
Gene Therapy Using Nanocarriers for Pancreatic Ductal Adenocarcinoma: Applications and Challenges in Cancer Therapeutics


Abstract
:1. Introduction
2. RNAi Therapy for Pancreatic Cancer
2.1. RNAi Using miRNAs
2.2. RNAi Using siRNAs
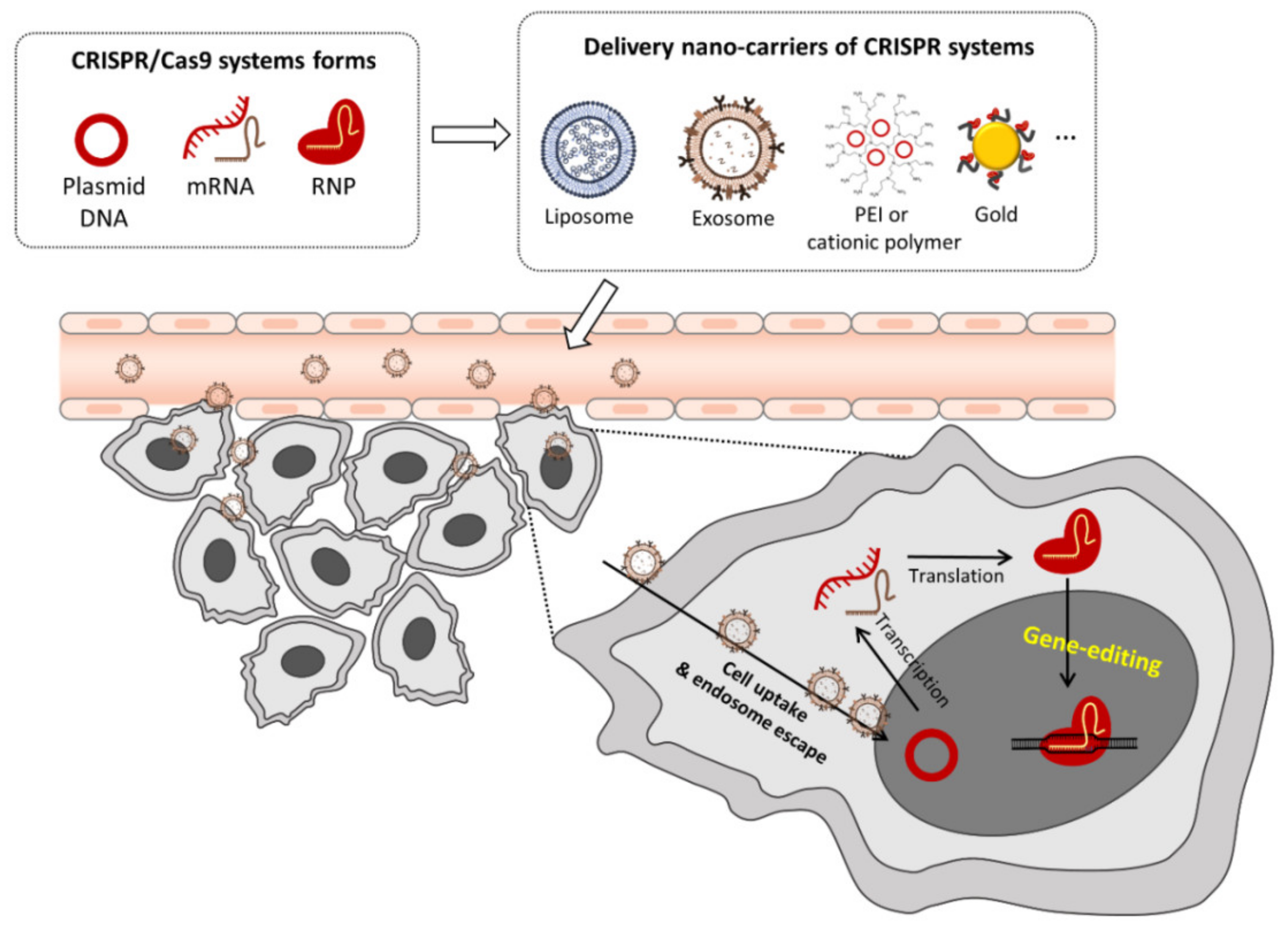
3. CRISPR-Cas Gene Editing
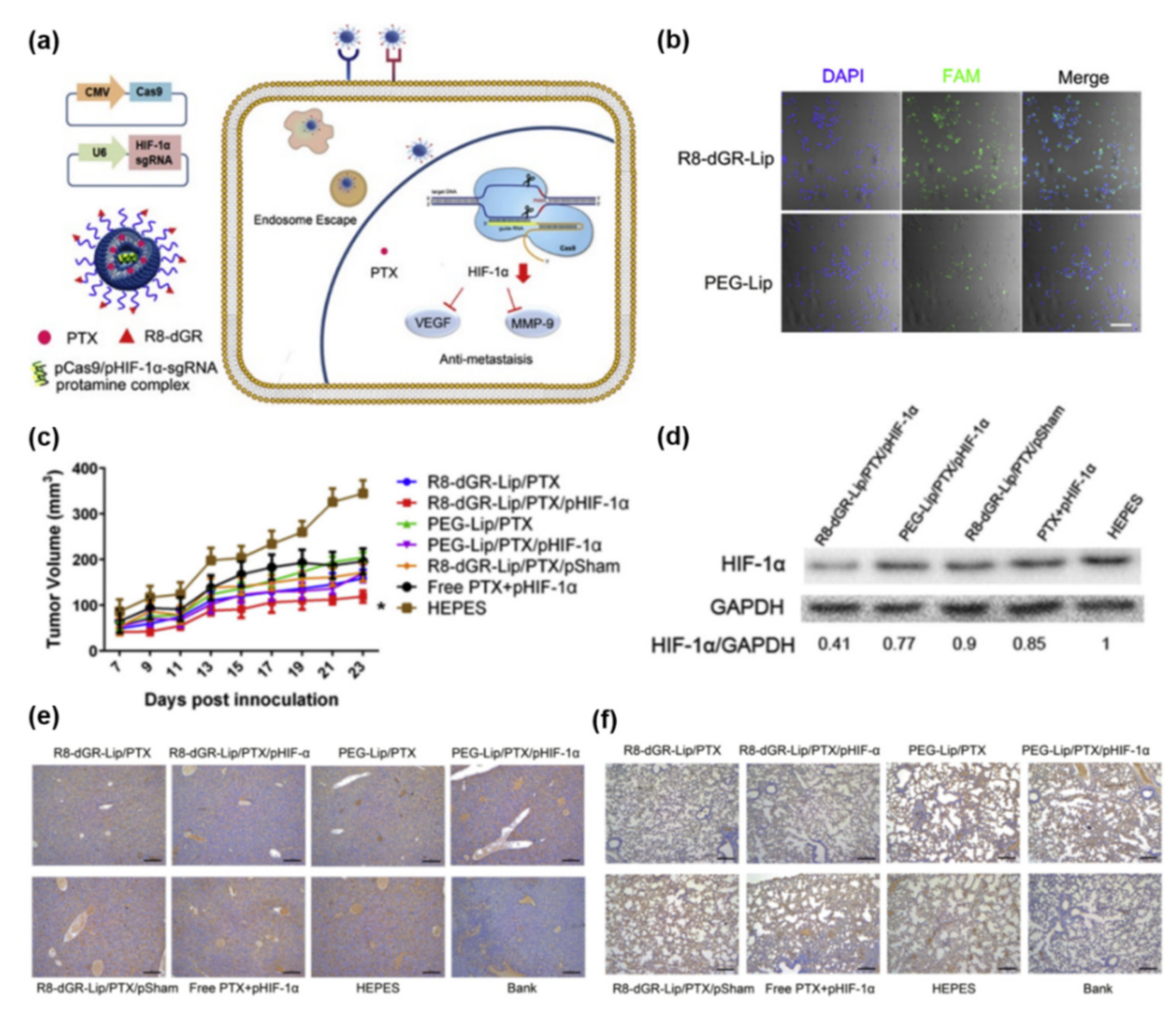
3.1. Plasmid DNA (Cas9/sgRNA Plasmid) Delivery
3.2. In Vitro Transcribed Cas9/sgRNA mRNA Delivery
3.3. Pre-Assembled Ribonucleoprotein (RNP) Complex Delivery
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Pook, H.; Pauklin, S. Mechanisms of Cancer Cell Death: Therapeutic Implications for Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 4834. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Cannon, A.; Thompson, C.; Hall, B.R.; Jain, M.; Kumar, S.; Batra, S.K. Desmoplasia in pancreatic ductal adenocarcinoma: Insight into pathological function and therapeutic potential. Genes Cancer 2018, 9, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef]
- El-Zahaby, S.A.; Elnaggar, Y.S.R.; Abdallah, O.Y. Reviewing two decades of nanomedicine implementations in targeted treatment and diagnosis of pancreatic cancer: An emphasis on state of art. J. Control. Release 2019, 293, 21–35. [Google Scholar] [CrossRef]
- Hu, X.; Xia, F.; Lee, J.; Li, F.; Lu, X.; Zhuo, X.; Nie, G.; Ling, D. Tailor-Made Nanomaterials for Diagnosis and Therapy of Pancreatic Ductal Adenocarcinoma. Adv. Sci. 2021, 8, 2002545. [Google Scholar] [CrossRef]
- Won, E.J.; Park, H.; Chang, S.H.; Kim, J.H.; Kwon, H.; Cho, Y.S.; Yoon, T.J. One-shot dual gene editing for drug-resistant pancreatic cancer therapy. Biomaterials 2021, 279, 121252. [Google Scholar] [CrossRef]
- Hannon, G.J. RNA interference. Nature 2002, 418, 244–251. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Hoy, S.M. Patisiran: First, Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef]
- Goodall, G.J.; Wickramasinghe, V.O. RNA in cancer. Nat. Rev. Cancer 2021, 21, 22–36. [Google Scholar] [CrossRef]
- Hernandez, Y.G.; Lucas, A.L. MicroRNA in pancreatic ductal adenocarcinoma and its precursor lesions. World J. Gastrointest. Oncol. 2016, 8, 18–29. [Google Scholar] [CrossRef]
- Gilles, M.E.; Hao, L.; Huang, L.; Rupaimoole, R.; Lopez-Casas, P.P.; Pulver, E.; Jeong, J.C.; Muthuswamy, S.K.; Hidalgo, M.; Bhatia, S.N.; et al. Personalized RNA Medicine for Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 1734–1747. [Google Scholar] [CrossRef] [Green Version]
- Goswami, C.P.; Nakshatri, H. PROGmiR: A tool for identifying prognostic miRNA biomarkers in multiple cancers using publicly available data. J. Clin. Bioinform. 2012, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, Y.; Li, J.; Zhang, Z.; Huang, C.; Lian, G.; Yang, K.; Chen, S.; Lin, Y.; Wang, L.; et al. Co-delivery of microRNA-21 antisense oligonucleotides and gemcitabine using nanomedicine for pancreatic cancer therapy. Cancer Sci. 2017, 108, 1493–1503. [Google Scholar] [CrossRef]
- Xie, Y.; Hang, Y.; Wang, Y.; Sleightholm, R.; Prajapati, D.R.; Bader, J.; Yu, A.; Tang, W.; Jaramillo, L.; Li, J.; et al. Stromal Modulation and Treatment of Metastatic Pancreatic Cancer with Local Intraperitoneal Triple miRNA/siRNA Nanotherapy. ACS Nano 2020, 14, 255–271. [Google Scholar] [CrossRef]
- Wu, Y.; Tang, Y.; Xie, S.; Zheng, X.; Zhang, S.; Mao, J.; Wang, B.; Hou, Y.; Hu, L.; Chai, K.; et al. Chimeric peptide supramolecular nanoparticles for plectin-1 targeted miRNA-9 delivery in pancreatic cancer. Theranostics 2020, 10, 1151–1165. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Bhardwaj, A.; Singh, S.; Arora, S.; Wang, B.; Grizzle, W.E.; Singh, A.P. MicroRNA-150 directly targets MUC4 and suppresses growth and malignant behavior of pancreatic cancer cells. Carcinogenesis 2011, 32, 1832–1839. [Google Scholar] [CrossRef]
- Arora, S.; Swaminathan, S.K.; Kirtane, A.; Srivastava, S.K.; Bhardwaj, A.; Singh, S.; Panyam, J.; Singh, A.P. Synthesis, characterization, and evaluation of poly (D,L-lactide-co-glycolide)-based nanoformulation of miRNA-150: Potential implications for pancreatic cancer therapy. Int. J. Nanomed. 2014, 9, 2933–2942. [Google Scholar]
- Zhang, S.; Cheng, Z.; Wang, Y.; Han, T. The Risks of miRNA Therapeutics: In a Drug Target Perspective. Drug Des. Dev. Ther. 2021, 15, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug Delivery: Is the Enhanced Permeability and Retention Effect Sufficient for Curing Cancer? Bioconjug. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef] [PubMed]
- Hattab, D.; Gazzali, A.M.; Bakhtiar, A. Clinical Advances of siRNA-Based Nanotherapeutics for Cancer Treatment. Pharmaceutics 2021, 13, 1009. [Google Scholar] [CrossRef]
- Zuckerman, J.E.; Gritli, I.; Tolcher, A.; Heidel, J.D.; Lim, D.; Morgan, R.; Chmielowski, B.; Ribas, A.; Davis, M.E.; Yen, Y. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc. Natl. Acad. Sci. USA 2014, 111, 11449–11454. [Google Scholar] [CrossRef] [Green Version]
- Schultheis, B.; Strumberg, D.; Santel, A.; Vank, C.; Gebhardt, F.; Keil, O.; Lange, C.; Giese, K.; Kaufmann, J.; Khan, M.; et al. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 4141–4148. [Google Scholar] [CrossRef]
- Schultheis, B.; Strumberg, D.; Kuhlmann, J.; Wolf, M.; Link, K.; Seufferlein, T.; Kaufmann, J.; Feist, M.; Gebhardt, F.; Khan, M.; et al. Safety, Efficacy and Pharcacokinetics of Targeted Therapy with The Liposomal RNA Interference Therapeutic Atu027 Combined with Gemcitabine in Patients with Pancreatic Adenocarcinoma. A Randomized Phase Ib/IIa Study. Cancers 2020, 12, 3130. [Google Scholar] [CrossRef]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [Green Version]
- Judge, A.D.; Robbins, M.; Tavakoli, I.; Levi, J.; Hu, L.; Fronda, A.; Ambegia, E.; McClintock, K.; MacLachlan, I. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J. Clin. Investig. 2009, 119, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- O’Brien, Z.; Wang, L.; Majeti, B.; Clamme, J.; Baclig, R.; Chu, J.; Fong, S.; Harborth, J.; Ibarra, J.; Yin, H.; et al. A novel lipid nanoparticle (NBF-006) encapsulating glutathione Stransferase P (GSTP) siRNA for the treatment of KRAS-driven non-small cell lung cancer. Cancer Res. 2018, 78, 5917. [Google Scholar]
- Chatterjee, A.; Gupta, S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018, 433, 33–42. [Google Scholar] [CrossRef]
- Khvalevsky, E.Z.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [Green Version]
- Aye, Y.; Li, M.; Long, M.J.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2015, 34, 2011–2021. [Google Scholar] [CrossRef]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef]
- Aleku, M.; Schulz, P.; Keil, O.; Santel, A.; Schaeper, U.; Dieckhoff, B.; Janke, O.; Endruschat, J.; Durieux, B.; Röder, N.; et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008, 68, 9788–9798. [Google Scholar] [CrossRef] [Green Version]
- Santel, A.; Aleku, M.; Röder, N.; Möpert, K.; Durieux, B.; Janke, O.; Keil, O.; Endruschat, J.; Dames, S.; Lange, C.; et al. Atu027 prevents pulmonary metastasis in experimental and spontaneous mouse metastasis models. Clin. Cancer. Res. 2010, 16, 5469–5480. [Google Scholar] [CrossRef] [Green Version]
- Strebhardt, K.; Ullrich, A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 2006, 6, 321–330. [Google Scholar] [CrossRef]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 392. [Google Scholar] [CrossRef]
- Zhao, X.; Li, F.; Li, Y.; Wang, H.; Ren, H.; Chen, J.; Nie, G.; Hao, J. Co-delivery of HIF1α siRNA and gemcitabine via biocompatible lipid-polymer hybrid nanoparticles for effective treatment of pancreatic cancer. Biomaterials 2015, 46, 13–25. [Google Scholar] [CrossRef]
- Pan, X.; Zhu, Q.; Sun, Y.; Li, L.; Zhu, Y.; Zhao, Z.; Zuo, J.; Fang, W.; Li, K. PLGA/poloxamer nanoparticles loaded with EPAS1 siRNA for the treatment of pancreatic cancer in vitro and in vivo. Int. J. Mol. Med. 2015, 35, 995–1002. [Google Scholar] [CrossRef]
- McCarroll, J.A.; Sharbeen, G.; Liu, J.; Youkhana, J.; Goldstein, D.; McCarthy, N.; Limbri, L.F.; Dischl, D.; Ceyhan, G.O.; Erkan, M.; et al. βIII-tubulin: A novel mediator of chemoresistance and metastases in pancreatic cancer. Oncotarget 2015, 6, 2235–2249. [Google Scholar] [CrossRef] [Green Version]
- Teo, J.; McCarroll, J.A.; Boyer, C.; Youkhana, J.; Sagnella, S.M.; Duong, H.T.; Liu, J.; Sharbeen, G.; Goldstein, D.; Davis, T.P.; et al. A Rationally Optimized Nanoparticle System for the Delivery of RNA Interference Therapeutics into Pancreatic Tumors In Vivo. Biomacromolecules 2016, 17, 2337–2351. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., III. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborn, M.J.; Belanto, J.J.; Tolar, J.; Voytas, D.F. Gene editing and its application for hematological diseases. Int. J. Hematol. 2016, 104, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Kaboli, S.; Babazada, H. CRISPR Mediated Genome Engineering and its Application in Industry. Curr. Issues Mol. Biol. 2018, 26, 81–92. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.B.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Nelson, C.E.; Gersbach, C.A. Engineering Delivery Vehicles for Genome Editing. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 637–662. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Kauffman, K.J.; Anderson, D.G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017, 16, 387–399. [Google Scholar] [CrossRef]
- Li, L.; Hu, S.; Chen, X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials 2018, 171, 207–218. [Google Scholar] [CrossRef]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Rapti, K.; Louis-Jeune, V.; Kohlbrenner, E.; Ishikawa, K.; Ladage, D.; Zolotukhin, S.; Hajjar, R.J.; Weber, T. Neutralizing antibodies against AAV serotypes 1, 2, 6, and 9 in sera of commonly used animal models. Mol. Ther. 2012, 20, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; He, Z.Y.; Wei, X.W.; Gao, G.P.; Wei, Y.Q. Challenges in CRISPR/CAS9 Delivery: Potential Roles of Nonviral Vectors. Hum. Gene Ther. 2015, 26, 452–462. [Google Scholar] [CrossRef]
- Wang, D.; Mou, H.; Li, S.; Li, Y.; Hough, S.; Tran, K.; Li, J.; Yin, H.; Anderson, D.G.; Sontheimer, E.J.; et al. Adenovirus-Mediated Somatic Genome Editing of Pten by CRISPR/Cas9 in Mouse Liver in Spite of Cas9-Specific Immune Responses. Hum. Gene Ther. 2015, 26, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Lao, Y.H.; Li, M.; Gao, M.A.; Shao, D.; Chi, C.W.; Huang, D.; Chakraborty, S.; Ho, T.C.; Jiang, W.; Wang, H.X.; et al. HPV Oncogene Manipulation Using Nonvirally Delivered CRISPR/Cas9 or Natronobacterium gregoryi Argonaute. Adv. Sci. 2018, 5, 1700540. [Google Scholar] [CrossRef]
- Li, M.; Xie, H.; Liu, Y.; Xia, C.; Cun, X.; Long, Y.; Chen, X.; Deng, M.; Guo, R.; Zhang, Z.; et al. Knockdown of hypoxia-inducible factor-1 alpha by tumor targeted delivery of CRISPR/Cas9 system suppressed the metastasis of pancreatic cancer. J. Control. Release 2019, 304, 204–215. [Google Scholar] [CrossRef]
- Luo, Y.L.; Xu, C.F.; Li, H.J.; Cao, Z.T.; Liu, J.; Wang, J.L.; Du, X.J.; Yang, X.Z.; Gu, Z.; Wang, J. Macrophage-Specific in Vivo Gene Editing Using Cationic Lipid-Assisted Polymeric Nanoparticles. ACS Nano 2018, 12, 994–1005. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, L.; Zheng, W.; Cong, L.; Guo, Z.; Xie, Y.; Wang, L.; Tang, R.; Feng, Q.; Hamada, Y.; et al. Thermo-triggered Release of CRISPR-Cas9 System by Lipid-Encapsulated Gold Nanoparticles for Tumor Therapy. Angew. Chem. Int. Ed. Engl. 2018, 57, 1491–1496. [Google Scholar] [CrossRef]
- Schuh, R.S.; de Carvalho, T.G.; Giugliani, R.; Matte, U.; Baldo, G.; Teixeira, H.F. Gene editing of MPS I human fibroblasts by co-delivery of a CRISPR/Cas9 plasmid and a donor oligonucleotide using nanoemulsions as nonviral carriers. Eur. J. Pharm. Biopharm. 2018, 122, 158–166. [Google Scholar] [CrossRef]
- Wang, H.X.; Song, Z.; Lao, Y.H.; Xu, X.; Gong, J.; Cheng, D.; Chakraborty, S.; Park, J.S.; Li, M.; Huang, D.; et al. Nonviral gene editing via CRISPR/Cas9 delivery by membrane-disruptive and endosomolytic helical polypeptide. Proc. Natl. Acad. Sci. USA 2018, 115, 4903–4908. [Google Scholar] [CrossRef] [Green Version]
- Ryu, N.; Kim, M.A.; Park, D.; Lee, B.; Kim, Y.R.; Kim, K.H.; Baek, J.I.; Kim, W.J.; Lee, K.Y.; Kim, U.K. Effective PEI-mediated delivery of CRISPR-Cas9 complex for targeted gene therapy. Nanomedicine 2018, 14, 2095–2102. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, P.; Feng, Q.; Wang, N.; Chen, Z.; Huang, Y.; Zheng, W.; Jiang, X. Lipid nanoparticle-mediated efficient delivery of CRISPR/Cas9 for tumor therapy. NPG Asia Mater. 2017, 9, e441. [Google Scholar] [CrossRef]
- Ding, Y.; Jiang, Z.; Saha, K.; Kim, C.S.; Kim, S.T.; Landis, R.F.; Rotello, V.M. Gold nanoparticles for nucleic acid delivery. Mol. Ther. 2014, 22, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.; Tschida, B.; Moriarity, B.; et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 2015, 6, 7391. [Google Scholar] [CrossRef]
- Wang, H.X.; Li, M.; Lee, C.M.; Chakraborty, S.; Kim, H.W.; Bao, G.; Leong, K.W. CRISPR/Cas9-Based Genome Editing for Disease Modeling and Therapy: Challenges and Opportunities for Nonviral Delivery. Chem. Rev. 2017, 117, 9874–9906. [Google Scholar] [CrossRef] [PubMed]
- Ibraheem, D.; Elaissari, A.; Fessi, H. Gene therapy and DNA delivery systems. Int. J. Pharm. 2014, 459, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Zhang, Y.; Lu, Y.; Liu, L.; Shi, D.; Wen, Y.; Yang, L.; Ma, Q.; Liu, T.; Zhu, X.; et al. The HIF-1α/CXCR4 pathway supports hypoxia-induced metastasis of human osteosarcoma cells. Cancer Lett. 2015, 357, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Chan, Y.C.; Chang, W.M.; Lin, Y.F.; Yang, C.J.; Su, C.Y.; Huang, M.S.; Wu, A.T.; Hsiao, M. Feedback regulation of ALDOA activates the HIF-1α/MMP9 axis to promote lung cancer progression. Cancer Lett. 2017, 403, 28–36. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, 6478. [Google Scholar] [CrossRef]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3, e99263. [Google Scholar] [CrossRef]
- Liu, H.; Qiao, S.; Fan, X.; Gu, Y.; Zhang, Y.; Huang, S. Role of exosomes in pancreatic cancer. Oncol. Lett. 2021, 21, 298. [Google Scholar] [CrossRef]
- McAndrews, K.M.; Xiao, F.; Chronopoulos, A.; LeBleu, V.S.; Kugeratski, F.G.; Kalluri, R. Exosome-mediated delivery of CRISPR/Cas9 for targeting of oncogenic Kras(G12D) in pancreatic cancer. Life Sci. Alliance 2021, 4. [Google Scholar] [CrossRef]
- Glass, Z.; Lee, M.; Li, Y.; Xu, Q. Engineering the Delivery System for CRISPR-Based Genome Editing. Trends Biotechnol. 2018, 36, 173–185. [Google Scholar] [CrossRef]
- Liang, X.; Potter, J.; Kumar, S.; Zou, Y.; Quintanilla, R.; Sridharan, M.; Carte, J.; Chen, W.; Roark, N.; Ranganathan, S.; et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015, 208, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Kwaku Dad, A.B.; Beloor, J.; Gopalappa, R.; Lee, S.K.; Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014, 24, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Liang, X.; Xie, H.; Kumar, S.; Ravinder, N.; Potter, J.; du Jeu, X.D.M.; Chesnut, J.D. Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol. Lett. 2016, 38, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zuris, J.A.; Meng, F.; Rees, H.; Sun, S.; Deng, P.; Han, Y.; Gao, X.; Pouli, D.; Wu, Q.; et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Natl. Acad. Sci. USA 2016, 113, 2868–2873. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.Y.; Ryu, J.Y.; Lee, H.A.R.; Hong, S.H.; Park, H.S.; Hong, K.S.; Park, S.G.; Kim, H.P.; Yoon, T.J. Lecithin nano-liposomal particle as a CRISPR/Cas9 complex delivery system for treating type 2 diabetes. J. Nanobiotechnol. 2019, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.Y.; Choi, Y.J.; Won, E.J.; Hui, E.; Kim, H.S.; Cho, Y.S.; Yoon, T.J. Gene editing particle system as a therapeutic approach for drug-resistant colorectal cancer. Nano Res. 2020, 13, 1576–1585. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, L.; Lang, J.; Cheng, K.; Wang, Y.; Li, X.; Shi, J.; Wang, Y.; Nie, G. A CRISPR-Cas13a system for efficient and specific therapeutic targeting of mutant KRAS for pancreatic cancer treatment. Cancer Lett. 2018, 431, 171–181. [Google Scholar] [CrossRef]
- Binenbaum, Y.; Na’ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updates 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306. [Google Scholar] [CrossRef] [Green Version]
- Poon, E.; Harris, A.L.; Ashcroft, M. Targeting the hypoxia-inducible factor (HIF) pathway in cancer. Expert Rev. Mol. Med. 2009, 11, e26. [Google Scholar] [CrossRef] [Green Version]
- Aghamiri, S.; Raee, P.; Talaei, S.; Mohammadi-Yeganeh, S.; Bayat, S.; Rezaee, D.; Ghavidel, A.A.; Teymouri, A.; Roshanzamiri, S.; Farhadi, S.; et al. Nonviral siRNA delivery systems for pancreatic cancer therapy. Biotechnol. Bioeng. 2021, 118, 3669–3690. [Google Scholar] [CrossRef]
- Yin, H.; Xue, W.; Anderson, D.G. CRISPR-Cas: A tool for cancer research and therapeutics. Nat. Rev. Clin. Oncol. 2019, 16, 281–295. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar]
- Zhang, M.; Eshraghian, E.A.; Al Jammal, O.; Zhang, Z.; Zhu, X. CRISPR technology: The engine that drives cancer therapy. Biomed. Pharmacother. 2021, 133, 111007. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic siRNAs | Indication | Target Gene /Protein | Delivery Systems | Route of Administrations | Phase/Status | Reference |
|---|---|---|---|---|---|---|
| CALLA-01 | Cancer, Solid tumors | RPM2 | Cyclo-dextrin based polymer | Systemc/IV | I/Terminated | [32] |
| Atu027 | Pancreatic cancer | PKN3 | Liposomes (Lipoplexes, Cationic lipid) | Systemc/IV | II/Completed | [33,34] |
| siG12D LODER | Pancreatic cancer | KRASG12D mutation | Polymer matrix (LODER polymer) | Local/Surgical implantation | II/Recruiting | [35] |
| TKM 080301 | Solid tumors with liver involvement | PLK-1 | SNALP | Systemc/IV | II/Completed | [36] |
| iExosomes | Pancreatic cancer | KRASG12D mutation | Mesenchymal Stromal Cells-derived Exosomes | Systemc/IV | I/Recruiting | [37] |
| NBF 006 | Non-small cell lung, Colorectal, and Pancreatic | GSTP | Lipid nanoparticles | Systemc/IV | I/Recruiting | [38] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Won, E.-J.; Park, H.; Yoon, T.-J.; Cho, Y.-S. Gene Therapy Using Nanocarriers for Pancreatic Ductal Adenocarcinoma: Applications and Challenges in Cancer Therapeutics. Pharmaceutics 2022, 14, 137. https://doi.org/10.3390/pharmaceutics14010137
Won E-J, Park H, Yoon T-J, Cho Y-S. Gene Therapy Using Nanocarriers for Pancreatic Ductal Adenocarcinoma: Applications and Challenges in Cancer Therapeutics. Pharmaceutics. 2022; 14(1):137. https://doi.org/10.3390/pharmaceutics14010137
Chicago/Turabian StyleWon, Eun-Jeong, Hyeji Park, Tae-Jong Yoon, and Young-Seok Cho. 2022. "Gene Therapy Using Nanocarriers for Pancreatic Ductal Adenocarcinoma: Applications and Challenges in Cancer Therapeutics" Pharmaceutics 14, no. 1: 137. https://doi.org/10.3390/pharmaceutics14010137