1. Introduction
In the pharmaceutical product development field, there exist a wide variety of drug substances including potential drug candidates which are often limited by poor aqueous solubility, despite their high therapeutic efficacy. Thus, achieving desired drug concentration at the target site is a major challenge typically faced by the formulation and development scientists. Sertraline, a selective serotonin reuptake inhibitor, is currently used as an anti-depressant, belongs to biopharmaceutics classification system (BCS) class II, has poor aqueous solubility (3.5 mg/L), high lipophilicity (log P 5.1), and low oral bioavailability of approximately 44% [
1,
2]. Administration of drugs through the oral route is most favored over other modes of drug delivery chiefly due to convenience of self-administration, safety, pain avoidance, high patient compliance, low cost, and flexibility in accommodating different types of drugs [
3]. Sertraline is gradually absorbed after oral administration however coadministration with food was shown to increase maximum plasma drug concentration (C
max) by nearly 25% and decrease time to reach peak concentration (T
max). Moreover, sertraline undergoes extensive hepatic biotransformation via oxidative and glucuronidation pathways [
4].
Various approaches like solid dispersion [
5], micronization [
6], pH modification [
7], crystal modification [
8], and self-emulsifying drug delivery systems [
9] have been evaluated for enhancing the low solubility and dissolution rate of hydrophobic drugs. Modification of physicochemical properties using typical approaches such as salt formation has inherent demerits. Therefore, the probable reason for the low bioavailability of hydrochloride salt might be due to its conversion to a less soluble freebase which further leads to the formation of aggregates in the gastrointestinal tract, thereby hindering the oral absorption of the drug. Few attempts have been carried out to improve the therapeutic efficacy of sertraline by formulating it into solid lipid nanoparticles [
10] as well as loading it into inorganic micro and mesoporous materials [
11]. Alternatively, the transdermal delivery approach has also been investigated [
12,
13].
Lipid-based preparations such as self-emulsifying drug delivery system (SEDDS), self-microemulsifying drug delivery system, and self-nanoemulsifying drug delivery system (SNEDDS) have been largely explored in many studies to augment the bioavailability of highly lipophilic drugs [
14,
15,
16,
17]. The SNEDDS encompasses isotropic mixtures of oil, surfactants along with one or more hydrophilic co-solvents or co-surfactants [
18]. These nanoemulsion preconcentrates or anhydrous nanoemulsion systems instantaneously emulsify, when dispersed in an aqueous medium under mild agitation to form o/w nanoemulsion with globule size < 100 nm [
19]. The spontaneous emulsification process eventually reduces particle size, maximizing surface area hence resulting in enhancement of solubility, dissolution, and bioavailability. Furthermore, these lipid formulations in a liquid state can be readily converted into solid-state powder, granules, beads, and pellets by adsorbing on a solid carrier thereby enabling them to develop as unit dosage forms suitable for peroral administration [
20]. The major benefits of conversion to solid dosage forms are high dose precision, lowest variability, portability, stable and accurate dosing. Solidification of liquid SNEDDS (L-SNEDDS) provides additional benefits such as good thermodynamic stability thereby preventing supersaturation of drug [
21]. On the other hand, the nanoemulsions are thermodynamically unstable colloidal systems susceptible to time-bound physical stability problems similar to coarse emulsions viz., creaming, coalescence, and phase breaking [
22]. The stability of nanoemulsions is also influenced by the temperature as well as pH [
23]. It was reported that the chemical properties of various additives included in the formulation can potentially influence the stability as well as in vitro release profile of the nanoemulsions [
24].
In SNEDDS, suitable excipients such as oil, surfactant, and co-surfactant are chosen based on their ability to form nanoemulsion spontaneously in the gastrointestinal environment after oral administration [
25,
26]. Moreover, in situ solubilized drugs formed in the lumen of the GIT can be subsequently absorbed via the lymphatic system evading the hepatic first-pass metabolism [
27]. Adsorbing the L-SNEDDS in a suitable carrier can potentially convert the encapsulated drug molecule from crystalline to an amorphous state and hence an increase in the aqueous solubility of the drug [
28]. In contrast to other studies, the primary objective of the research was to conduct systematic and thorough investigations to examine the practical feasibility of SNEDDS to improve the oral bioavailability of sertraline. The liquid SNEDDS was later modified to solid SNEDDS (S-SNEDDS) and patient-friendly tablet dosage form based on a full-factorial statistical design of experiment technique for future clinical application via oral therapy. This novel methodology has the prospect of potential industrial scale-up due to the generation of flowable, coherent, and compactable powdered form, fewer manufacturing steps, and equipment, generally regarded as safe (GRAS) approved excipients, avoidance of organic solvents, and therefore less production expense. In addition, in vivo evaluation of selected formulation was studied in Wistar rats, and to assess the relative bioavailability, pharmacokinetic parameters were compared with pure sertraline.
2. Materials and Methods
2.1. Materials
Sertraline hydrochloride, polyvinylpyrrolidone K30, microcrystalline cellulose, croscarmellose sodium (Ac-Di-Sol®) were procured from Torrent Pharmaceuticals, Gandhinagar, India. Kollidon® VA 64 and Solutol HS 15 were received from BASF, Ludwigshafen, Germany. The β-cyclodextrin, castor oil, corn seed oil, crospovidone, glycerol monooleate, isopropyl myristate, L-hydroxypropyl cellulose, magnesium stearate, oleic acid, poloxamer 188, polyethylene glycol 200 (PEG 200), polyethylene glycol 400 (PEG 400), propylene glycol, sodium starch glycolate, sorbitan monolaurate (Span 20), sorbitan monoleate (Span 80), sunflower oil, and talc were commercially acquired from Central Drug House, Mumbai, India. Glycerol triacetate was purchased from HiMedia, Mumbai, India. LabrafacTM WL 1349 and Labrasol® were donated by Gattefosse, Saint-Priest Cedex, France. Lemon oil, orange oil, and coconut oil were procured from Astron Chemicals, Ahmedabad, India. Capryol® 90 and Cremophor® EL were donated by Piramal Healthcare, Mumbai, India. Soyabean oil, Tween 40, and Tween 80 were obtained from Chemdyes Corporation, Vadodara, India.
2.2. Quantification of Sertraline
Analysis of sertraline from various samples was carried out by minor modification of the previously reported high-performance liquid chromatography (HPLC) method [
29]. The system is comprised of the Shimadzu Prominence HPLC (DGU-20A5, Tokyo, Japan) attached with a monolithic C
18 HPLC column (Zorbax, 150 mm × 4.6 mm). The quantification of the analyte was carried out by connecting to a fluorescence detector maintained at an excitation wavelength of 260 nm and emission at 310 nm. The extraction of sertraline and subsequent analysis was performed using a solvent mixture constituted of acetonitrile and water (80:20%
v/
v) maintained at a flow of 1 mL/min. The volume of injection was set at 50 μL, and the retention time was noticed at 12.2 min. Linear regression analysis demonstrates acceptable linearity between the sertraline concentration of 5–500 ng/mL (r
2 = 0.9992). The method was validated as per ICH Q2 guidelines [
30], which demonstrated the limit of quantification (LOQ) and the limit of detection (LOD) as 7.90 ng/mL and 3.60 ng/mL, respectively. The coefficient of variation was estimated between 1.26–4.84% and the recovery of sertraline from plasma was found to be 96.35 ± 1.3%.
2.3. Liquid Self-Nanoemulsifying Drug Delivery System (L-SNEDDS)
2.3.1. Preliminary Studies for Components of L-SNEDDS
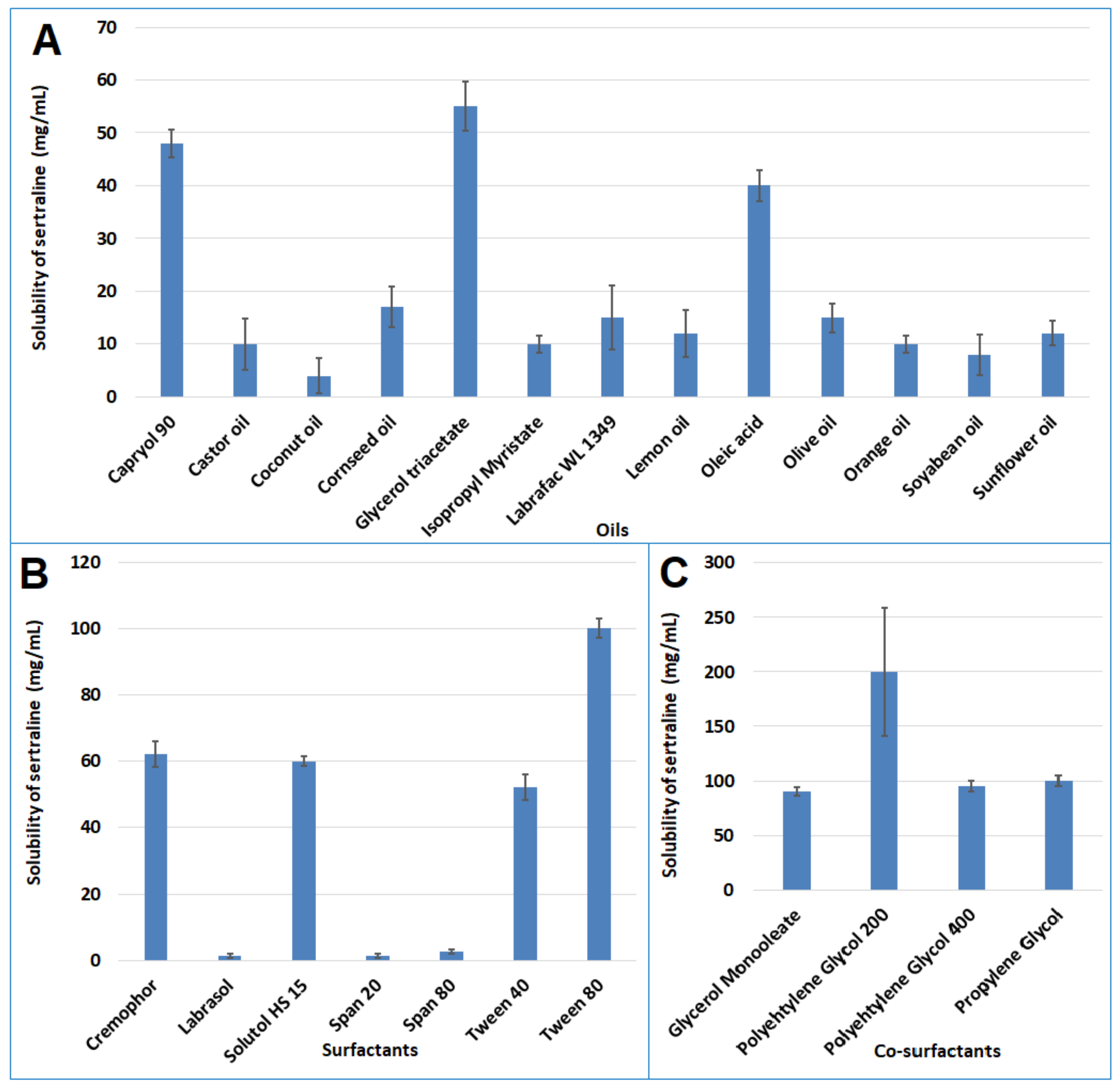
The solubility of sertraline in various oils (Capryol 90®, castor oil, coconut oil, corn seed oil, glycerol triacetate, isopropyl myristate, Labrafac™ WL 1349, lemon oil, oleic acid, olive oil, orange oil, soyabean oil, and sunflower oil), surfactants (Cremophor® EL, Labrasol®, solutol HS 15, Span 20, 80, Tween 40 and 80), and co-surfactants (glycerol monooleate, PEG 200, PEG 400, propylene glycol) was estimated by equilibrium solubility method. To experiment, an extra quantity of the drug was placed in vials holding 2 mL of each of the excipients. The vials were shaken using an orbital shaker at 40 rpm at 37 ± 0.2 °C for 72 h. After 72 h, the vials were centrifuged at 4000 rpm for 10 min, filtered, suitably diluted with methanol, and analyzed by HPLC.
2.3.2. Creation of Ternary Phase Diagram
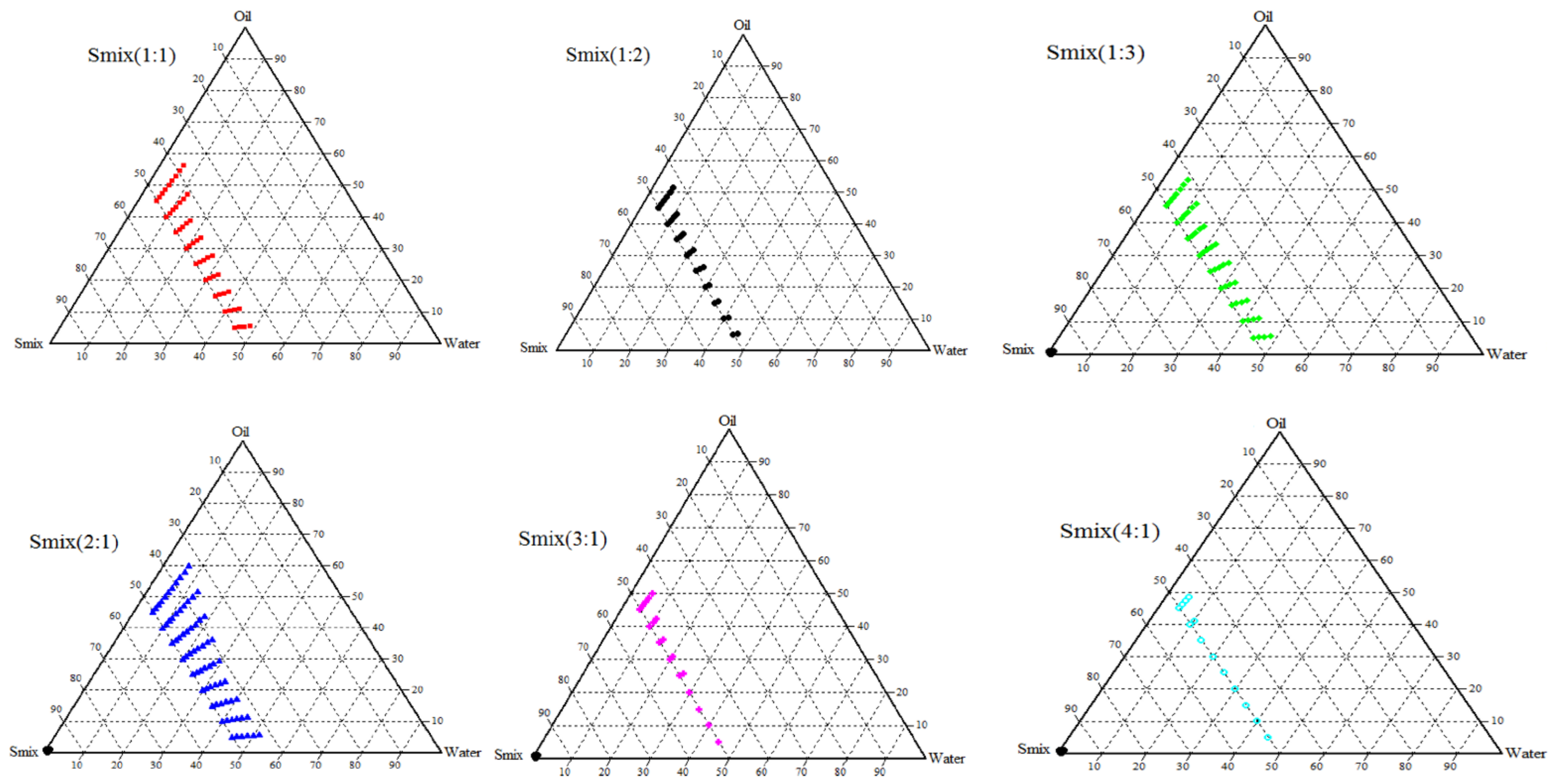
A ternary phase diagram was drawn using the titration method as mentioned elsewhere [
31]. The physical characteristics of the nanoemulsion were noted on the individual axis of the phase diagram depicting percentage water, oil, and S
mix at fixed weight ratios, respectively. To experiment, different ratios (1:1, 1:2, 1:3, 2:1, 3:1, 4:1) of S
mix combinations were prepared. A set of oil/water mixtures were prepared at all possible ratios (9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, 1:9) and titrated with S
mix in 5% increment up to 100% to establish the nanoemulsion region. The total volume of S
mix consumed was expressed as %
v/v and phase diagrams were created using Chemix School software (version 3.60, Bergen, Norway) to obtain the nanoemulsion region.
2.4. Thermodynamic Stability and Dispersibility Studies of L-SNEDDS Preparations
Thermodynamic stability studies were performed based on centrifugation test (5000 rpm for 30 min), heating-cooling cycle (45 °C and at 0 °C for 48 h), and freeze-thaw cycle (−21 °C and 21 °C for 24 h) [
32]. The lack of phase separation indicates the stability of the preparation. Dispersibility test which demonstrates the efficiency of self-emulsification tendency of SNEDDS was evaluated using a USP dissolution type II apparatus (Paddle), where 1 mL of SNEDDS was separately added to distilled water or 0.1 N HCl (500 mL) kept at 37 ± 0.5 °C and paddle rotated at 50 rpm [
33]. In vitro behavior of the preparations was visually checked based on the following grading system [
34]: (A) Spontaneous formation (<1 min) of nanoemulsion with a transparent or slight bluish look; (B) Spontaneously forming with low transparent microemulsion, with a bluish color; (C) Formation of milky emulsion (<2 min; (D) Dull, greyish white emulsion with somewhat oily appearance (>2 min); and (E), Formulation with low emulsification but had big oil globules appearing on top.
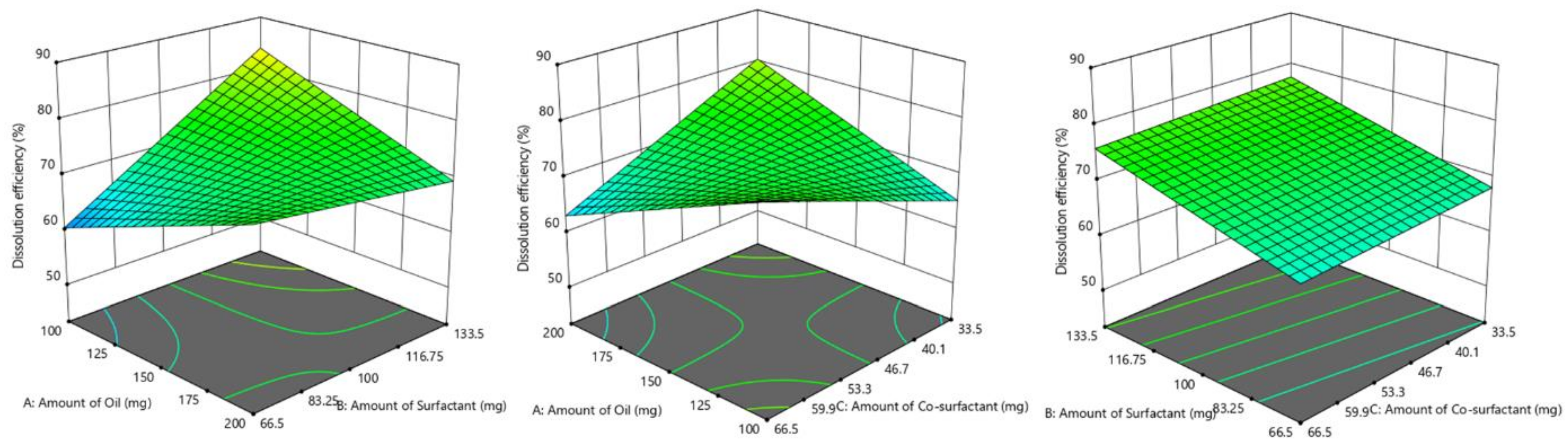
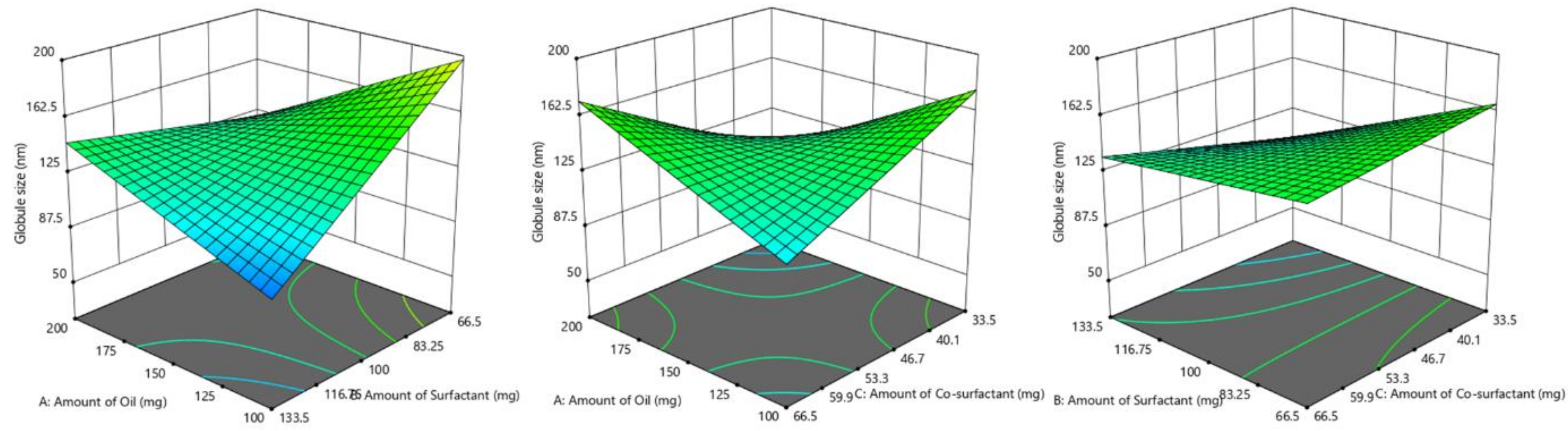
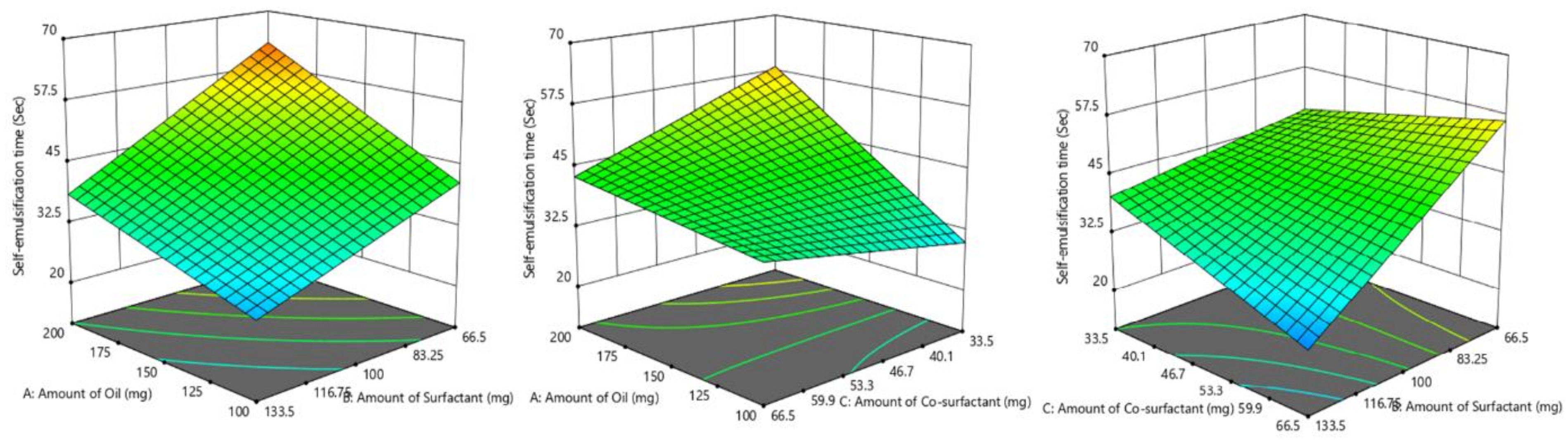
2.5. Screening of Formulations by Full Factorial Design
Based on the results of the ternary phase diagram and preliminary studies like screening of components, thermodynamic stability, and dispersibility studies, the independent variables with their effective concentration were identified. From the suitable statistical experimental designs, the full factorial design was selected. The L-SNEDDS were prepared based on the statistical design of the experiment applying a three-factor, two levels (2
3) full factorial design (FFD) using Design-Expert software (Stat-Ease, Version 12, Minneapolis, USA) by selecting the amount (milligram) of glycerol triacetate (X
1), Tween 80 (X
2) and PEG 200 (X
3) as independent variables, while dissolution efficiency % (Y
1), globule size in nm (Y
2) and self-emulsification time (SEF) in secs (Y
3) as responses and dependent variables (
Table 1).
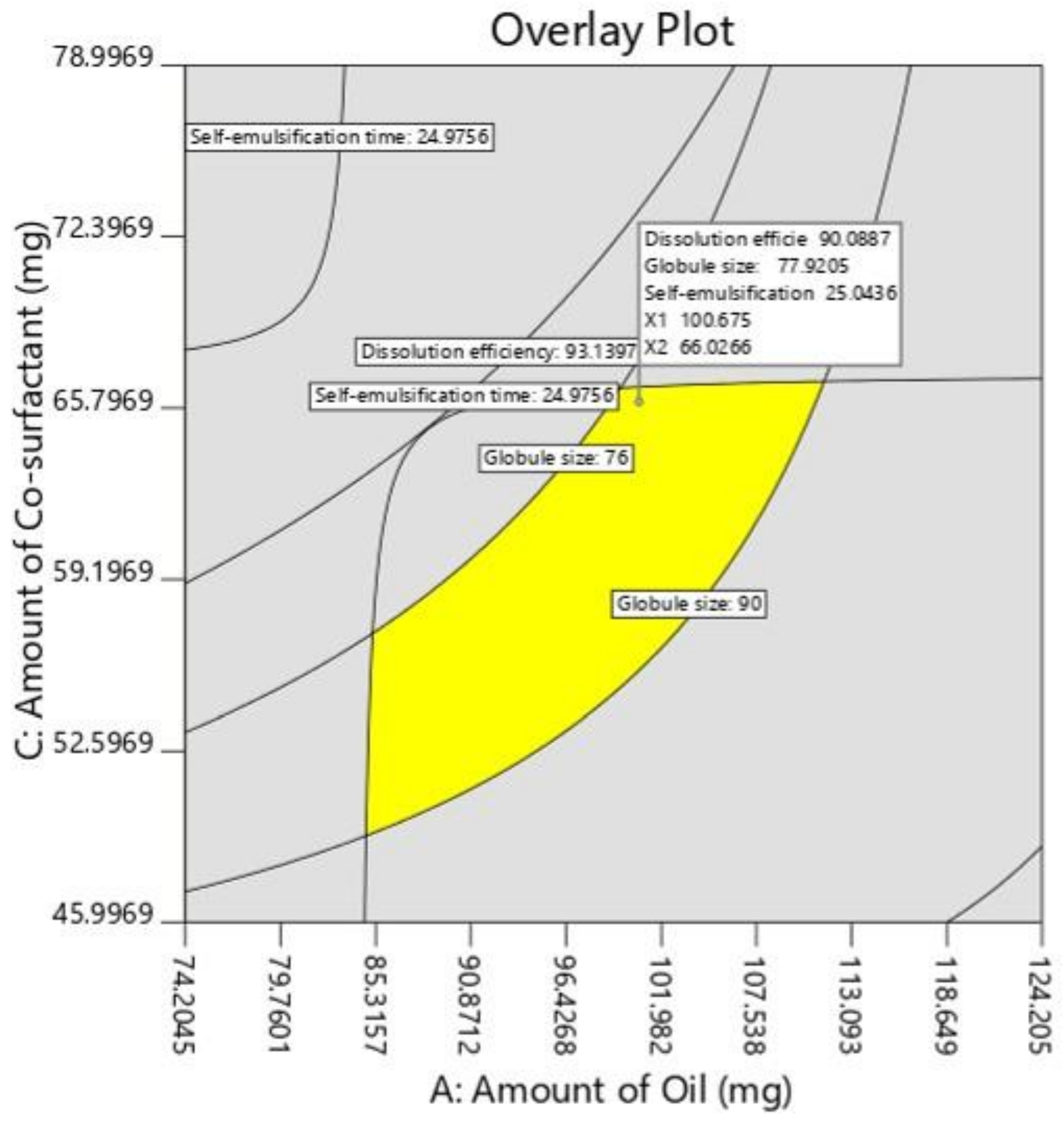
Response surface analyses were performed to find the influence of various independent factors on the observed dependent variables or responses. A set dose of sertraline hydrochloride (50 mg) was added to oil, Smix at room temperature under constant stirring in a vortex mixer to obtain a homogenous mixture. The responses were statistically analyzed utilizing the one-way ANOVA method. The statistical differences between data displaying p < 0.05 were selected as significant. The most favorable formulation was chosen by factorial design, which shows maximum % dissolution efficiency, low globule size, and less self-emulsification time. To evaluate the authenticity of the created mathematical model, the validation of the model was evaluated by checkpoint batch. For these formulations, all the three dependent variables (Y1–Y3) were evaluated as per established mathematical models and experimental techniques.
2.6. Characterization of Designed Batches of L-SNEDDS
2.6.1. Determination of Dissolution Efficiency
A dissolution efficiency test was performed in simulated intestinal fluid (250 mL, pH 6.8) with 10% Tween 80 to achieve sink condition and the dissolution medium was set at 37 ± 0.5 °C and agitated at 100 rpm as presented in
Figure 1. The SNEDDS formulation (1 mL) was placed in a previously hydrated dialysis bag (Spectra/Por 4, diameter 25 mm, MW cut-off: 12,000–14,000 Spectrum Inc., Los Angeles, CA, USA). Aliquot volume (1 mL) of the samples was removed at regular time points (5, 10, 15, 30, 45, and 60 min) and an equivalent amount of medium was replaced. The samples taken were diluted suitably, filtered through a 0.2 μm Millex syringe-driven filter unit, and analyzed by HPLC. The dissolution efficiency was calculated according to the equation described below [
35].
where, the dissolution efficiency is the area under the dissolution curve between time points t
1 and t
2 expressed as a percentage of the curve at maximum dissolution; y100, over the same period or the area under the dissolution curve up to a certain time; t, expressed as a percentage of the area of the rectangle described by 100% dissolution in the same time.
2.6.2. Globule Size
To carry out the investigation, 1 mL of L-SNEDDS was placed in a vial and diluted with 20 mL of water. The vial was mildly mixed to generate a fine emulsion and kept for 12 h at room temperature (25 ± 1 °C). The globule size of the nanoemulsion was measured employing a Malvern particle size analyzer (Nano ZS90, Malvern Instruments Ltd., Worcestershire, UK).
2.6.3. Determination of Self-Emulsification Time
To carry out the test, 1 mL from an individual L-SNEDDS preparation was dropped into 500 mL of distilled water taken in a glass beaker and maintained at a temperature of 37 ± 0.5 °C under mild agitation (50 rpm) using a magnetic mixer. The self-emulsification process was visually checked and recorded for the rate of emulsification and subsequent formation of nanoemulsion.
2.6.4. Determination of Viscosity, Zeta Potential, Percentage Transmittance
The viscosity of the undiluted batch (F4) nanoemulsion (0.25 g) was measured using a Brookfield viscometer (LVDVI prime, Middleborough, USA) at room temperature. The zeta potential, as well as polydispersity index, were measured employing a Malvern particle size analyzer (Nano ZS90, Malvern Instruments Ltd., Malvern, Worcestershire, UK). The optical clarity of the emulsion upon dilution was measured as percentage transmittance against double distilled as blank using UV-spectrophotometer (model UV-1800, Shimadzu Corporation, Kyoto, Japan) at 500 nm [
36].
2.7. Solidification of L-SNEDDS
Solidification of selected L-SNEDDS (Batch F4) was done by the solid carrier adsorption method. Briefly, 20 g of L-SNEDDS formulation was blended with 24 g of microcrystalline cellulose to obtain a wet mixture. Later, 6 g of Aerosil 200 was added to the wet mixture and mixed to obtain S-SNEDDS.
Flowability and Compressibility
The flow property of S-SNEDDS was estimated by the conventional static angle of repose (θ) method using Flodex apparatus (Erweka, Heusenstamm, Germany) according to the standard formulae [
37]. Similarly, percentage compressibility (Carr’s index) of powder was determined utilizing tap density apparatus (Labindia, Mumbai, India) according to the formulae mentioned in other studies [
38]. The flowability of the powder was also interpreted with Hausner’s ratio using the standard equation: Tapped density/bulk density.
2.8. Tablet Preparation of S-SNEDDS
For the preparation of S-SNEDDS loaded tablets, listed ingredients (
Table 2) were weighed accurately and sifted through sieve number 60. The ingredients were mixed until a uniform mixture was obtained and was again sieved. Lubricant and glidant were added, mixed and the mixture was directly compressed using a 16-station punching machine (Cadmach CMD4, Ahmedabad, India).
2.9. Characterization of S-SNEDDS Loaded Tablets
2.9.1. Thickness and Hardness
The thickness, as well as hardness of prepared tablets, were measured by Vernier caliper (1P-67, Mitutoyo, Tokyo, Japan), and tablet hardness tester (125 Series, Erweka Gmbh, Germany), respectively, using three tablets, and an average value was calculated [
39].
2.9.2. Friability
The friability of prepared formulations was determined by adding pre-weighed tablets in a Roche friabilator and was allowed to revolve (rotates at 25 ± 1 rpm) for one hundred times according to USP. The same tablets were weighed again after removing dust and calculated the percentage friability [
36].
2.9.3. Disintegration
The disintegration time (min) of the tablet was measured employing USP disintegration test apparatus (DT 1000, Labindia, Mumbai, India) in water at 37 ± 2 °C. The time at which the tablet disintegrates completely was noted down and the average time was calculated.
2.9.4. Drug Content
To analyze the drug content, ten tablets were pulverized and the quantity of powder proportionate to 50 mg of sertraline hydrochloride was taken and dispersed in methanol by stirring for 10 min. The solution was filtered using a 0.2 μm filter and assayed by HPLC. The average percentage of drug content was estimated and compared with the total dose.
2.9.5. Drug Release
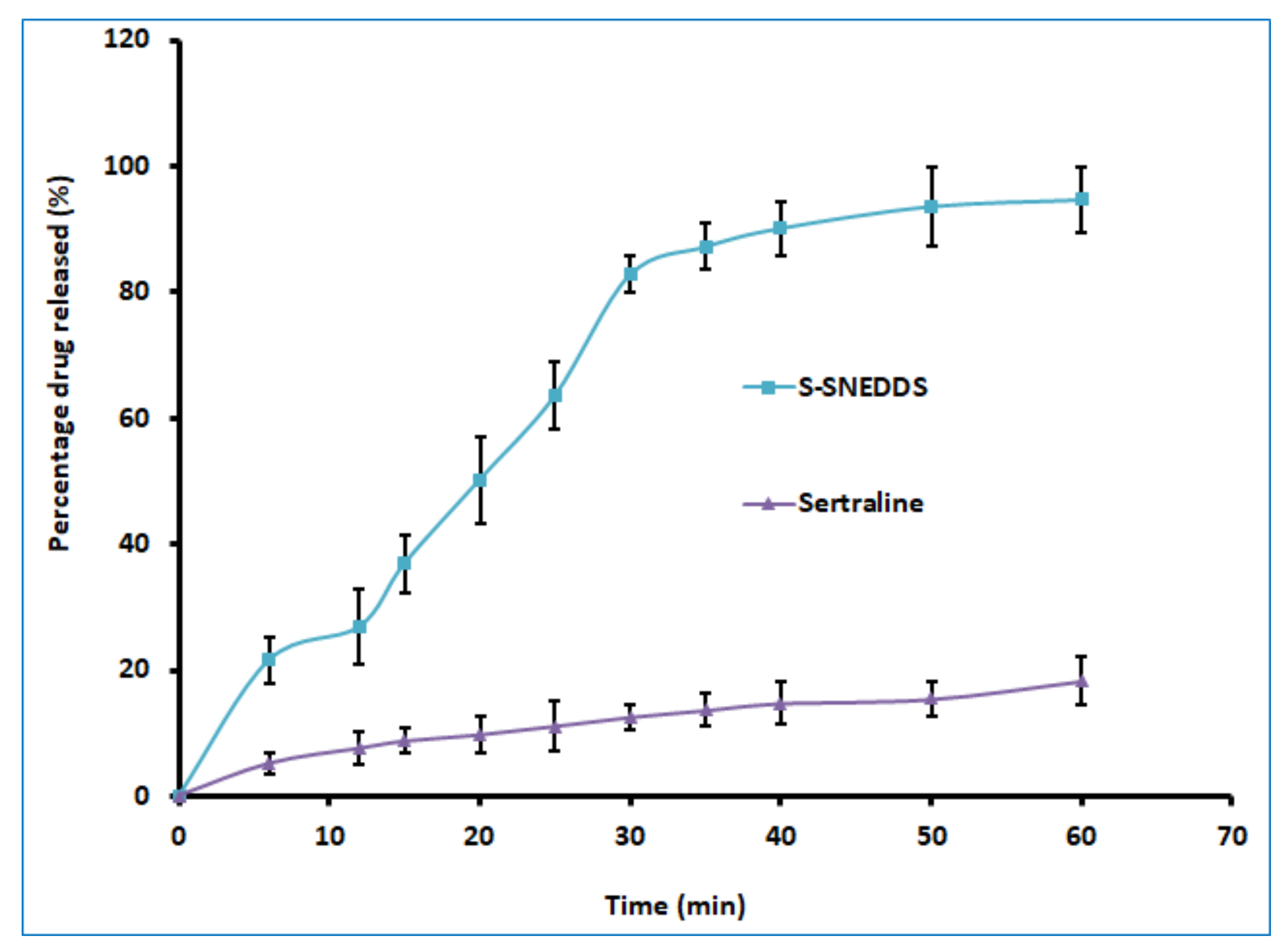
The percentage release rate of the S-SNEDDS loaded tablet and the pure drug was estimated employing USP dissolution testing apparatus type II (Labindia, Mumbai, India) operated at 75 rpm. The dissolution test was carried out in simulated intestinal fluid (900 mL; pH 6.8) [
40] with 10% Tween 80 to maintain sink condition, and the temperature of the dissolution medium was set at 37 ± 0.5 °C using a thermostatically controlled water bath. The selected formulation of sertraline L-SNEDDS, S-SNEDDS, and plain drug equivalent to 50 mg sertraline were used for the comparative evaluation of dissolution data. Aliquot volume (5 mL) of the sample was withdrawn at specified time intervals of 6, 12, 15, 20, 25, 30, 35, 40, 50, and 60 min, and replacements were made with 5 mL of release medium. Each sample was filtered using a 0.2 μm filter and analyzed for sertraline content by HPLC.
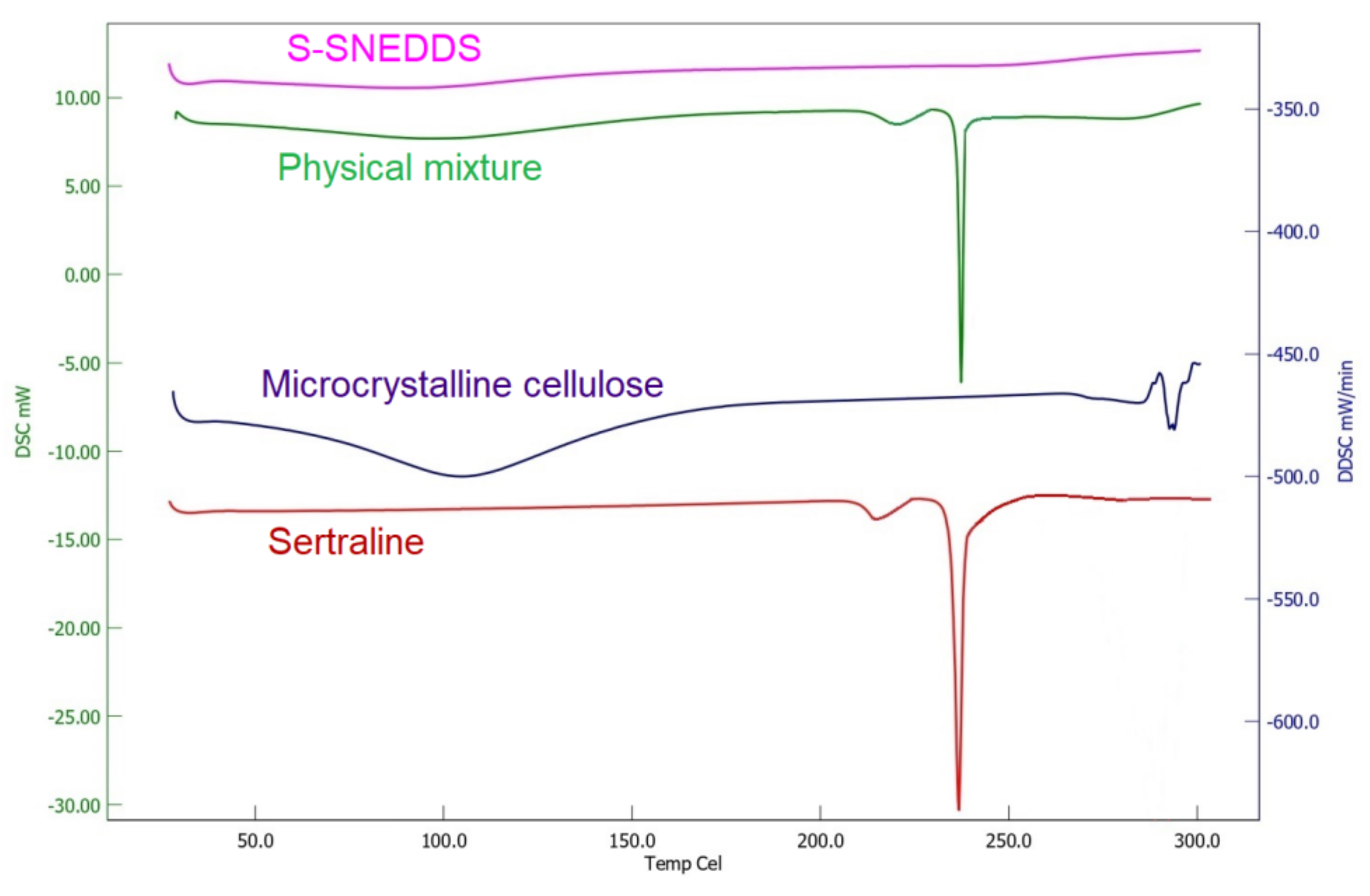
2.9.6. Differential Scanning Calorimetry (DSC)
DSC curves of the pure sertraline hydrochloride, microcrystalline cellulose, physical mixture, and S-SNEDDS were obtained by employing a scanning calorimeter furnished with a thermal analysis data system (DSC 60 Shimadzu, Tokyo, Japan). Thermal scanning of the samples placed in hermetically sealed pans was carried out at a temperature ranging between 10 to 300 °C at a heating rate of 20 °C/min using a blank aluminum pan as the reference standard [
41].
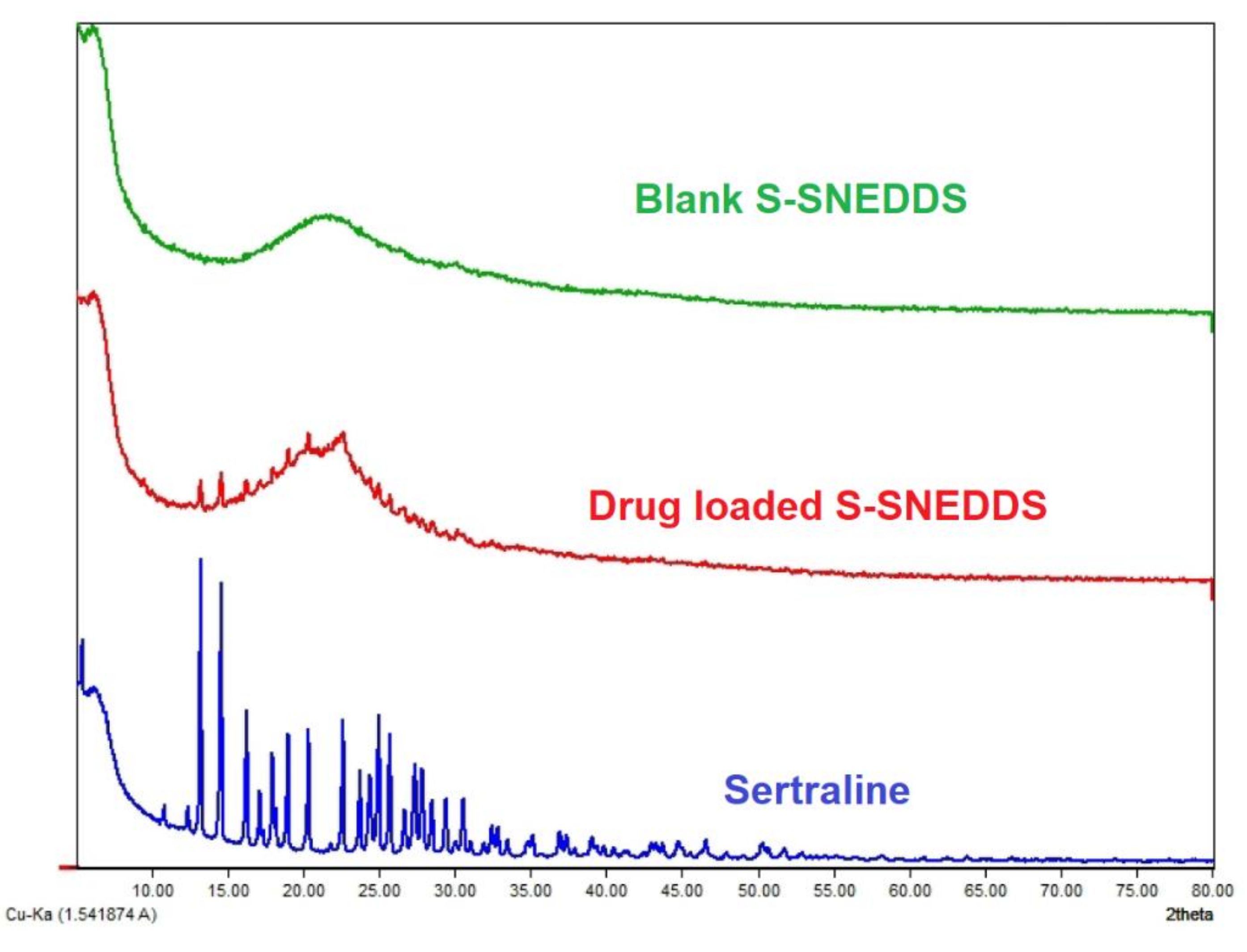
2.9.7. X-ray Diffraction (XRD)
Diffraction patterns exploring the physical state of the sertraline, blank S-SNEDDS, and drug-loaded S-SNEDDS were studied with the utilization of Bruker AXS D8 Focus P-XRD (Billerica, MA, USA). The drug or formulations were powdered separately and placed in an aluminum holder. The spectral scanning was carried using CU Kα radiation at a similar voltage and current (40 mA) between the range of 2θ angles from 10–40°, with a slow angle scan of 0.01°/min at a sampling interval of 0.02°/s [
42].
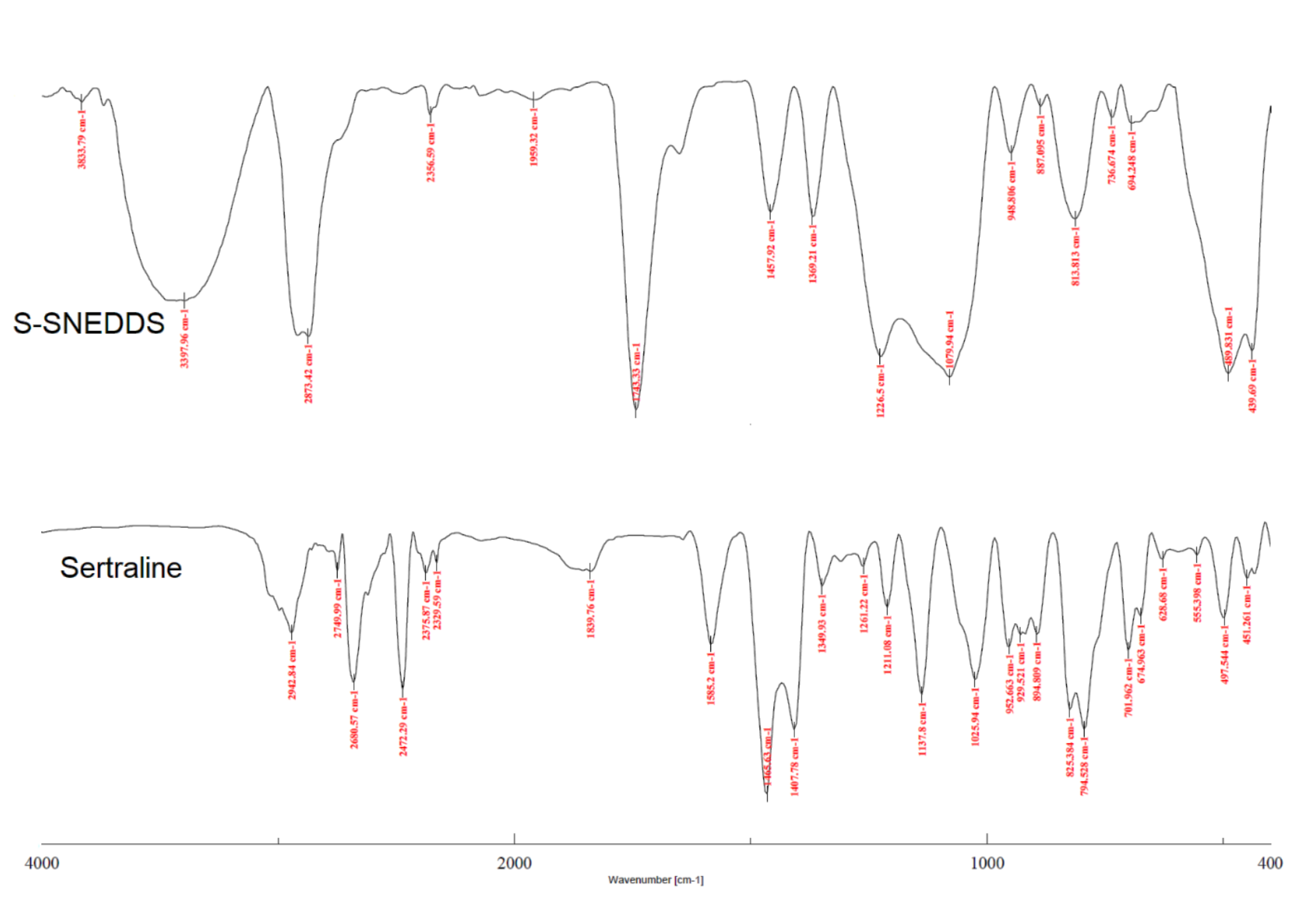
2.9.8. Fourier Transform Infrared (FTIR)
FTIR spectra of powder samples of pure sertraline and S-SNEDDS were recorded on a Spectrum-GX FTIR spectrophotometer (Perkin Elmer, Waltham, MA, USA). Samples were compressed with potassium bromide (1:10 ratio) at 1 ton/cm
2 to obtain the disc by employing a hydraulic punching machine [
43]. The spectral scanning was performed between 4000–400 cm
−1.
2.9.9. Scanning Electron Microscopy (SEM)
The morphological characteristics of the powder sample were recorded using an SEM (ESEM EDAX XL-30, Philips, Eindhoven, The Netherlands). Before observation, samples were retained on an aluminum dock using a double-sided adhesive tape which was further coated with gold (~20 nm) to make it electrically conductive in a vacuum. The scanning operation of the SEM was conducted at an acceleration voltage of 15 kV.
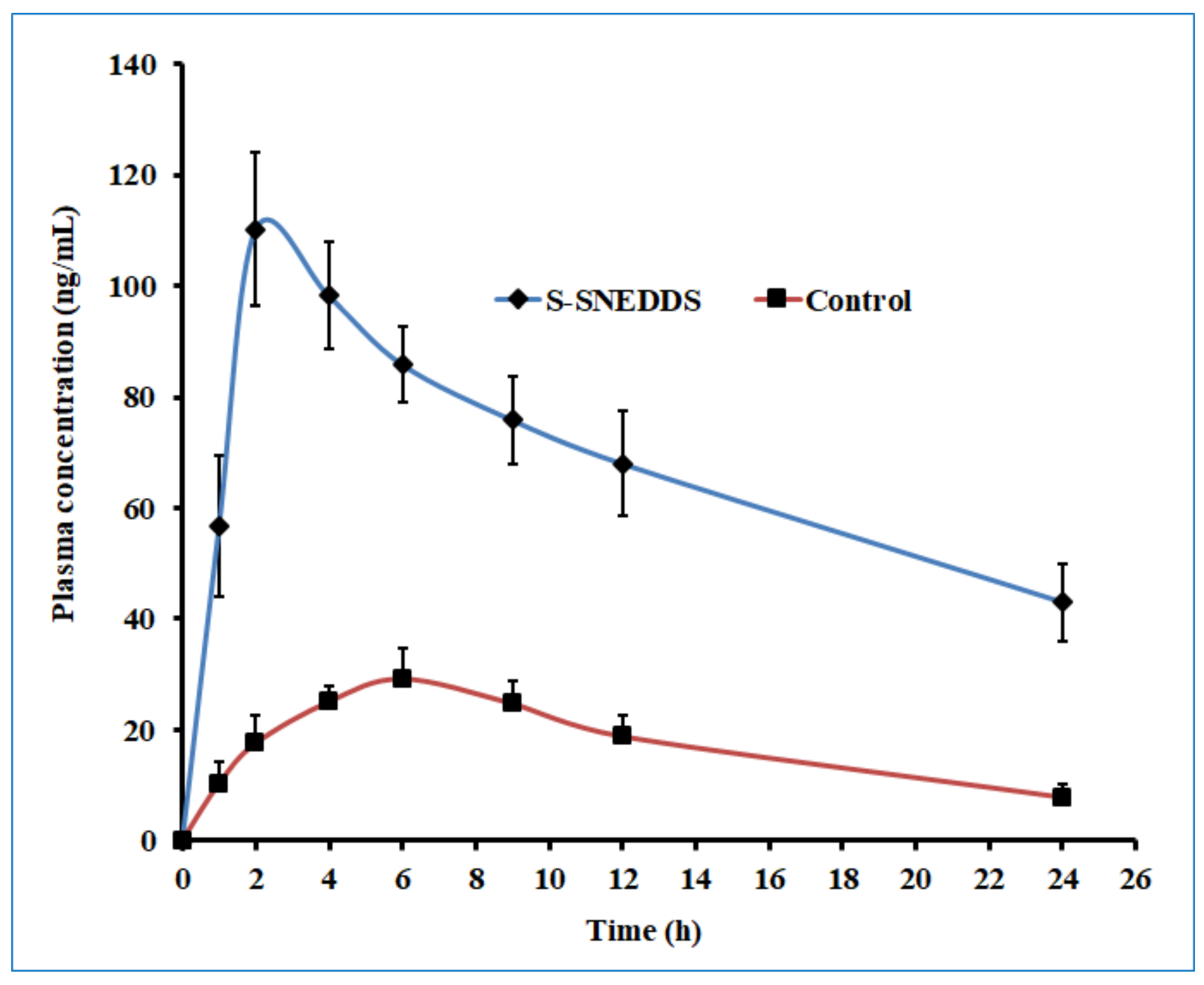
2.10. Oral Bioavailability Studies
The pharmacokinetic evaluation of sertraline was investigated on male albino Wistar rats (200–250 g) to assess the oral bioavailability differences between the S-SNEDDS and pure drug (control). Animals were caged individually in a well-ventilated room that was maintained at constant temperature (20–24 °C), specified photoperiod (12-h light/12-h dark cycle), and unrestricted access to food and water. Animals fasted for 12 h were classified into two groups (group I and II), each consisting of six rats. Animal experiments were performed following the institutional ethical committee guidelines of animal care (Protocol No. IP/PCEU/FAC/29/2021/40; dated 18/09/2021). A dose equivalent to 5 mg/kg of sertraline or S-SNEDDS was administered as calculated from the standard daily human dose of 50 mg utilizing the equation recommended in the literature [
44]. The formulation was prepared as a suspension in 0.5%
w/v carboxymethyl cellulose and administered to rats orally as a single dose by intragastric gavage in both groups. A blood sample (~200 µL) was drawn at predefined time points (1, 2, 4, 6, 9, 12, and 24 h) post-dosing from retro-orbital plexus of individual rat under anesthesia using isoflurane [
45]. Samples were collected in heparin pre-coated tubes and proteins were subsequently precipitated with a similar volume of acetonitrile [
46]. It was then centrifuged at 12,000 rpm for 15 min and the supernatant fraction was membrane filtered (0.2 μm). The filtrate (50 μL) was injected into the HPLC system whilst the sample taken at zero time was considered as the baseline value during analysis. The pharmacokinetic parameters of interest included area under the concentration-time curve (AUC
0–
t), peak concentration (C
max), and time to reach peak concentration (T
max) and were determined by non-compartmental analysis described elsewhere [
45].
4. Conclusions
To improve the solubility of the poorly water-soluble drug, sertraline, the S-SNEDDS approach was employed. Initially, screening of nanoemulsion formulation was carried out by examining the influence of the quantity of oil, surfactant, and co-surfactant on dissolution efficiency, globule size, and self-emulsification time. The selected liquid SNEDDS formulation (batch F4) was converted to a flowable powder using microcrystalline cellulose as a solid carrier and Aerosil 200 as coating material. The powder was compressed into tablet dosage to integrate the bioavailability enhancement of sertraline besides solving the formulation challenges typically associated with liquid lipid formulations. A rapid and complete drug release noticed in S-SNEDDS formulation met the acceptance criteria specified in the USP dissolution specification guidance for immediate release dosage forms. DSC and XRD data signify the complete transformation of crystalline sertraline in the prepared formulation, which might be contributing towards the improvement of solubility and thereby dissolution enhancement of the drug. The FTIR study confirms no interaction between the drug and excipients used. In vivo data demonstrate a significant (p < 0.0001) difference in the Cmax, and AUC0-α by prepared S-SNEDDS as compared to control, indicating improvement in oral absorption of sertraline. In conclusion, the results obtained here demonstrated the feasibility of S-SNEDDS to enhance the oral bioavailability and thereby the clinical efficacy of sertraline.


 ,
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}