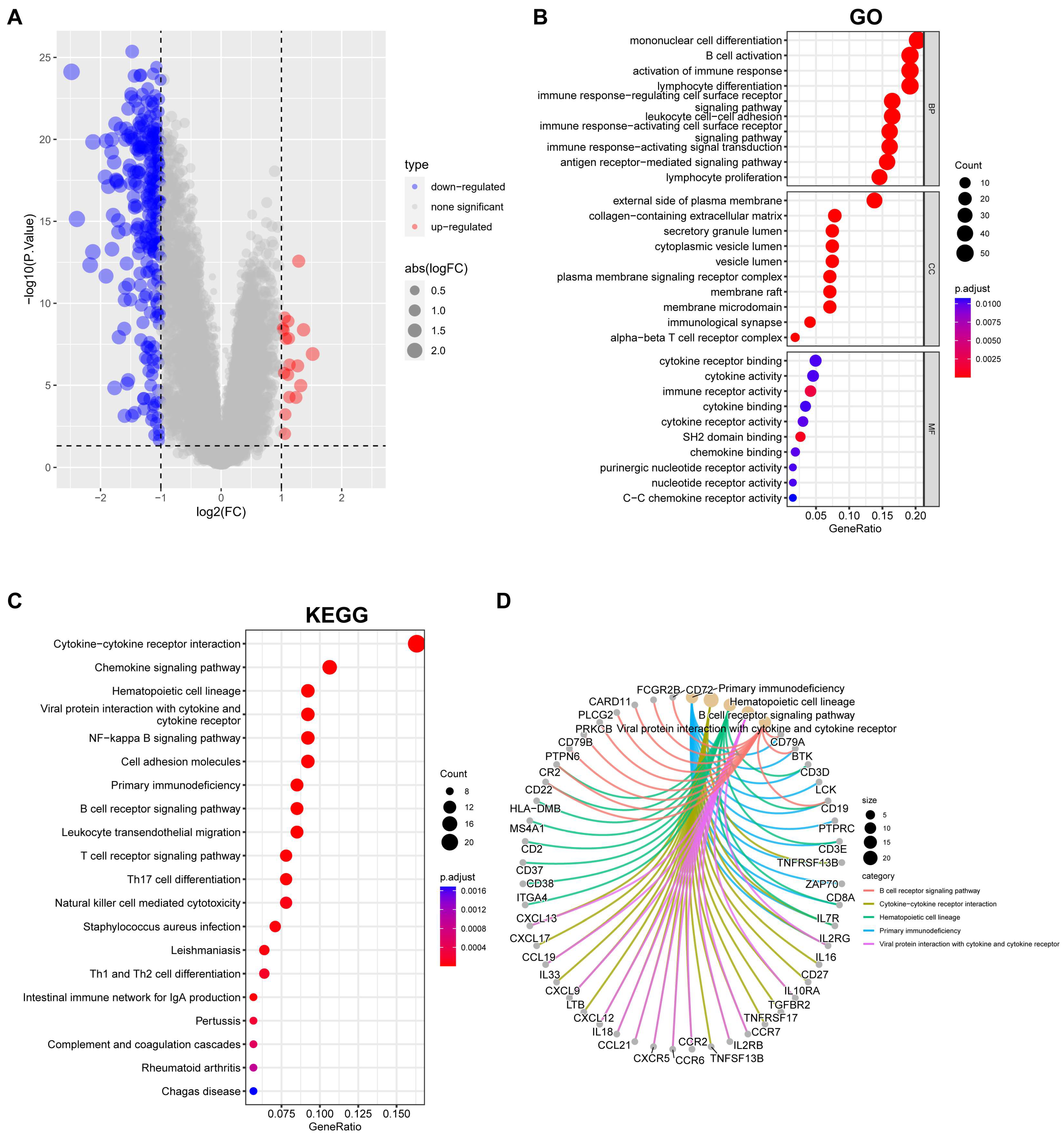
2.2. Functional Enrichment Analysis
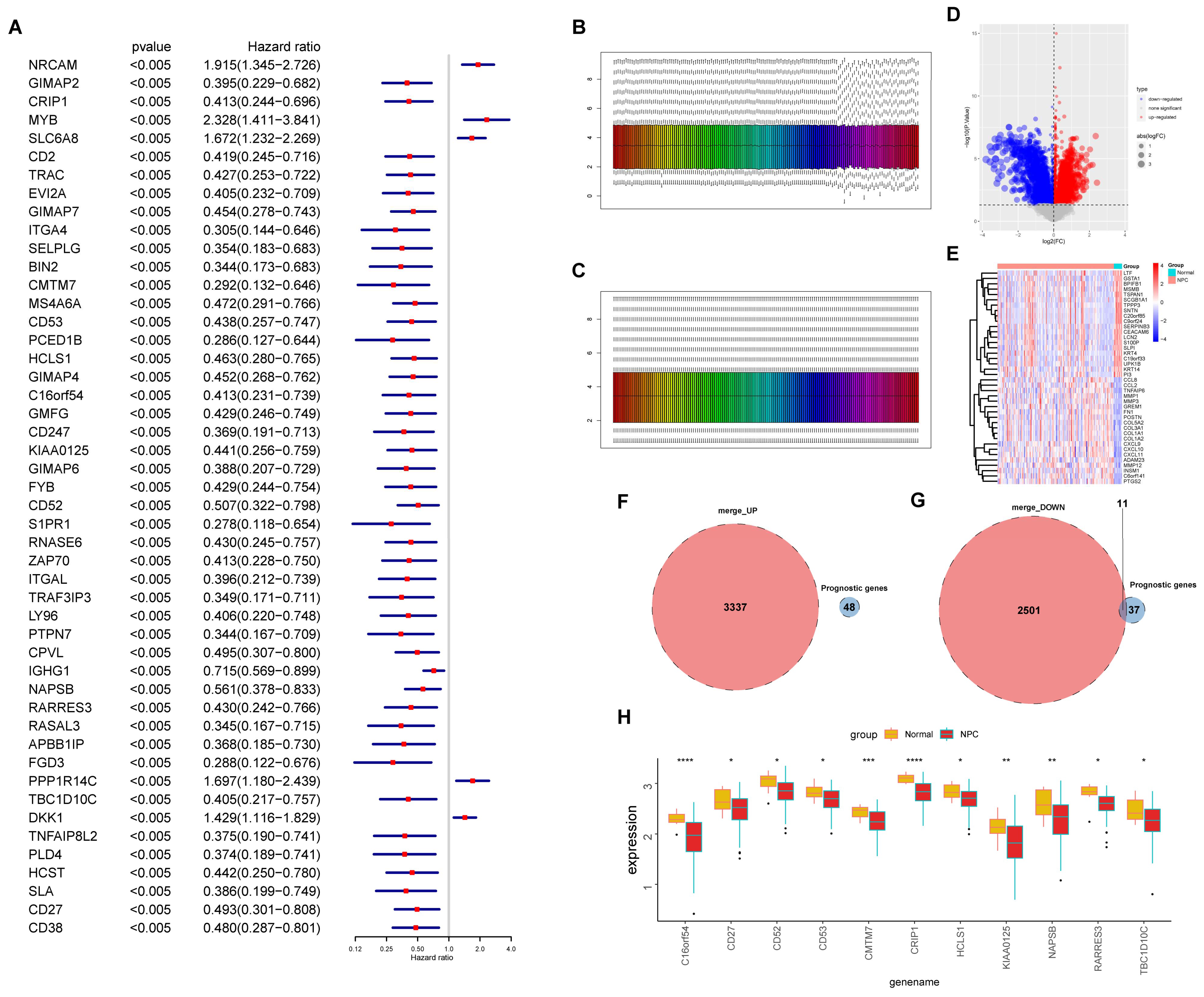
2.3. Univariate Cox Regression and Survival Analysis
2.4. Unsupervised Hierarchical Clustering
2.5. Gene Set Variation Analysis (GSVA) in EBV-Associated NPC Subtypes
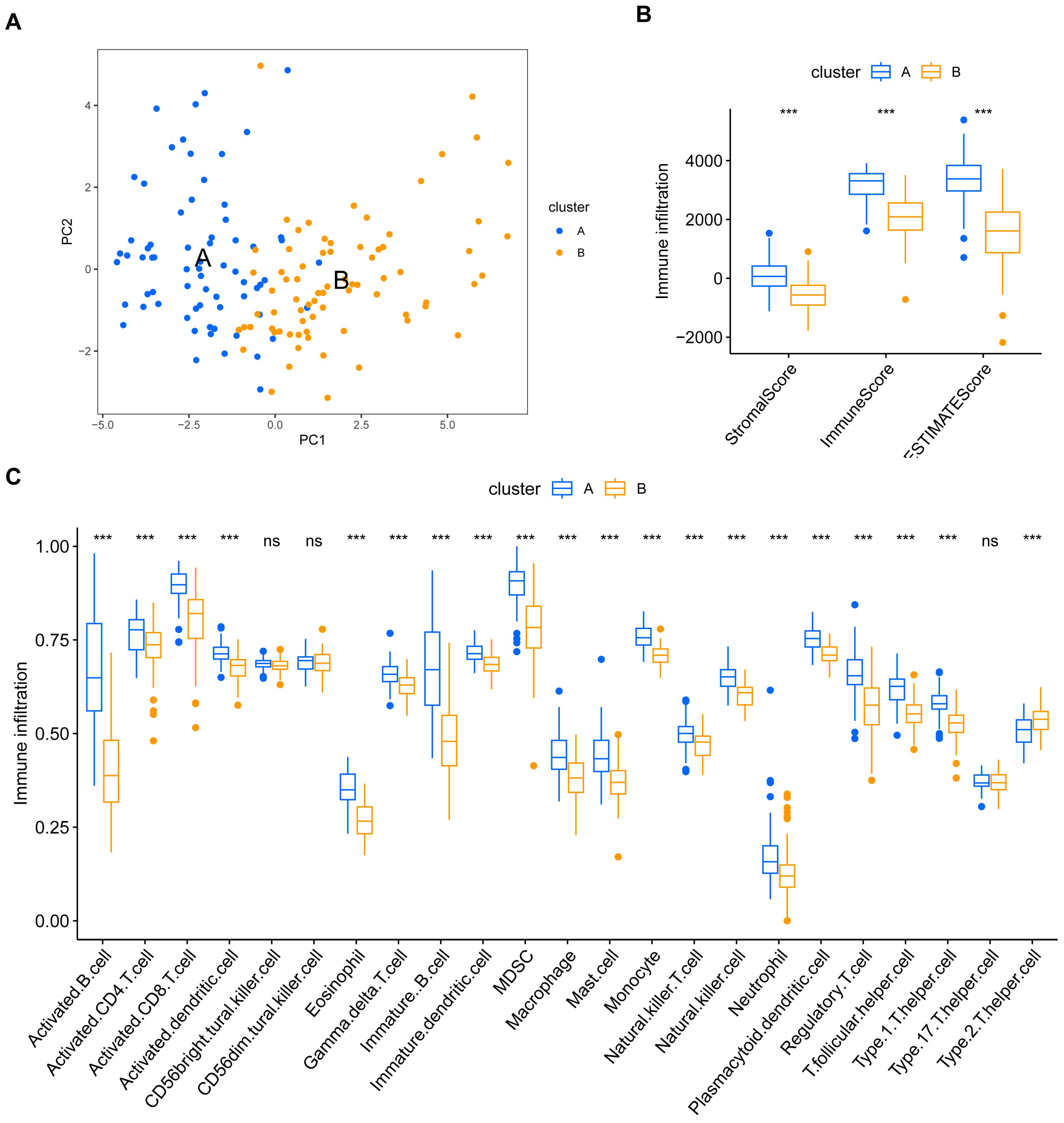
2.6. Immune Infiltration Analysis in EBV-Associated NPC Subtypes
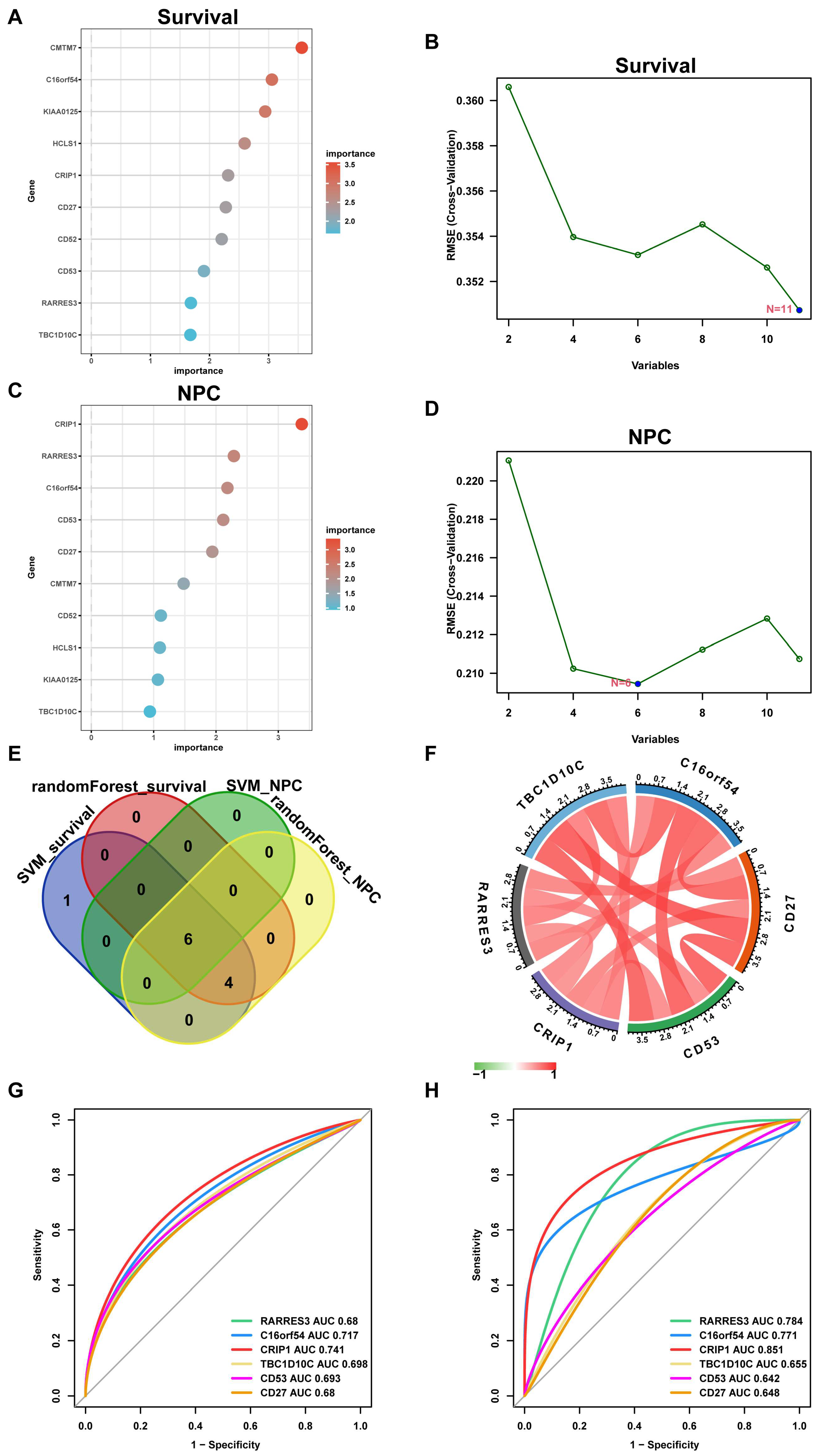
2.7. Supervised Learning Approaches to Identify EBV-Associated Hub Genes
2.8. Correlation Analysis
2.9. Prediction of Upstream Regulatory Networks of Hub Genes
2.10. Statistical Analysis
3. Results
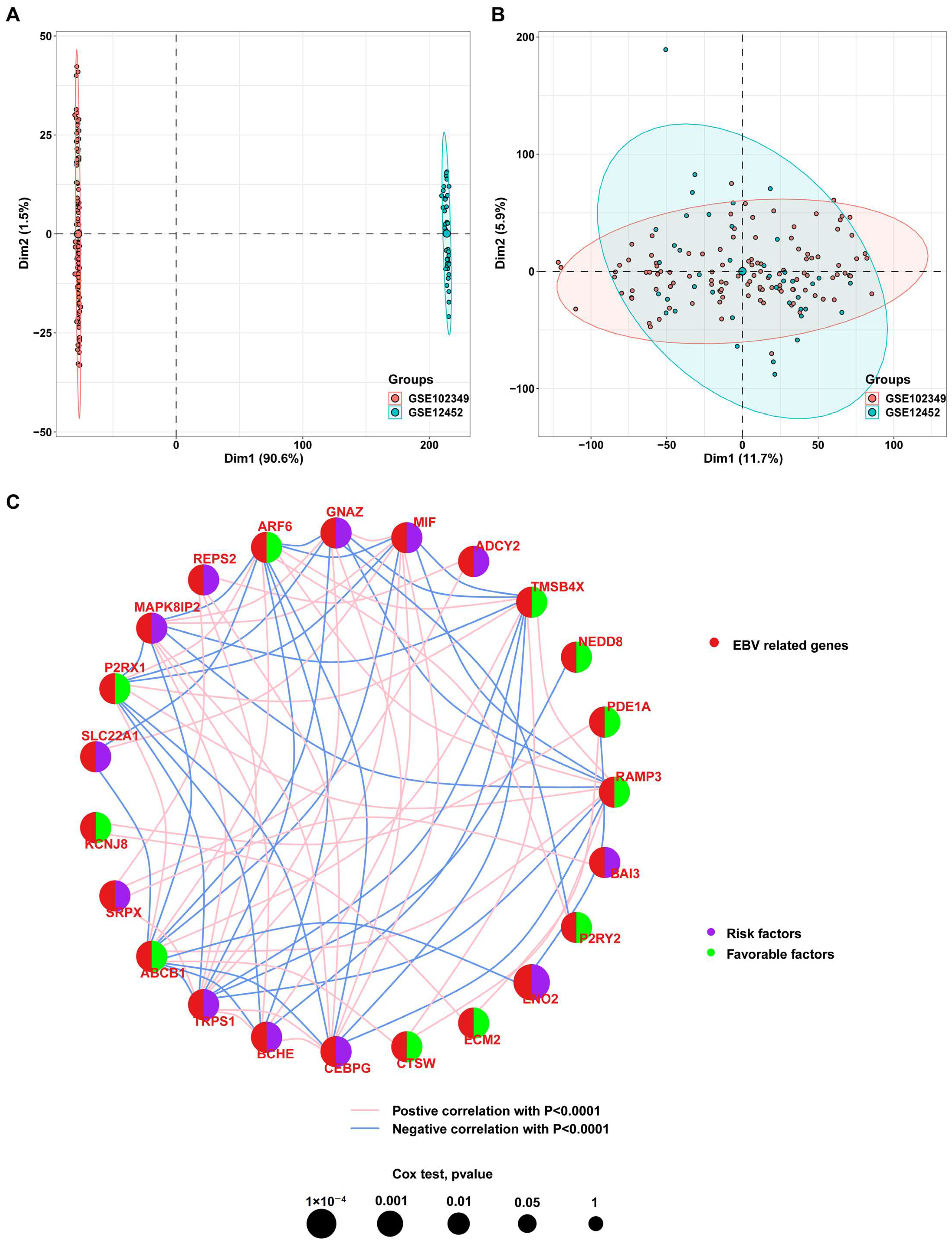
3.1. Identification of Genes Associated with EBV Infection in NPC Cells
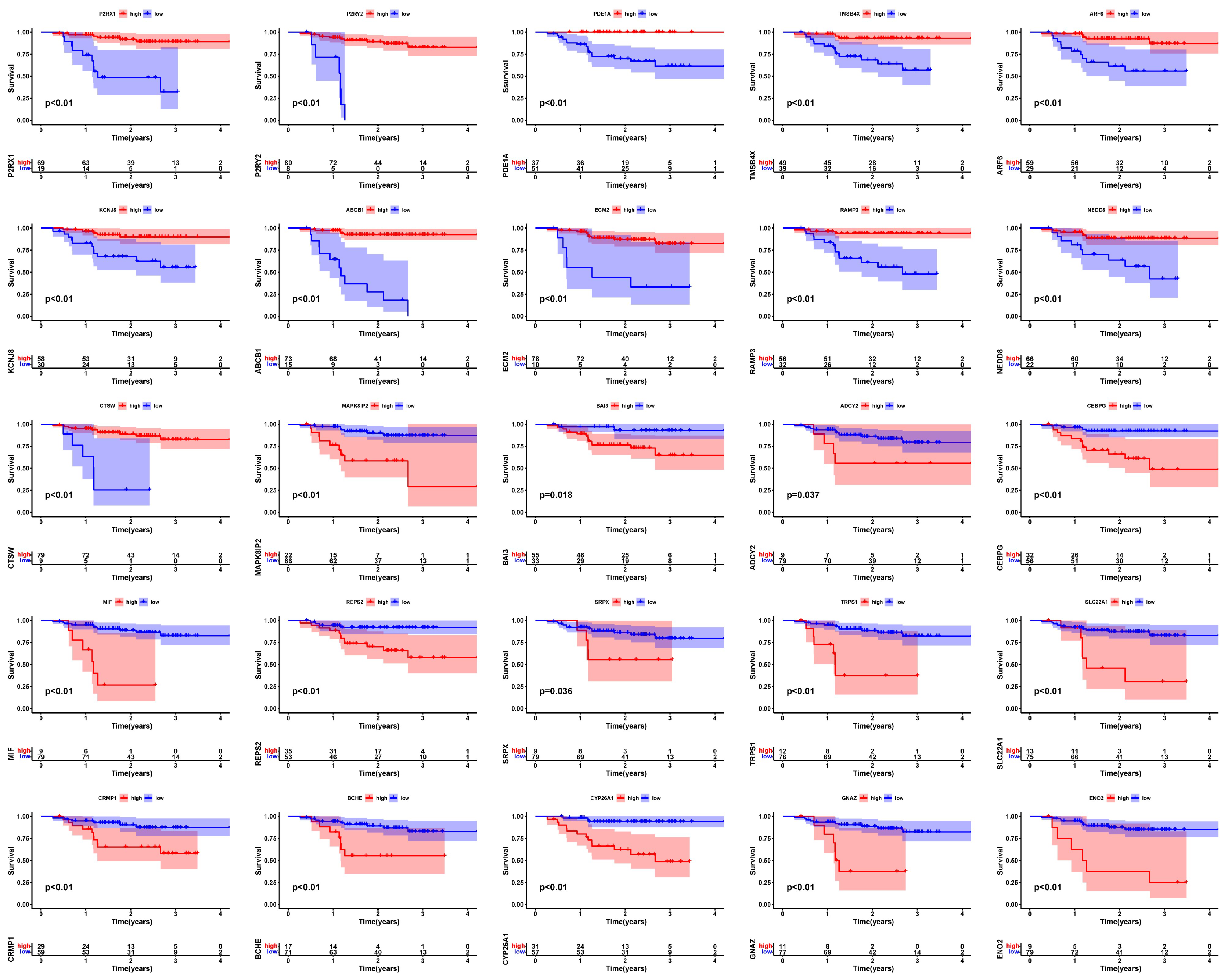
3.2. Screening of Prognosis-Associated Genes among the EBV-DEGs
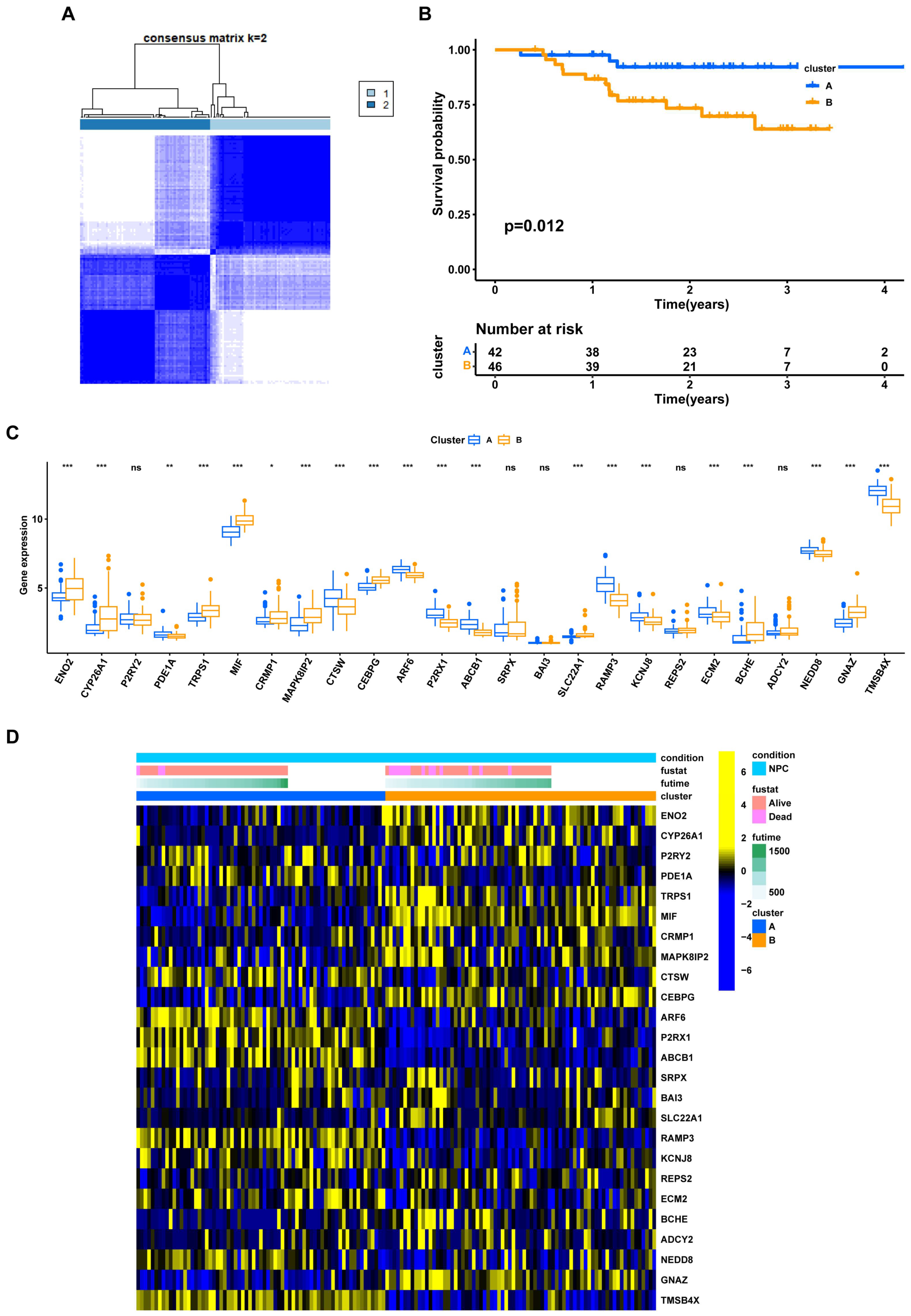
3.3. NPC Prognostic Risk Stratification Based on the EBV-DEGs
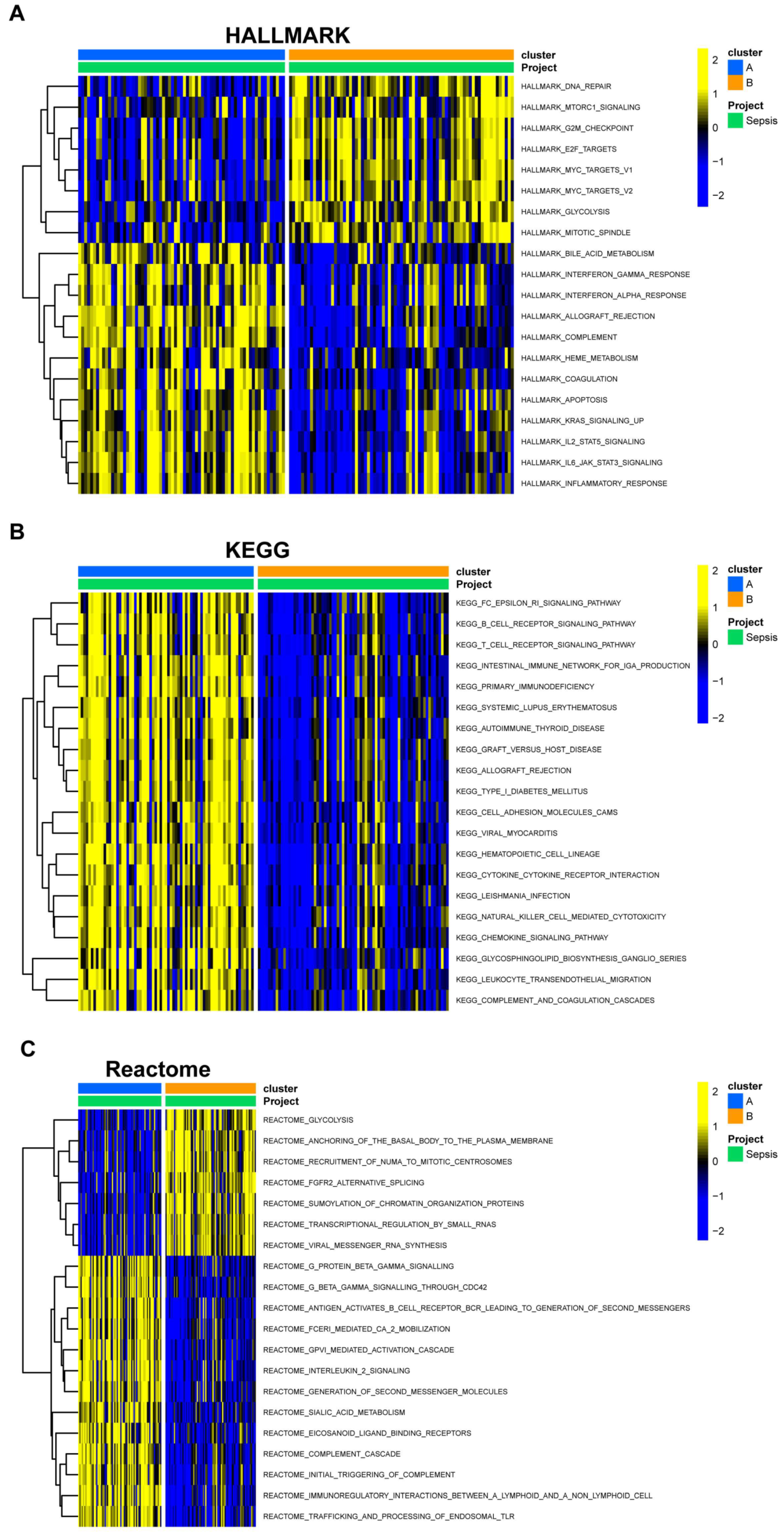
3.4. Gene Set Variation Analysis and Immune Infiltration Differences in EBV-Associated NPC Clusters
3.5. Overview of Gene Expression and Pathway Enrichment in EBV-Associated NPC Clusters
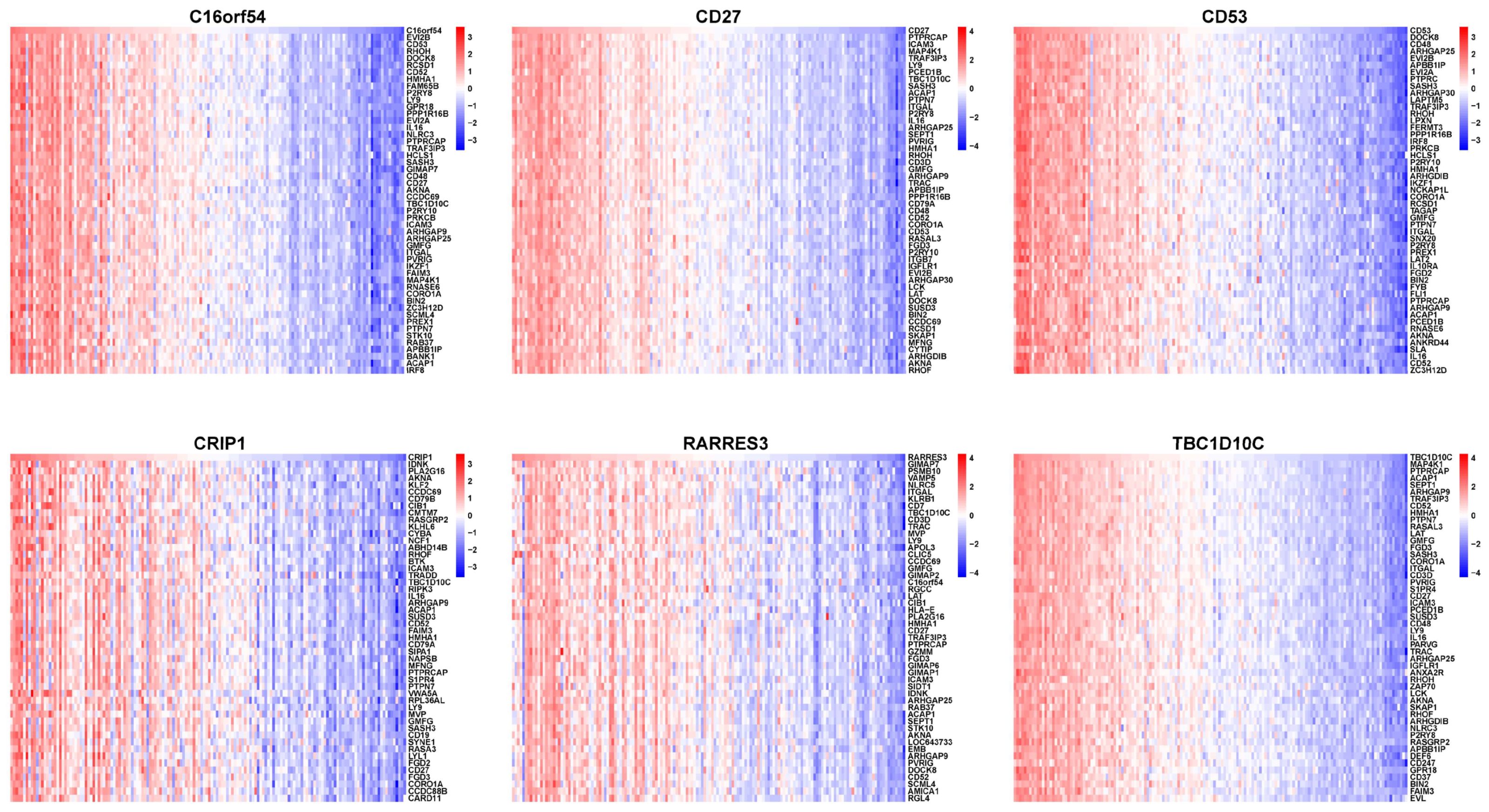
3.6. Selecting Hub Genes from the EBV-typeDEGs
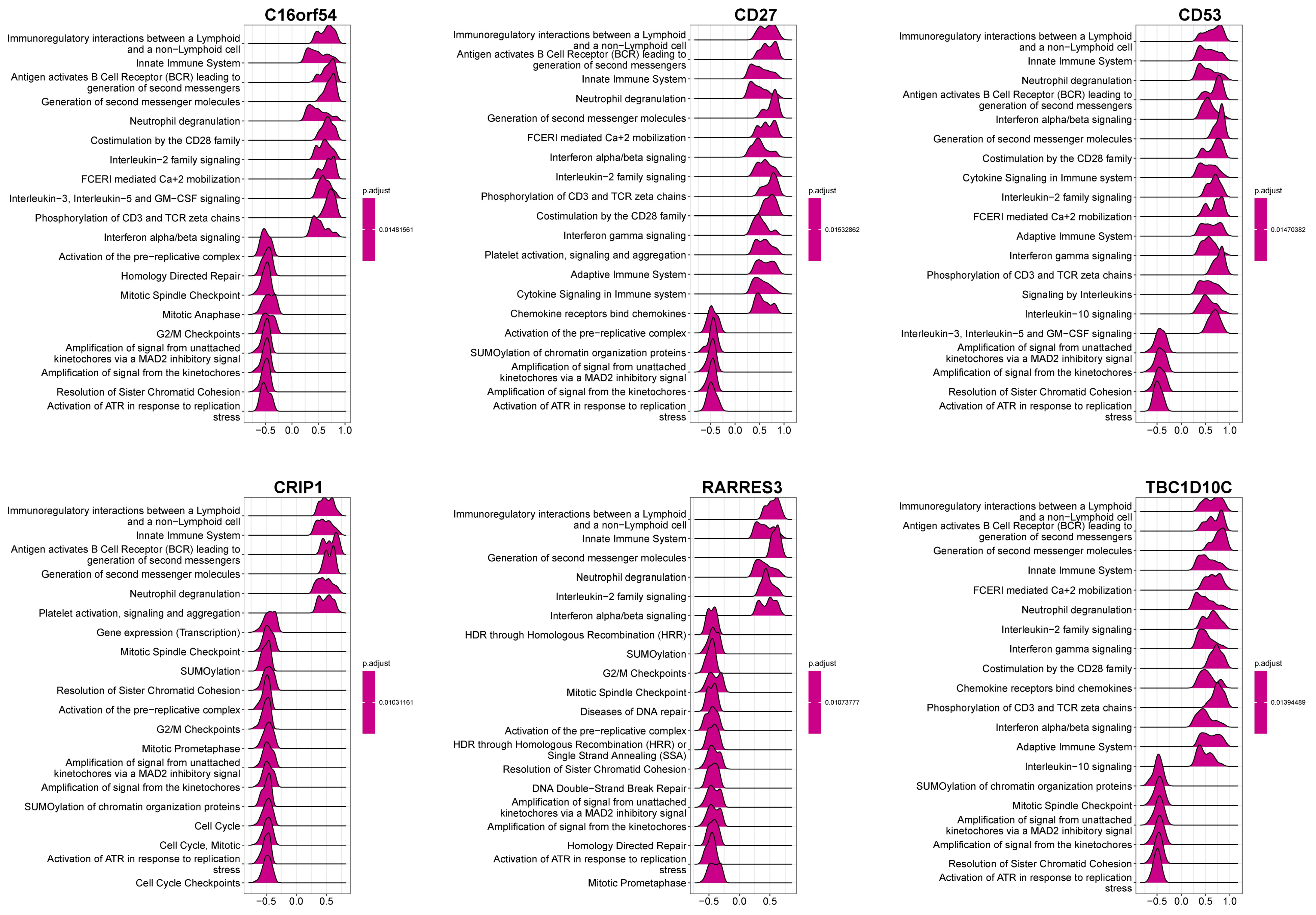
3.7. The Relationship between Hub Genes with Immune Infiltration and Gene Set Enrichment Analysis (GSEA)
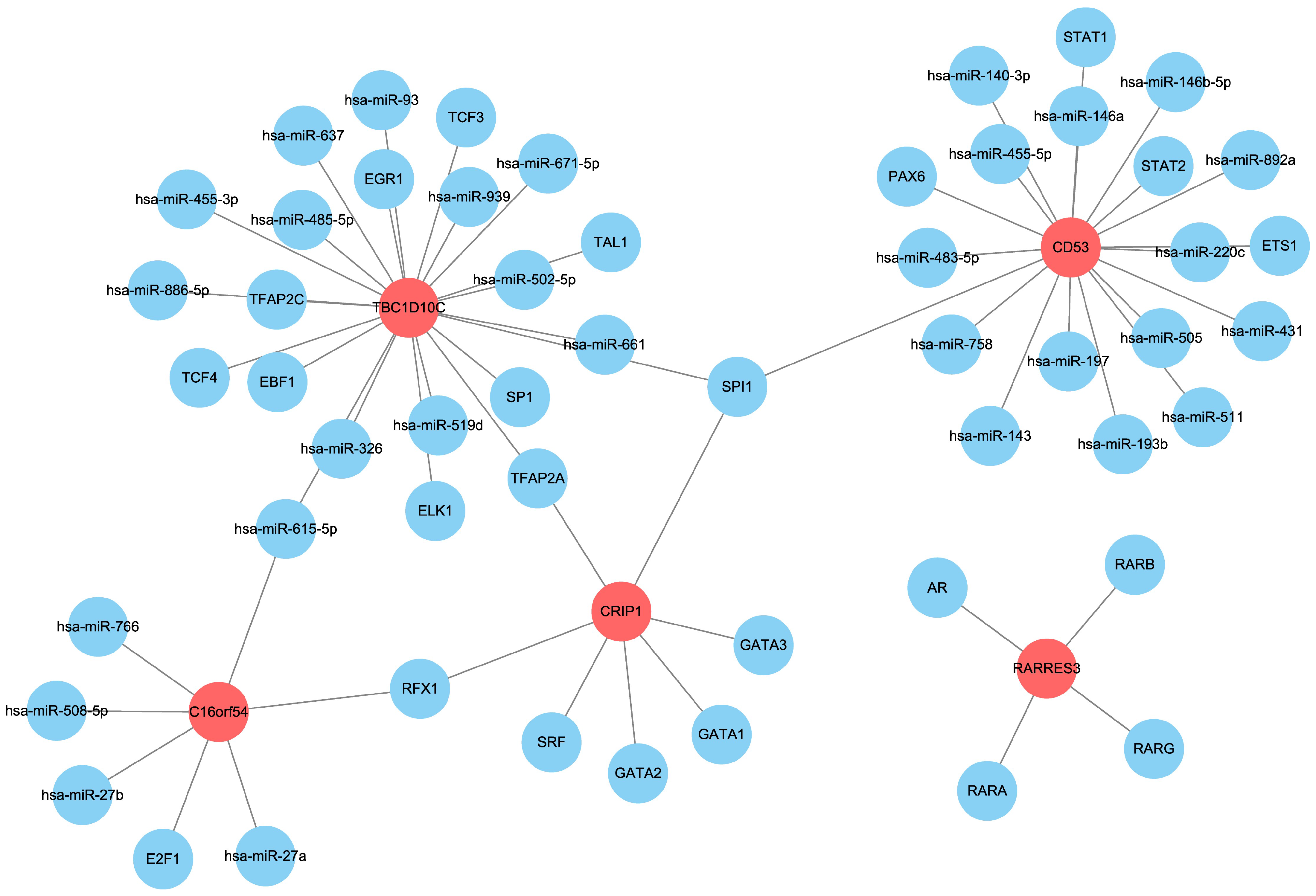
3.8. Prediction of Upstream miRNAs and Transcription Factors of the Hub Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, M.; Zhang, X.-L.; You, R.; Liu, Y.-P.; Cai, H.-M.; Liu, L.-Z.; Liu, X.-F.; Zou, X.; Xie, Y.-L.; Zou, R.-H.; et al. Evolutionary route of nasopharyngeal carcinoma metastasis and its clinical significance. Nat. Commun. 2023, 14, 610. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.K.J.; King, A.D.; Miller, J.A.; Liu, Z.; Yu, K.J.; Chua, M.L.K.; Ma, B.B.Y.; Chen, M.Y.; Pinsky, B.A.; Lou, P.-J.; et al. Recommendations for Epstein-Barr virus–based screening for nasopharyngeal cancer in high- and intermediate-risk regions. J. Natl. Cancer Inst. 2023, 115, 355–364. [Google Scholar] [CrossRef]
- Society, A.C. Key Statistics for Nasopharyngeal Cancer 2021. Available online: https://www.cancer.org/cancer/nasopharyngeal-cancer/about/key-statistics.html (accessed on 1 August 2022).
- Cancer.Net. Nasopharyngeal Cancer: Statistics. 2023 [updated by the Cancer.Net Editorial Board, 03/2023]. Available online: https://www.cancer.net/cancer-types/nasopharyngeal-cancer/statistics (accessed on 21 April 2023).
- Mo, Y.; Wang, Y.; Zhang, S.; Xiong, F.; Yan, Q.; Jiang, X.; Deng, X.; Wang, Y.; Fan, C.; Tang, L.; et al. Circular RNA circRNF13 inhibits proliferation and metastasis of nasopharyngeal carcinoma via SUMO2. Mol. Cancer 2021, 20, 112. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Long, Z.; Qiu, Q.; Liu, F.; Xu, Y.; Zhang, T.; Guo, R.; Zhong, W. Graphene quantum dots-based targeted nanoprobes detecting drug delivery, imaging, and enhanced chemotherapy of nasopharyngeal carcinoma. Bioeng. Transl. Med. 2022, 7, e10270. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.C.W.; Hui, E.P.; Lo, K.-W.; Lam, W.K.J.; Johnson, D.; Li, L.; Tao, Q.; Chan, K.C.A.; To, K.-F.; King, A.D.; et al. Nasopharyngeal carcinoma: An evolving paradigm. Nat. Rev. Clin. Oncol. 2021, 18, 679–695. [Google Scholar] [CrossRef]
- Gong, L.; Luo, J.; Zhang, Y.; Yang, Y.; Li, S.; Fang, X.; Zhang, B.; Huang, J.; Chow, L.K.-Y.; Chung, D.; et al. Nasopharyngeal carcinoma cells promote regulatory T cell development and suppressive activity via CD70-CD27 interaction. Nat. Commun. 2023, 14, 1912. [Google Scholar] [CrossRef] [PubMed]
- Kutok, J.; Wang, F. Spectrum of Epstein-Barr virus-associated diseases. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 375–404. [Google Scholar] [CrossRef]
- Guo, Y.; Kamara, S.; Zhang, J.; Wen, H.; Zheng, M.; Liu, Y.; Zhou, L.; Chen, J.; Zhu, S.; Zhang, L. EBV LMP1-C terminal binding affibody molecule downregulates MEK/ERK/p90RSK pathway and inhibits the proliferation of nasopharyngeal carcinoma cells in mouse tumor xenograft models. Front. Cell. Infect. Microbiol. 2022, 12, 1078504. [Google Scholar] [CrossRef]
- Kondo, S.; Okabe, A.; Nakagawa, T.; Matsusaka, K.; Fukuyo, M.; Rahmutulla, B.; Dochi, H.; Mizokami, H.; Kitagawa, Y.; Kurokawa, T.; et al. Repression of DERL3 via DNA methylation by Epstein-Barr virus latent membrane protein 1 in nasopharyngeal carcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166598. [Google Scholar] [CrossRef]
- Cheung, C.-C.; Chung, G.-T.; Lun, S.-W.; To, K.-F.; Choy, K.-W.; Lau, K.-M.; Siu, S.P.-K.; Guan, X.-Y.; Ngan, R.K.-C.; Yip, T.T.-C.; et al. miR-31 is consistently inactivated in EBV-associated nasopharyngeal carcinoma and contributes to its tumorigenesis. Mol. Cancer 2014, 13, 184. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Ye, Y.; Jiang, Q.; Chen, Y.; Lyu, X.; Li, J.; Wang, S.; Liu, T.; Cai, H.; Yao, K.; et al. Epstein–Barr virus-encoded microRNA BART1 induces tumour metastasis by regulating PTEN-dependent pathways in nasopharyngeal carcinoma. Nat. Commun. 2015, 6, 7353. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.N.; Tsai, C.L.; Tse, K.P.; Chang, H.Y.; Chang, Y.S. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc. Natl. Acad. Sci. USA 2002, 99, 10084–10089. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Wei, J.; Luo, Y.; Deng, Y.; Que, T.; Zhang, X.; Liu, F.; Zhang, J.; Luo, X. Epstein-Barr Virus Promotes Tumor Angiogenesis by Activating STIM1-Dependent Ca2+ Signaling in Nasopharyngeal Carcinoma. Pathogens 2021, 10, 1275. [Google Scholar] [CrossRef] [PubMed]
- Tao, D.; Zhang, N.; Huang, Q.; Ge, C.; Li, Q.; Li, S.; Weng, K.; Guo, Q.; Sui, J.; Wang, C.; et al. Association of Epstein-Barr virus infection with peripheral immune parameters and clinical outcome in advanced nasopharyngeal carcinoma. Sci. Rep. 2020, 10, 21976. [Google Scholar] [CrossRef]
- Miller, J.A.; Sahoo, M.K.; Yamamoto, F.; Huang, C.; Wang, H.; Zehnder, J.L.; Le, Q.-T.; Pinsky, B.A. Multiplex Epstein-Barr virus BALF2 genotyping detects high-risk variants in plasma for population screening of nasopharyngeal carcinoma. Mol. Cancer 2022, 21, 154. [Google Scholar] [CrossRef]
- Chan, D.; Lam, W.; Hui, E.; Ma, B.; Chan, C.; Lee, V.; Cheng, S.; Gai, W.; Jiang, P.; Wong, K.; et al. Improved risk stratification of nasopharyngeal cancer by targeted sequencing of Epstein–Barr virus DNA in post-treatment plasma. Ann. Oncol. 2022, 33, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.; Chan, K.; Ma, B.; Hui, E.; Mo, F.; Chow, K.; Leung, L.; Chu, K.; Zee, B.; Lo, Y.; et al. Plasma Epstein–Barr viral DNA load at midpoint of radiotherapy course predicts outcome in advanced-stage nasopharyngeal carcinoma. Ann. Oncol. 2014, 25, 1204–1208. [Google Scholar] [CrossRef]
- Chen, Y.-P.; Lv, J.-W.; Mao, Y.-P.; Li, X.-M.; Li, J.-Y.; Wang, Y.-Q.; Xu, C.; Li, Y.-Q.; He, Q.-M.; Yang, X.-J.; et al. Unraveling tumour microenvironment heterogeneity in nasopharyngeal carcinoma identifies biologically distinct immune subtypes predicting prognosis and immunotherapy responses. Mol. Cancer 2021, 20, 14. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Hwang, Y.-C.; Chen, K.-C.; Lin, Y.-S.; Huang, D.-Y.; Huang, T.W.; Huang, T.-W.; Kao, C.-W.; Wu, H.-C.; Lin, C.-T.; et al. Effect of Epstein-Barr virus infection on global gene expression in nasopharyngeal carcinoma. Funct. Integr. Genom. 2007, 7, 79–93. [Google Scholar] [CrossRef]
- Sengupta, S.; den Boon, J.A.; Chen, I.-H.; Newton, M.A.; Dahl, D.B.; Chen, M.; Cheng, Y.-J.; Westra, W.H.; Chen, C.-J.; Hildesheim, A.; et al. Genome-Wide Expression Profiling Reveals EBV-Associated Inhibition of MHC Class I Expression in Nasopharyngeal Carcinoma. Cancer Res. 2006, 66, 7999–8006. [Google Scholar] [CrossRef]
- Zhang, L.; MacIsaac, K.D.; Zhou, T.; Huang, P.-Y.; Xin, C.L.; Dobson, J.R.; Yu, K.; Chiang, D.Y.; Fan, Y.; Pelletier, M.; et al. Genomic Analysis of Nasopharyngeal Carcinoma Reveals TME-Based Subtypes. Mol. Cancer Res. 2017, 15, 1722–1732. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K.; Arihiro, K. Tumor-Driven Evolution of Immunosuppressive Networks during Malignant Progression. Cancer Res. 2006, 66, 5527–5536. [Google Scholar] [CrossRef]
- Cohen, J.I. Epstein-Barr virus infection. N. Engl. J. Med. 2000, 343, 481–492. [Google Scholar] [CrossRef]
- Kam, N.-W.; Laczka, O.; Li, X.; Wilkinson, J.; Hung, D.; Lai, S.P.H.; Wu, K.C.; Tsao, S.W.; Dai, W.; Che, C.M.; et al. ENOX2 inhibition enhances infiltration of effector memory T-cell and mediates response to chemotherapy in immune-quiescent nasopharyngeal carcinoma. J. Adv. Res. 2023, S2090-1232(23)00093-0. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ding, Q.; Lin, T.; Sun, Y.; Huang, Z.; Li, Y.; Hong, W.; Chen, X.; Wang, D.; Qiu, S. An immune-related prognostic model predicts neoplasm-immunity interactions for metastatic nasopharyngeal carcinoma. Front. Immunol. 2023, 14, 1109503. [Google Scholar] [CrossRef]
- Cousins, R.J.; Lanningham-Foster, L. Regulation of cysteine-rich intestinal protein, a zinc finger protein, by mediators of the immune response. J. Infect. Dis. 2000, 182 (Suppl. 1), S81–S84. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhou, R.; Zheng, Y.; Wen, Z.; Zhang, D.; Zeng, D.; Wu, J.; Huang, Z.; Rong, X.; Huang, N.; et al. CRIP1 cooperates with BRCA2 to drive the nuclear enrichment of RAD51 and to facilitate homologous repair upon DNA damage induced by chemotherapy. Oncogene 2021, 40, 5342–5355. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, Y.; Zhang, D.; Zhao, W.; Lu, Y.; Liu, C.; Lin, W.; Zhang, Y.; Chen, K.; Wang, H.; et al. CRIP1 suppresses BBOX1 -mediated carnitine metabolism to promote stemness in hepatocellular carcinoma. EMBO J. 2022, 41, e110218. [Google Scholar] [CrossRef]
- Wang, Q.; Williamson, M.; Bott, S.; Brookman-Amissah, N.; Freeman, A.; Nariculam, J.; Hubank, M.J.F.; Ahmed, A.; Masters, J.R. Hypomethylation of WNT5A, CRIP1 and S100P in prostate cancer. Oncogene 2007, 26, 6560–6565. [Google Scholar] [CrossRef]
- Zhang, L.Z.; Huang, L.-Y.; Huang, A.-L.; Liu, J.-X.; Yang, F. CRIP1 promotes cell migration, invasion and epithelial-mesenchymal transition of cervical cancer by activating the Wnt/beta-catenin signaling pathway. Life Sci. 2018, 207, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Ludyga, N.; Englert, S.; Pflieger, K.; Rauser, S.; Braselmann, H.; Walch, A.; Auer, G.; Höfler, H.; Aubele, M. The impact of Cysteine-Rich Intestinal Protein 1 (CRIP1) in human breast cancer. Mol. Cancer 2013, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Balluff, B.; Rauser, S.; Meding, S.; Elsner, M.; Schöne, C.; Feuchtinger, A.; Schuhmacher, C.; Novotny, A.; Jütting, U.; Maccarrone, G.; et al. MALDI Imaging Identifies Prognostic Seven-Protein Signature of Novel Tissue Markers in Intestinal-Type Gastric Cancer. Am. J. Pathol. 2011, 179, 2720–2729. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Xia, W.; Fan, W.; Shen, X.; Wu, H.; Zhang, H. Integrated Analysis of C16orf54 as a Potential Prognostic, Diagnostic, and Immune Marker across Pan-Cancer. Dis. Markers 2022, 2022, 9365046. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, L.; Hu, J.; Fan, R.; Zhou, J.; Wang, L.; Zhong, J. RARRES3 suppressed metastasis through suppression of MTDH to regulate epithelial-mesenchymal transition in colorectal cancer. Am. J. Cancer Res. 2015, 5, 1988–1999. [Google Scholar] [PubMed]
- Qiao, H.; Lv, R.; Pang, Y.; Yao, Z.; Zhou, X.; Zhu, W.; Zhou, W. Weighted Gene Coexpression Network Analysis Identifies TBC1D10C as a New Prognostic Biomarker for Breast Cancer. Anal. Cell. Pathol. 2022, 2022, 5259187. [Google Scholar] [CrossRef]
- Starzer, A.M.; Berghoff, A.S. New emerging targets in cancer immunotherapy: CD27 (TNFRSF7). ESMO Open 2019, 4 (Suppl. 3), e000629. [Google Scholar] [CrossRef]
- Dunlock, V.E. Tetraspanin CD53: An overlooked regulator of immune cell function. Med. Microbiol. Immunol. 2020, 209, 545–552. [Google Scholar] [CrossRef]
- Feng, Z.; Wang, H.; Zhou, R.; Li, J.; Xu, B. Enzyme-Instructed Assembly and Disassembly Processes for Targeting Downregulation in Cancer Cells. J. Am. Chem. Soc. 2017, 139, 3950–3953. [Google Scholar] [CrossRef]
- Jin, S.; Li, R.; Chen, M.-Y.; Yu, C.; Tang, L.-Q.; Liu, Y.-M.; Li, J.-P.; Liu, Y.-N.; Luo, Y.-L.; Zhao, Y.; et al. Single-cell transcriptomic analysis defines the interplay between tumor cells, viral infection, and the microenvironment in nasopharyngeal carcinoma. Cell Res. 2020, 30, 950–965. [Google Scholar] [CrossRef]
- Chen, Y.-P.; Yin, J.-H.; Li, W.-F.; Li, H.-J.; Chen, D.-P.; Zhang, C.-J.; Lv, J.-W.; Wang, Y.-Q.; Li, X.-M.; Li, J.-Y.; et al. Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma. Cell Res. 2020, 30, 1024–1042. [Google Scholar] [CrossRef]
- Gong, L.; Kwong, D.L.-W.; Dai, W.; Wu, P.; Li, S.; Yan, Q.; Zhang, Y.; Zhang, B.; Fang, X.; Liu, L.; et al. Comprehensive single-cell sequencing reveals the stromal dynamics and tumor-specific characteristics in the microenvironment of nasopharyngeal carcinoma. Nat. Commun. 2021, 12, 1540. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, S.; Wang, X.-L.; Peng, W.; Chen, Q.-Y.; Chi, D.-M.; Chen, J.-R.; Han, B.-W.; Lin, G.-W.; Li, Y.-Q.; et al. Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat. Commun. 2021, 12, 741. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, T.; Zhang, Y.; Ren, Z.; Cong, Y.; Long, J.; Peng, M.; Faleti, O.D.; Yang, Y.; Li, X.; Lyu, X. EBV-Associated Hub Genes as Potential Biomarkers for Predicting the Prognosis of Nasopharyngeal Carcinoma. Viruses 2023, 15, 1915. https://doi.org/10.3390/v15091915
Ding T, Zhang Y, Ren Z, Cong Y, Long J, Peng M, Faleti OD, Yang Y, Li X, Lyu X. EBV-Associated Hub Genes as Potential Biomarkers for Predicting the Prognosis of Nasopharyngeal Carcinoma. Viruses. 2023; 15(9):1915. https://doi.org/10.3390/v15091915
Chicago/Turabian StyleDing, Tengteng, Yuanbin Zhang, Zhixuan Ren, Ying Cong, Jingyi Long, Manli Peng, Oluwasijibomi Damola Faleti, Yinggui Yang, Xin Li, and Xiaoming Lyu. 2023. "EBV-Associated Hub Genes as Potential Biomarkers for Predicting the Prognosis of Nasopharyngeal Carcinoma" Viruses 15, no. 9: 1915. https://doi.org/10.3390/v15091915