
Development of Extended-Release Mini-Tablets Containing Metoprolol Supported by Design of Experiments and Physiologically Based Biopharmaceutics Modeling

 ,
, 

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Mini-Tablets
2.2.1. Characterization of Mini-Tablets
2.2.2. Coating of Mini-Tablets
2.3. In Vitro Studies
2.3.1. Dissolution Method
2.3.2. Drug Quantification in the Dissolution Test
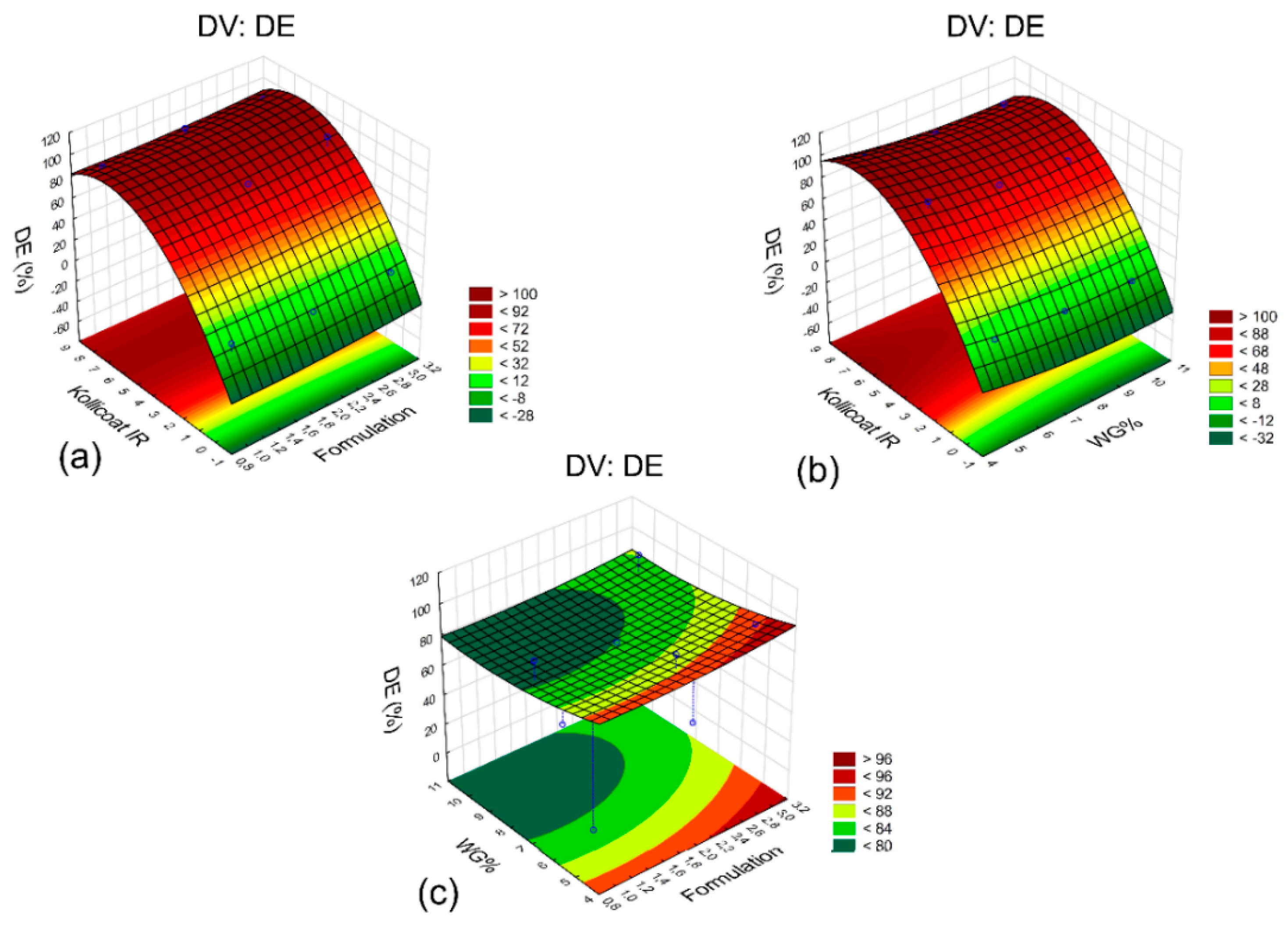
2.4. Statistical Analysis of the Design of Experiments
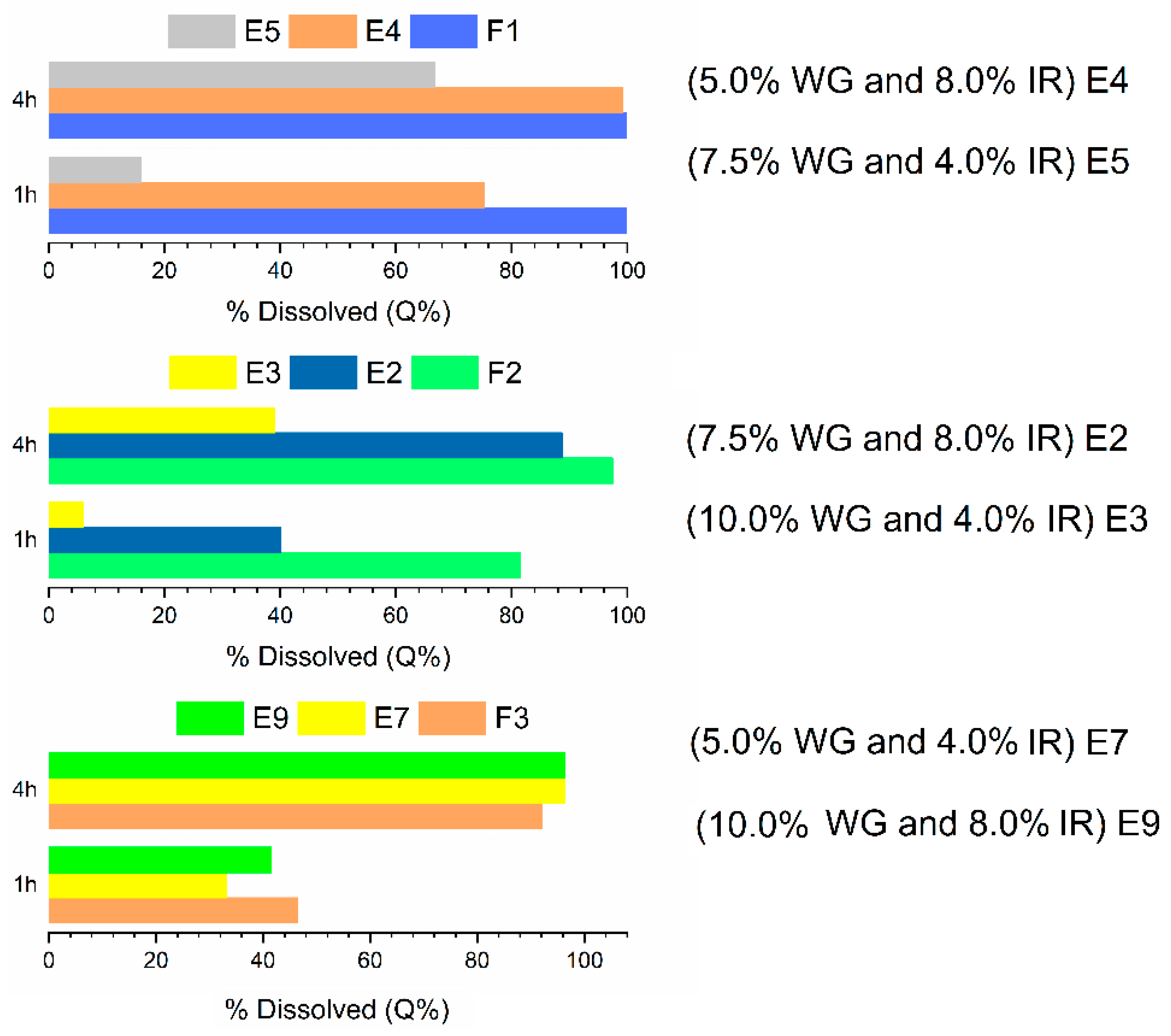
2.5. Additional Formulations
2.6. Formulation Optimization
- Q%Mtc: percentage dissolved of the mixture at its collection time (tc);
- Q%F1tc: percentage dissolved of formulation F1 at time tc;
- Q%E11tc: percentage dissolved of E11 formulation at time tc;
- Q%E12tc: percentage dissolved of E12 formulation at time tc;
- n: total amount of mini-tablets;
- nF1: number of mini-tablets of the formulation F1;
- nE11: number of mini-tablets of the formulation E11;
- nE12: number of mini-tablets of the formulation E12.
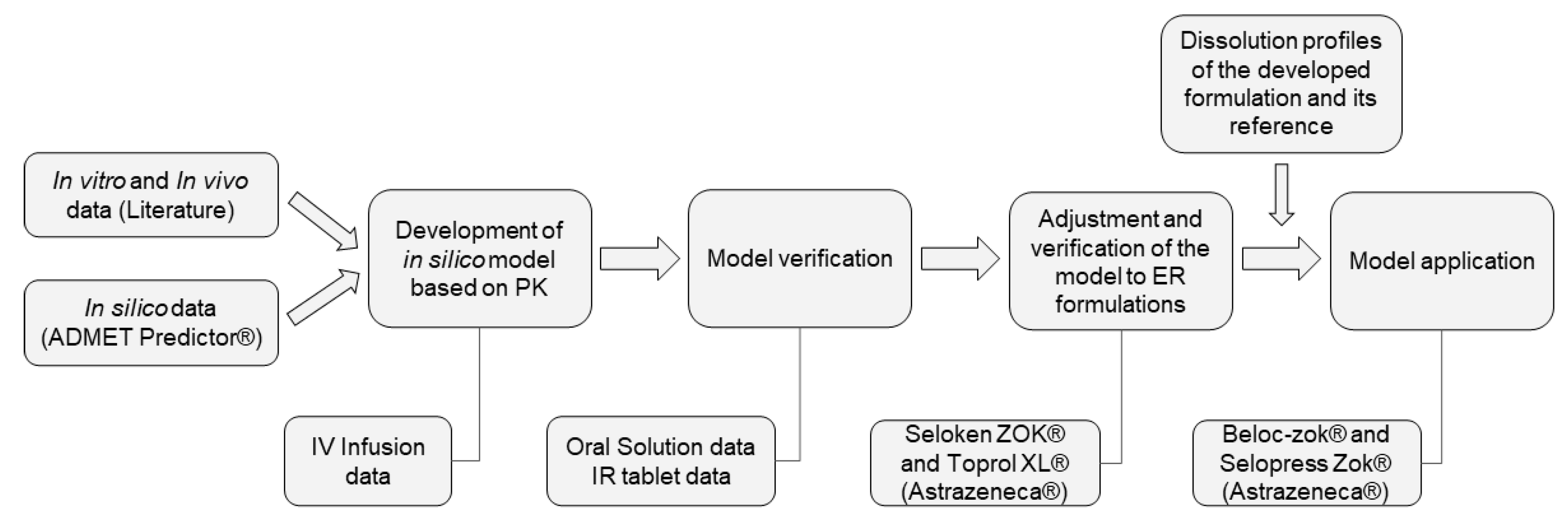
2.7. Development, Verification, and Application of a Pharmacokinetic Model for Metoprolol
2.7.1. Development and Verification of the Pharmacokinetic Model
2.7.2. Model Verification and Adjustments to ER Formulations
2.7.3. Evaluation of the Predictability of the Model
- = average of the parameter;
- = number of subjects.
2.7.4. Application of the PK Model to the Developed Formulation
2.8. Virtual Bioequivalence Study
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parrott, N.; Suarez-Sharp, S.; Kesisoglou, F.; Pathak, S.M.; Good, D.; Wagner, C.; Dallmann, A.; Mullin, J.; Patel, N.; Riedmaier, A.E.; et al. Best Practices in the Development and Validation of Physiologically Based Biopharmaceutics Modeling. A Workshop Summary Report. J. Pharm. Sci. 2021, 110, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Almukainzi, M.; Okumu, A.; Wei, H.; Löbenberg, R. Simulation of in vitro dissolution behavior using DDDPlus™. AAPS PharMSiTech 2015, 16, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandner, P.; Ziegelbauer, K. Product-related research: How research can contribute to successful life-cycle management. Drug Discov. Today 2008, 13, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.; Noack, R.M. Application of design of experiments to pharmaceutical formulation development. Int. J. Exp. Des. Process Optim. 2011, 2, 58–65. [Google Scholar] [CrossRef]
- Aleksovski, A.; Lustrik, M.; Sibanc, R.; Dreu, R. Design and evaluation of a specific, bi-phase extended release system based on differently coated mini-tablets. Eur. J. Pharm. Sci. 2015, 75, 114–122. [Google Scholar] [CrossRef]
- Mohamed, F.A.A.; Roberts, M.; Seton, L.; Ford, J.L.; Levina, M.; Rajabi-Siahboomi, A.R. Film-coated matrix mini-tablets for the extended release of a water-soluble drug. Drug Dev. Ind. Pharm. 2015, 41, 623–630. [Google Scholar] [CrossRef]
- Tissen, C.; Woertz, K.; Breitkreutz, J.; Kleinebudde, P. Development of mini-tablets with 1 mm and 2 mm diameter. Int. J. Pharm. 2011, 416, 164–170. [Google Scholar] [CrossRef]
- Zerbini, A.P.N.A.; Ferraz, H.G. Sistemas multiparticulados: Minicomprimidos. Rev. Bras. Cienc. Farm. Basic Appl. 2011, 32, 149–158. [Google Scholar]
- Ajam, T.; Ajam, S.; Devaraj, S.; Mohammed, K.; Sawada, S.; Kamalesh, M. Effect of carvedilol vs metoprolol succinate on mortality in heart failure with reduced ejection fraction. Am. Heart J. 2018, 199, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Rigby, J.W.; Scott, A.K.; Hawksworth, G.M.; Petrie, J.C. A comparison of the pharmacokinetics of atenolol, metoprolol, oxprenolol and propranolol in elderly hypertensive and young healthy subjects. Brit. J. Clin. Pharmacol. 1985, 20, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Bermejo, M.; Hens, B.; Dickens, J.; Mudie, D.; Paixão, P.; Tsume, Y.; Shedden, K.; Amidon, G.L. A mechanistic physiologically-based biopharmaceutics modeling (PBBM) approach to assess the in vivo performance of an orally administered drug product: Form IVIVC to IVIVP. Pharmaceutics 2020, 12, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckernäs, E.; Tannergren, C. Physiologically based biopharmaceutics modeling of regional and colon absorption in dogs. Mol. Pharm. 2021, 18, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Yang, H.; Fang, L.; Gonzalez-Sales, M.; Zhao, L.; Trame, M.N.; Lesko, L.; Schmidt, S. Physiologically based pharmacokinetic modeling to evaluate formulation factors influencing bioequivalence of metoprolol extended-release products. J. Clin. Pharmacol. 2019, 59, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.G.; Pessole, L.; Takahashi, A.I.; Andréo Filho, N.; Ferraz, H.G. Physicochemical and dissolution profile characterization of pellets containing different binders obtained by the extrusion-spheronization process. Braz. J. Pharm. Sci. 2012, 48, 379–388. [Google Scholar] [CrossRef] [Green Version]
- FDA-Recommended Dissolution Methods. U.S. Department of Health and Human Services. Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Updated 31 October 2013. Available online: https://www.accessdata.fda.gov/scripts/cder/dissolution/dsp_getallData.cfm (accessed on 23 August 2021).
- Kannan, K.; Manikandan, M.; Periyasamy, G.; Manavalan, R. Design, development and evaluation of metoprolol succinate and hydrochlorothiazide bilayer tablets. J. Pharm. Sci. Res. 2012, 4, 1827–1835. [Google Scholar]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Regårdh, C.G.; Borg, K.O.; Johansson, R.; Johnsson, G.; Palmer, L. Pharmacokinetic studies on the selective beta1-receptor antagonist metoprolol in man. J. Pharmacokinet. Biopharm. 1974, 2, 347–364. [Google Scholar] [CrossRef]
- Wang, P.-H.; Lien, E.J. Effects of different buffer species on partition coefficients of drugs used in quantitative structure-activity relationships. J. Pharm. Sci. 1980, 69, 662–668. [Google Scholar] [CrossRef]
- Tubic-Grozdanis, M.; Bolger, M.B.; Langguth, P. Application of gastrointestinal simulation for extensions for biowaivers of highly permeable compounds. AAPS J. 2008, 10, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Kasim, N.A.; Whitehouse, M.; Ramachandran, C.; Bermejo, M.; Lennernäs, H.; Hussain, A.S.; Junginger, H.E.; Stavchansky, S.A.; Midha, K.K.; Shah, V.P.; et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [CrossRef]
- Lukacova, V.; Woltosz, W.S.; Bolger, M.B. Prediction of modified release pharmacokinetics and pharmacodynamics from in vitro, immediate release, and intravenous data. AAPS J. 2009, 11, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Eddington, N.D.; Marroum, P.; Uppoor, R.; Hussain, A.; Augsburger, L. Development and internal validation of an in vitro-in vivo correlation for a hydrophilic metoprolol tartrate extended release tablet formulation. Pharm. Res. 1998, 15, 466–473. [Google Scholar] [CrossRef] [PubMed]
- SÚLK. State Institute for Drug Control, Public Assessment Report for Paediatric Studies Submitted in Accordance with Article 45 of Regulation (EC) No1901/2006, Metoprolol Succinate, Selokeen ZOK Controlled Release Tablets, NL/W/0037/pdWS/001. 2013. Available online: https://www.sukl.cz>file>85715_1_1 (accessed on 11 August 2021).
- FDA. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Application Number ANDA 076640, Bioequivalence Reviews. 2006. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/076640Orig1s000BioeqR.pdf (accessed on 24 August 2021).
- Ravishankar, H.; Patil, P.; Samel, A.; Petereit, H.U.; Lizio, R.; Iyer-Chavan, J. Modulated release metoprolol succinate formulation based on ionic interactions: In vivo proof of concept. J. Control. Release 2006, 111, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, V.; Arora, S.; Tamás Katona, M.; Modhave, D.; Fröhlich, E.; Paudel, A. On absorption modeling and food effect prediction of rivaroxaban, a SCB II drug orally administered as an immediate-release tablet. Pharmaceutics 2021, 13, 283. [Google Scholar] [CrossRef]
- Abduljalil, K.; Cain, T.; Humphries, H.; Rostami-Hodjegan, A. Deciding on success criteria for predictability of pharmacokinetic parameters from in vitro studies: An analysis based on in vivo observations. Drug Metab. Dispos. 2014, 42, 1478–1484. [Google Scholar] [CrossRef]
- Lopes, C.M.; Lobo, J.M.S.; Pinto, J.F.; Costa, P. Compressed mini-tablets as a biphasic delivery system. Int. J. Pharm. 2006, 323, 93–100. [Google Scholar] [CrossRef]
- Lopes, C.M.; Lobo, J.M.S.; Costa, P.; Pinto, J.F. Directly compressed mini matrix tablets containing ibuprofen. Preparation and evaluation of sustained release. Drug Dev. Ind. Pharm. 2006, 32, 95–106. [Google Scholar] [CrossRef]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders: Surface are, Pore Size and Density; Kluwer Academic Publisher: Dordrecht, The Netherlands, 2004; p. 347. [Google Scholar]
- Rowe, R.C.; Sheskey, P.J.; Owen, S.C. Handbook of Pharmaceutical Excipients, 5th ed.; Pharmaceutical Press: London, UK, 2006; p. 918. [Google Scholar]
- Nokhodchi, A.; Ford, J.L.; Rowe, P.H.; Rubistein, M.H. The effects of compression rate and force on the compaction properties of different viscosity grades of hydroxypropylmethylcellulose. Int. J. Pharm. 1996, 129, 21–31. [Google Scholar] [CrossRef]
- Bühler, V. Kollidon® Polyninylpyrrolidone Excipients for the Pharmaceutical Industry, 9th ed.; Basf SE: Ludwigshafen, Germany, 2008; p. 326. [Google Scholar]
- Calado, V.; Montgomery, D.C. Planejamento de Experimentos Usando o Statistica; E-Papers Serviços Editoriais: Rio de Janeiro, Brazil, 2003; p. 260. [Google Scholar]
- Vueba, M.L.; Carvalho, L.A.E.B.; Sousa, F.V.J.J.; Pina, M.E. In vitro release of ketoprofen from hydrophilic matrix tablets containing cellulose polymer mixtures. Drug. Dev. Ind. Pharm. 2013, 39, 1651–1662. [Google Scholar] [CrossRef]
- USP, United States Pharmacopeia, 34th ed.; United States Pharmacopeial Convention: Rockville, MD, USA, 2011.
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Greb, E. The hour of the particle. Pharm. Technol. 2010, 34, 38–42. [Google Scholar]
- Duque, M.D.; Kreidel, R.N.; Taqueda, M.E.S.; Baby, A.R.; Kaneko, T.M.; Velasco, M.V.R.; Consiglieri, V.O. Optimization of primaquine diphosphate tablet formulation for controlled release using the mixture experimental design. Pharm. Dev. Technol. 2013, 18, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Paterakis, P.G.; Korakianiti, E.S.; Dallas, P.P.; Rekkas, D.M. Evaluation and simultaneous optimization of some pellets characteristics using a 33 factorial design and a desirability function. Int. J. Pharm. 2002, 248, 51–60. [Google Scholar] [CrossRef]
- Zuo, J.; Gao, Y.; Bou-Chacra, N.; Löbenberg, R. Evaluation of the DDSolver software applications. BioMed Res. Int. 2014, 2014, 204925. [Google Scholar] [CrossRef] [Green Version]
- Thummel, K.E.; Shen, D.D.; Isoherranen, N. Design and optimization of dosage regimens: Pharmacokinetic date. In Goodmann & Gilman’s the Pharmacological Basis of Therapeutics, 12th ed.; Brunton, L.L., Chabner, B.A., Knollmann, B.C., Eds.; Table AII-1; McGraw Hill Medical: New York, NY, USA, 2011; p. 1953. [Google Scholar]
- Sirisuth, N.; Eddington, N.D. The influence of first pass metabolism on the development and validation of an IVIVC for metoprolol extended release tablets. Eur. J. Pharm. Biopharm. 2002, 53, 301–309. [Google Scholar] [CrossRef]
- FDA, Food and Drug Administration. Guidance for Industry: Statistical Approaches to Establishing Bioequivalence. U.S. Department of Health and Human Services, Center for Drug Evaluation and Research (CDER). 2001. Available online: https://www.fda.gov/media/70958/download (accessed on 11 October 2021).
- BRASIL, National Health Surveillance Agency-Agência Nacional de Vigilância Sanitária (ANVISA). Resolução RE nº 1170 de 19 de abril de 2006, Guia Para Provas de Biodisponibilidade Relativa/Bioequivalência de Medicamentos. CFAR/GTFAR/GGMED/ANVISA. 2006. Available online: http://antigo.anvisa.gov.br/documents/33880/2568070/RE_1170_2006.pdf/153aa760-5abc-4325-8ec5-95e6b43b35ad?version=1.0 (accessed on 11 October 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | F1 | F2 | F3 |
|---|---|---|---|
| MS | 40% | 40% | 40% |
| Microcrystalline cellulose 102 | 58% | 18% | 18% |
| Kollidon® SR | - | 40% | - |
| Methocel® K100M | - | - | 40% |
| Colloidal silicon dioxide | 1% | 1% | 1% |
| Magnesium stearate | 1% | 1% | 1% |
| Assay | Core Formulation | Coating Weight Gain (%) | Kollicoat IR (%) |
|---|---|---|---|
| E1 | F2 | 5 | 0 |
| E2 | F2 | 7.5 | 8 |
| E3 | F2 | 10 | 4 |
| E4 | F1 | 5 | 8 |
| E5 | F1 | 7.5 | 4 |
| E6 | F1 | 10 | 0 |
| E7 | F3 | 5 | 4 |
| E8 | F3 | 7.5 | 0 |
| E9 | F3 | 10 | 8 |
| Test | E11 | E12 | F1 |
|---|---|---|---|
| M1 | 5 | 10 | 1 |
| M2 | 1 | 14 | 1 |
| M3 | 3 | 12 | 1 |
| M4 | 4 | 11 | 1 |
| M5 | 2 | 13 | 1 |
| Parameters | Value | Source |
|---|---|---|
| Molecular weight (g/mol) | 267.37 | ADMET Predictor® |
| LogD | −1.72 at pH 4.0 | [19] |
| Solubility (mg/mL) | 171 (pH = 6.5) | [20] |
| pKa | 9.7 (Base) | [18] |
| Peff (cm/s × 10−4) | 1.34 (Human) | [21] |
| Fup (%) | 89 | [22] |
| Blood-to-plasma ratio | 1.13 | [18] |
| Mean precipitation time (s) | 900 | Default setting |
| Test | Core | Coating Weight Gain (WG%) | Kollicoat® IR (%) | DE (%) |
|---|---|---|---|---|
| E1 | F2 | 5 | 0 | 6.7 |
| E2 | F2 | 7.5 | 8 | 91.7 |
| E3 | F2 | 10 | 4 | 75.9 |
| E4 | F1 | 5 | 8 | 96.4 |
| E5 | F1 | 7.5 | 4 | 80.4 |
| E6 | F1 | 10 | 0 | 0.1 |
| E7 | F3 | 5 | 4 | 92.8 |
| E8 | F3 | 7.5 | 0 | 2.2 |
| E9 | F3 | 10 | 8 | 93.7 |
| Reference | - | - | - | 64.8 |
| Assay | IR * | Q%1h | Q%4h | Q%8h | Q%20h | DE (%) |
|---|---|---|---|---|---|---|
| E10 | 1% | 0.8 ± 0.0 | 2.0 ± 1.5 | 4.6 ± 2.7 | 22.3 ± 3.8 | 11.0 |
| E11 | 2% | 0.9 ± 1.0 | 7.5 ± 2.3 | 28.9 ± 1.4 | 79.9 ± 4.8 | 47.3 |
| E12 | 3% | 0.5 ± 0.3 | 18.9 ± 1.3 | 56.3 ± 2.9 | 94.2 ± 2.7 | 63.3 |
| Reference | - | 9.4 ± 1.5 | 26.9 ± 1.4 | 51.6 ± 1.9 | 96.4 ± 3.5 | 64.8 |
| USP specification [30] | - | Not less than 25% | Between 20–40% | Between 40–60% | Not less than 80% | - |
| Assay | F1 | E11 | E12 | Q%2h | Q%4h | Q%10h | Q%20h |
|---|---|---|---|---|---|---|---|
| M1 | 1 | 5 | 10 | 9.5 ± 0.2 | 20.1 ± 1.2 | 63.4 ± 1.7 | 71.8 ± 1.6 |
| M2 | 1 | 1 | 14 | 9.8 ± 0.2 | 22.8 ± 1.3 | 70.5 ± 2.2 | 77.9 ± 2.0 |
| M3 | 1 | 3 | 12 | 9.6 ± 0.4 | 21.5 ± 1.2 | 67.0 ± 2.0 | 74.8 ± 1.8 |
| M4 | 1 | 4 | 11 | 9.6 ± 0.6 | 20.8 ± 1.2 | 65.2 ± 1.8 | 73.3 ± 1.7 |
| M5 | 1 | 2 | 13 | 9.7 ± 0.3 | 22.1 ± 1.2 | 68.8 ± 2.1 | 76.4 ± 1.9 |
| Parameter | Reference | Optimization |
|---|---|---|
| Q%2h | 14.8 ± 0.8 | Maximum |
| Q%4h | 51.6 ± 1.9 | Maximum |
| Q%10h | 63.5 ± 2.2 | Within the range |
| Q%20h | 96.4 ± 3.5 | Maximum |
| Zero-Order | Reference | FO |
| ko | 0.08 | 0.08 |
| R2adj. | 0.9186 | 0.8861 |
| AIC | 86.93 | 91.16 |
| MSC | 2.34 | 2.00 |
| Higuchi | Reference | FO |
| kH | 2.56 | 2.57 |
| R2adj. | 0.9415 | 0.9223 |
| AIC | 82.96 | 86.48 |
| MSC | 2.67 | 2.40 |
| Korsmeyer–Peppas | Reference | FO |
| kKP | 0.35 | 0.54 |
| n | 0.80 | 0.73 |
| R2adj. | 0.9600 | 0.9787 |
| AIC | 79.25 | 82.30 |
| MSC | 2.98 | 2.74 |
| Parameters | Value |
|---|---|
| Clearance, CL (L/h) | 76.67 |
| Central compartment volume, Vc (L/Kg) | 2.85 |
| Elimination half-life, T1/2 (h) | 3.05 |
| Distribution rate constant from C1 to C2, K12 (h−1) | 1.80 |
| Distribution rate constant from C2 to C1, K21 (h−1) | 2.50 |
| Distribution volume of second compartment, V2 (L/Kg) | 2.05 |
| Drug Record | IV 5 mg-10 min | IV 10 mg-5 min | IR-T 50 mg | OS 50 mg | |
|---|---|---|---|---|---|
| Cmax (µg/mL) | Observed | 0.021 | 0.045 | 0.055 | 0.055 |
| Predicted | 0.022 | 0.048 | 0.050 | 0.055 | |
| %PE (%) | 5.24 | 6.67 | −9.09 | 0.01 | |
| AUC0–t (µg·h/mL) | Observed | 0.057 | 0.084 | 0.312 | 0.351 |
| Predicted | 0.058 | 0.108 | 0.298 | 0.331 | |
| %PE (%) | 1.75 | 28.57 | −4.49 | −5.70 | |
| Drug Record | Fasted | Fed | ||||
|---|---|---|---|---|---|---|
| A 25 mg | B 95 mg | C 200 mg | D 200 mg | E 200 mg | ||
| Cmax (µg/mL) | Predicted | 0.007 | 0.032 | 0.057 | 0.043 | 0.037 |
| LL—UL | 0.005–0.011 | 0.023–0.046 | 0.041–0.069 | 0.039–0.057 | 0.031–0.044 | |
| AUC0−t (µg·h/mL) | Predicted | 0.119 | 0.529 | 1.111 | 0.889 | 0.925 |
| LL—UL | 0.066–0.180 | 0.336–0.801 | 0.854–1.586 | 0.770–1.342 | 0.718–1.096 | |
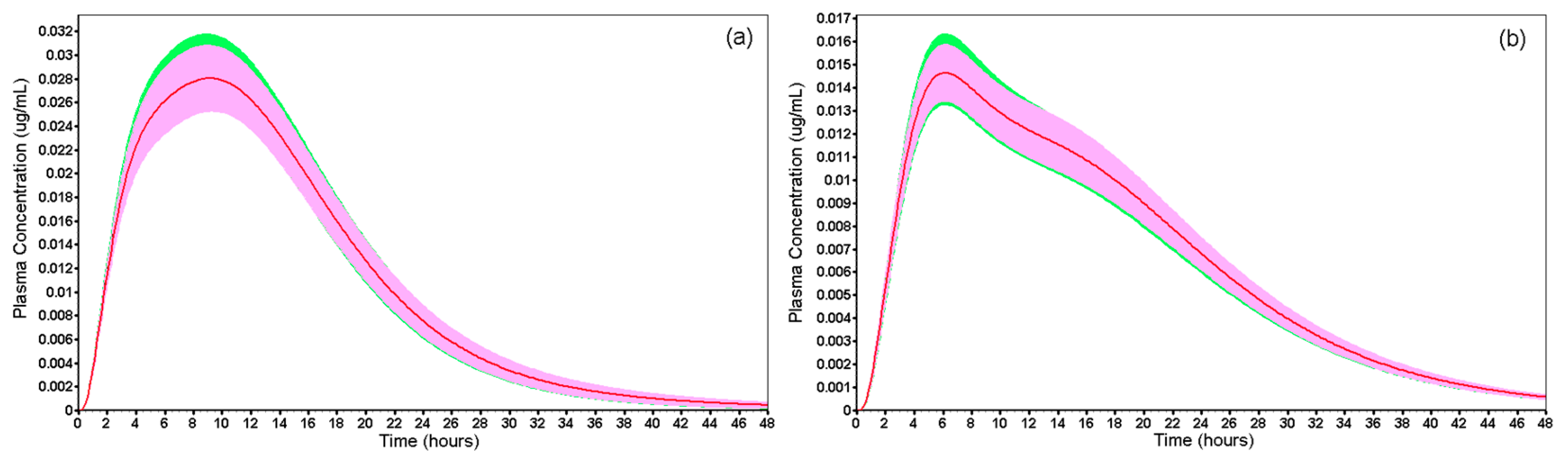
| Drug Record | Fasted | Fed | |||
|---|---|---|---|---|---|
| FO | Reference | FO | Reference | ||
| Cmax (µg/mL) | Mean | 0.031 | 0.030 | 0.016 | 0.015 |
| 90% CI | 0.027–0.033 87.31–105.73 | 0.014–0.017 89.60–107.99 | |||
| AUC0–t (µg·h/mL) | Mean | 0.518 | 0.510 | 0.311 | 0.313 |
| 90% CI | 0.461–0.560 88.86–108.11 | 0.288–0.338 92.60–108.88 | |||
| AUC0–inf (µg·h/mL) | Mean | 0.524 | 0.516 | 0.316 | 0.318 |
| 90% CI | 0.466–0.567 88.84–108.27 | 0.293–0.344 92.66–108.77 | |||
| Average AUC0–t/AUC0–inf | 0.989 | 0.988 | 0.984 | 0.984 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Issa, M.G.; de Souza, N.V.; Jou, B.W.C.; Duque, M.D.; Ferraz, H.G. Development of Extended-Release Mini-Tablets Containing Metoprolol Supported by Design of Experiments and Physiologically Based Biopharmaceutics Modeling. Pharmaceutics 2022, 14, 892. https://doi.org/10.3390/pharmaceutics14050892
Issa MG, de Souza NV, Jou BWC, Duque MD, Ferraz HG. Development of Extended-Release Mini-Tablets Containing Metoprolol Supported by Design of Experiments and Physiologically Based Biopharmaceutics Modeling. Pharmaceutics. 2022; 14(5):892. https://doi.org/10.3390/pharmaceutics14050892
Chicago/Turabian StyleIssa, Michele Georges, Natalia Vieira de Souza, Bruna Wenyi Chuang Jou, Marcelo Dutra Duque, and Humberto Gomes Ferraz. 2022. "Development of Extended-Release Mini-Tablets Containing Metoprolol Supported by Design of Experiments and Physiologically Based Biopharmaceutics Modeling" Pharmaceutics 14, no. 5: 892. https://doi.org/10.3390/pharmaceutics14050892