
Gene and Cellular Therapies for Leukodystrophies



Abstract
:1. Leukodystrophies
2. Classification and Treatment of Leukodystrophies

{kind=link}
{kind=link}
| Disease | Affected Gene/ Protein | Inheritance | Prevalence | Affected System | Historical (Radiographical) Classification [14] | Functional Classification |
|---|---|---|---|---|---|---|
| Inborn errors of metabolism | ||||||
| X-linked adrenoleuko- dystrophy (X-ALD) [15] | ABCD1/ATP binding cassette, subunit D | X-linked (female carriers may be affected) | 1/14,000–17,000 in males | Peroxisome, lipid metabolism | demyelination | myelin disorder (demyelination) |
| Globoid cell leukodystrophy (Krabbe) | GALC/ Galactosyl- ceramidase | Autosomal recessive | 1–5/100,000 | Lysosome, lipid metabolism | demyelination, myelin vacuolization | myelin disorder (demyelination) |
| Metachromatic Leukodystrophy (MLD) | ARSA/ Arylsulfatase A | Autosomal recessive | 1/40,000–160,000 | Lysosome, lipid metabolism | demyelination | myelin disorder (demyelination) |
| Fabry disease (FD) [16] | GLA/ α-galactosidase A | X-linked (female carriers may be affected) | 1/20,000–40,000 | Lysosome, Lipid metabolism | hypomyelination | Secondary (glycosphingo- lipid deposition) |
| Cerebrotendinous Xanthomatosis (CTX) [17] | CYP27A1/ mitochondrial enzyme sterol 27-hydroxylase | Autosomal recessive | 1/50,000–1,000,000 | Lipid (Cholesterol) metabolism | cerebellar and cerebral atrophy | Secondary (cholesterol- derivative accumulation) |
| Sjögren-Larsson Syndrome (SLS) [18] | FALDH (ALDH3A2)/ Fatty aldehyde dehydrogenase | Autosomal recessive | 1/250,000 | Lipid metabolism | cerebral atrophy | Secondary (accumulation of fatty alcohols and fatty aldehydes) |
| Pompe disease [19] | GAA/ acid α-glucosidase | Autosomal recessive | 1/40,000 | Lysosome, Glycogen metabolism | demyelination | Secondary (glycogen accumulation) |
| Canavan disease | ASPA/ aspartoacylase | Autosomal recessive | 1/100,000 | Absence of myelin lipid synthesis | spongiform (myelin vacuolization) | myelin disorder (vacuolization) |
| Peroxisomal biogenesis disorders (Zellweger syndrome, neonatal leukodys-trophy and infantile Refsum disease) [20] | PEX1/Peroxisomal biogenesis factor 1 | Autosomal recessive | 1/50,000 | Peroxisome assembly | demyelination | myelin disorder |
| Disorders of RNA/DNA Transcription/Translation | ||||||
| Congenital peripheral hypomyelinating neuropathy, central dys- myelination and Waardenburg–Hirschsprung (PCHW) [21] | SOX10/SOX10 | Autosomal dominant | <1/ 1,000,000 | Myelin development | hypo- myelination | myelin disorder [22] |
| Aicardi-Goutieres syndrome [23] | ADAR, TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, IFIH1 | Autosomal recessive or dominant | 1–5/10,000 | Nuclease genes | intracerebral calcifications, cerebral atrophy, temporal cysts | astrocytopathy |
| Childhood ataxia with CNS hypomyelination/ Vanishing White Matter disease (CACH/VWM) [24] | EIF2B1-5/translation initiation factor eIF2B subunits | Autosomal recessive | 1–4/ 1,000,000 births | isolated oligodendrocyte and astrocyte cell death | hypo- myelinating | astrocytopathy |
| Cytoskeletal | ||||||
| Alexander disease [25,26,27] | GFAP/glial fibrillary acidic protein | Autosomal dominant | 1/ 1,000,000 births | accumulation of GFAP in Rosenthal fibers | hypo- myelination, spongiform | astrocytopathy |
| Hypomyelinating leukodystrophy with atrophy of the basal ganglia and cerebellum (H-ABC) [28] | TUBB4A/tubulin β-4A | Autosomal dominant | unknown | alteration of microtubule dynamics or stability | hypomyelination, small or absent putamen, cerebral and cerebellar atrophy | leuko-axonopathy |
| Myelin disorders | ||||||
| Pelizaeus-Merzbacher disease (PMD, HLD1) | PLP1/proteolipid protein | X-linked | 1/100,000 | Myelin protein disorder | diffuse hypo- myelination, spongiform | myelin disorder (hypomyelination) |
| Pelizaeus-Merzbacher-like disease (PMLD) [22] | GJC2/gap-junction protein, gamma-2 and others (HSPD1, FAM126A, POLR3A, POLR3B, RARS, PYCR2, POLR1C, VPS11, SLC16A2) | Autosomal recessive | unknown | Gap junctions | diffuse hypo- myelination | Myelin disorder (hypomyelination) |
| Hypomyelinating leukodystrophy (HLD3) [29] | AIMP1/ARS-interacting multifunctional protein 1 | Autosomal recessive | unknown | Hyperphosphorylation of neurofilament proteins | cerebral atrophy, hypomyelination | leuko-axono- pathy |
| Fucosidosis [30] | FUCA1/alpha fucosidase | Autosomal recessive | <1/200,000 births | Lysosome | hypomyelination, cerebral and cerebellar atrophy | leuko-axono- pathy (oligo- dendrocyte) |
| Hypomyelination with congenital cataract (HCC) [31,32] | FAM126A (DRCTNNB1A)/ Hyccin | Autosomal recessive | Very rare, unknown | Myelin production | hypomyelination, white matter atrophy | leuko-axono- pathy (oligo- dendrocyte) |
| Clinical Trial | Title/Year | Country | Vector/ Transgene | NTC No |
|---|---|---|---|---|
| Ex vivo gene therapy | ||||
| Phase I/II | Gene therapy for metachromatic leukodystrophy (MLD) (2010–2025) | Italy | SIN-LV-ARSA (Libmeldy) | NCT01560182 |
| Phase II/III completed | A Study of the efficacy and safety of Hematopoietic Stem Cells transduced with Lenti-D lentiviral vector for the treatment of cerebral adrenoleukodystrophy (CALD) (2013–2021) | USA | Lenti-D-ABCD1 (Skysona) | NCT01896102 |
| Phase I/II recruiting | Autologous Hematopoietic Stem Cell Gene Therapy for Metachromatic Leukodystrophy and Adrenoleukodystrophy (2015–2025) | China | LV-ARSA/ LV-ABCD1 | NCT02559830 |
| Phase II | A Safety and Efficacy Study of Cryopreserved OTL-200 for Treatment of MLD (2018–2028) | Italy | LV-ARSA | NCT03392987 |
| Phase I/II | Lentiviral gene therapy for MLD (2018–2020) | China | LV-TYF-ARSA | NCT03725670 |
| Phase I/II | Lentiviral Gene Therapy for X-ALD (2018–2020) | China | LV-TYF-ABCD1 | NCT03727555 |
| Phase III | A Clinical Study to Assess the Efficacy and Safety of Gene Therapy for the Treatment of Cerebral Adrenoleukodystrophy (CALD) (2019–2023) | USA | Lenti-D-ABCD1 (Skysona) | NCT03852498 |
| Phase III recruiting | OTL-200 in Patients With Late Juvenile Metachromatic Leukodystrophy (MLD) (2022–2031) | Italy | SIN-LV-ARSA (Libmeldy) | NCT04283227 |
| In vivo gene therapy | ||||
| Phase I/II | Intracerebral Gene Therapy for Children With Early-Onset Forms of MLD (TG-MLD) (2014–2029) | France | AAVrh10-ARSA | NCT01801709 |
| Phase I/II | rAAV-Olig001-ASPA Gene Therapy for Treatment of Children With Typical Canavan Disease (CAN-GT) (2021–2024) | USA | rAAV-Olig001-ASPA | NCT04833907 |
| Phase I/II recruiting | Gene Transfer Clinical Trial for Krabbe Disease (RESKUE) (2021–2024) | USA | AAVrh10-GALC | NCT04693598 |
| Phase I/II recruiting | A Study of AAV9 Gene Therapy in Participants With Canavan Disease (CANaspire) (2021–2028) | USA | rAAV9-ASPA | NCT04998396 |
| Phase I/II | Study of Safety, Tolerability and Efficacy of PBKR03 in Pediatric Subjects With Early Infantile Krabbe Disease (GALax-C) (2022–2030) | USA | AAVHu68-GALC | NCT04771416 |
| Phase I/II recruiting | Gene Transfer Clinical Trial for Infantile and Late Infantile Krabbe Disease Treated in the Past With HSCT (REKLAIM) (2023–2025) | USA | AAVrh10-GALC | NCT05739643 |
2.1. X-Linked Adrenoleukodystrophy (ALD)
2.2. Metachromatic Leukodystrophy (MLD)
2.3. Globoid Cell Leukodystrophy (GCL, Krabbe)
3. Gene Therapeutic Approaches to Treat Early-Onset Leukodystrophies
3.1. Issues Regarding the Development of HSPC Gene Therapy for the Treatment of Leukodystrophies
3.2. rAAV Vector Design and Serotypes for In Vivo Targeting of the Central Nervous System
3.3. rAAV Vectors in Leukodystrophies
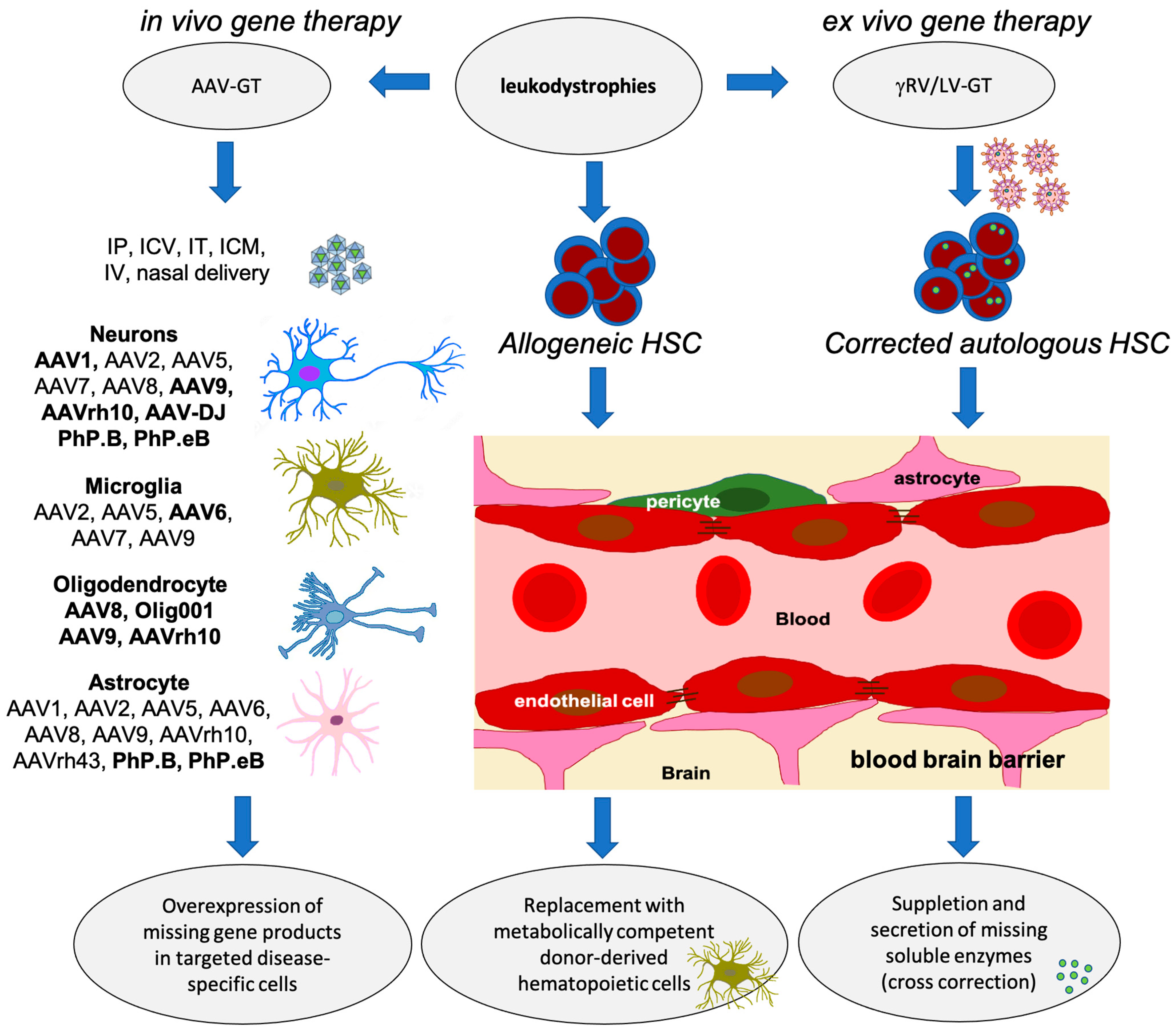
4. Use of Novel Technologies, CRISPR/Cas9 and Base-Editors
4.1. CRISPR/Cas9 Targeted Genome Editing
4.2. Base-Editing Techniques
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van der Knaap, M.S.; Bugiani, M. Leukodystrophies: A proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 2017, 134, 351–382. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.R.; Mortgart, K.; May, A. Introduction to leukodystrophy. Curr. Probl. Pediatr. Adolesc. Health Care 2022, 52, 101312. [Google Scholar] [CrossRef] [PubMed]
- Kevelam, S.H.; Steenweg, M.E.; Srivastava, S.; Helman, G.; Naidu, S.; Schiffmann, R.; Blaser, S.; Vanderver, A.; Wolf, N.I.; van der Knaap, M.S. Update on Leukodystrophies: A Historical Perspective and Adapted Definition. Neuropediatrics 2016, 47, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.T. Leukodystrophies. Continuum 2018, 24, 130–149. [Google Scholar] [CrossRef]
- Gordon-Lipkin, E.; Fatemi, A. Current Therapeutic Approaches in Leukodystrophies: A Review. J. Child Neurol. 2018, 33, 861–868. [Google Scholar] [CrossRef]
- Perlman, S.J.; Mar, S. Leukodystrophies. Adv. Exp. Med. Biol. 2012, 724, 154–171. [Google Scholar] [CrossRef]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; De Oliveira, S.; Thrasher, A.J.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef]
- van den Broek, B.T.A.; Page, K.; Paviglianiti, A.; Hol, J.; Allewelt, H.; Volt, F.; Michel, G.; Diaz, M.A.; Bordon, V.; O’Brien, T.; et al. Early and late outcomes after cord blood transplantation for pediatric patients with inherited leukodystrophies. Blood Adv. 2018, 2, 49–60. [Google Scholar] [CrossRef]
- Martin, P.L.; Carter, S.L.; Kernan, N.A.; Sahdev, I.; Wall, D.; Pietryga, D.; Wagner, J.E.; Kurtzberg, J. Results of the cord blood transplantation study (COBLT): Outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with lysosomal and peroxisomal storage diseases. Biol. Blood Marrow Transplant. 2006, 12, 184–194. [Google Scholar] [CrossRef]
- Prasad, V.K.; Mendizabal, A.; Parikh, S.H.; Szabolcs, P.; Driscoll, T.A.; Page, K.; Lakshminarayanan, S.; Allison, J.; Wood, S.; Semmel, D.; et al. Unrelated donor umbilical cord blood transplantation for inherited metabolic disorders in 159 pediatric patients from a single center: Influence of cellular composition of the graft on transplantation outcomes. Blood 2008, 112, 2979–2989. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef]
- Wolf, N.I.; Breur, M.; Plug, B.; Beerepoot, S.; Westerveld, A.S.R.; van Rappard, D.F.; de Vries, S.I.; Kole, M.H.P.; Vanderver, A.; van der Knaap, M.S.; et al. Metachromatic leukodystrophy and transplantation: Remyelination, no cross-correction. Ann. Clin. Transl. Neurol. 2020, 7, 169–180. [Google Scholar] [CrossRef]
- Gene Therapy Clinical Trials Worldwide. Available online: https://a873679.fmphost.com/fmi/webd/GTCT (accessed on 14 September 2023).
- Gaillard, F.; Jones, J.; Saber, M. Leukodystrophies. 2023. [Google Scholar] [CrossRef]
- Gupta, A.O.; Raymond, G.; Pierpont, E.I.; Kemp, S.; McIvor, R.S.; Rayannavar, A.; Miller, B.; Lund, T.C.; Orchard, P.J. Treatment of cerebral adrenoleukodystrophy: Allogeneic transplantation and lentiviral gene therapy. Expert Opin. Biol. Ther. 2022, 22, 1151–1162. [Google Scholar] [CrossRef]
- Nowacki, J.C.; Fields, A.M.; Fu, M.M. Emerging cellular themes in leukodystrophies. Front. Cell Dev. Biol. 2022, 10, 902261. [Google Scholar] [CrossRef]
- Vanrietvelde, F.; Lemmerling, M.; Mespreuve, M.; Crevits, L.; De Reuck, J.; Kunnen, M. MRI of the brain in cerebrotendinous xanthomatosis (van Bogaert-Scherer-Epstein disease). Eur. Radiol. 2000, 10, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Bindu, P.S. Sjogren-Larsson Syndrome: Mechanisms and Management. Appl. Clin. Genet. 2020, 13, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Broomfield, A.; Fletcher, J.; Hensman, P.; Wright, R.; Prunty, H.; Pavaine, J.; Jones, S.A. Rapidly Progressive White Matter Involvement in Early Childhood: The Expanding Phenotype of Infantile Onset Pompe? JIMD Rep. 2018, 39, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, C.; D’Agostino, M.D.; Braverman, N. Peroxisome biogenesis disorders. Transl. Sci. Rare Dis. 2016, 1, 111–144. [Google Scholar] [CrossRef]
- Inoue, K.; Shilo, K.; Boerkoel, C.F.; Crowe, C.; Sawady, J.; Lupski, J.R.; Agamanolis, D.P. Congenital hypomyelinating neuropathy, central dysmyelination, and Waardenburg-Hirschsprung disease: Phenotypes linked by SOX10 mutation. Ann. Neurol. 2002, 52, 836–842. [Google Scholar] [CrossRef]
- Charzewska, A.; Wierzba, J.; Izycka-Swieszewska, E.; Bekiesinska-Figatowska, M.; Jurek, M.; Gintowt, A.; Klosowska, A.; Bal, J.; Hoffman-Zacharska, D. Hypomyelinating leukodystrophies—A molecular insight into the white matter pathology. Clin. Genet. 2016, 90, 293–304. [Google Scholar] [CrossRef]
- van Heteren, J.T.; Rozenberg, F.; Aronica, E.; Troost, D.; Lebon, P.; Kuijpers, T.W. Astrocytes produce interferon-alpha and CXCL10, but not IL-6 or CXCL8, in Aicardi-Goutieres syndrome. Glia 2008, 56, 568–578. [Google Scholar] [CrossRef]
- van der Knaap, M.S.; Fogli, A.; Boespflug-Tanguy, O.; Abbink, T.E.M.; Schiffmann, R. Childhood Ataxia with Central Nervous System Hypomyelination/Vanishing White Matter. In GeneReviews® [Internet]; 2003 Feb 20 [updated 2019 Apr 4], 1993–2023; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2023. [Google Scholar]
- Li, R.; Messing, A.; Goldman, J.E.; Brenner, M. GFAP mutations in Alexander disease. Int. J. Dev. Neurosci. 2002, 20, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Sosunov, A.; Olabarria, M.; Goldman, J.E. Alexander disease: An astrocytopathy that produces a leukodystrophy. Brain Pathol. 2018, 28, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Messing, A.; Brenner, M.; Feany, M.B.; Nedergaard, M.; Goldman, J.E. Alexander disease. J. Neurosci. 2012, 32, 5017–5023. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.M.; Polder, E.; Vanderver, A.; Naidu, S.; Schiffmann, R.; Fisher, K.; Raguz, A.B.; Blumkin, L.; Group, H.A.R.; van Berkel, C.G.; et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: Further delineation of the phenotype and genotype-phenotype correlation. Brain 2014, 137, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, M.; Markus, B.; Noyman, I.; Shalev, H.; Flusser, H.; Shelef, I.; Liani-Leibson, K.; Shorer, Z.; Cohen, I.; Khateeb, S.; et al. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am. J. Hum. Genet. 2010, 87, 820–828. [Google Scholar] [CrossRef] [PubMed]
- van der Knaap, M.; Valk, J. Chapter 12: Fucosidosis. In Magnetic Resonance of Myelination and Myelin Disorders; Springer: Berlin, Heidelberg, 2005; pp. 119–122. [Google Scholar]
- Gazzerro, E.; Baldassari, S.; Giacomini, C.; Musante, V.; Fruscione, F.; La Padula, V.; Biancheri, R.; Scarfi, S.; Prada, V.; Sotgia, F.; et al. Hyccin, the molecule mutated in the leukodystrophy hypomyelination and congenital cataract (HCC), is a neuronal protein. PLoS ONE 2012, 7, e32180. [Google Scholar] [CrossRef]
- Wolf, N.I.; Biancheri, R.; Zara, F.; Bruno, C.; Gazzerro, E.; Rossi, A.; van der Knaap, M.S.; Minetti, C. Hypomyelination and Congenital Cataract. In GeneReviews® [Internet]; 2008 Oct 14 [updated 2021 Jan 14], 1993–2023; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2023. [Google Scholar]
- Heubner, O. Über diffuse Hirnsclerose. Charité Ann. 1897, 22, 298–310. [Google Scholar]
- Migeon, B.R.; Moser, H.W.; Moser, A.B.; Axelman, J.; Sillence, D.; Norum, R.A. Adrenoleukodystrophy: Evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc. Natl. Acad. Sci. USA 1981, 78, 5066–5070. [Google Scholar] [CrossRef]
- Mosser, J.; Douar, A.M.; Sarde, C.O.; Kioschis, P.; Feil, R.; Moser, H.; Poustka, A.M.; Mandel, J.L.; Aubourg, P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 1993, 361, 726–730. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Bougneres, P.; Schmidt, M.; Kalle, C.V.; Fischer, A.; Cavazzana-Calvo, M.; Aubourg, P. Lentiviral hematopoietic cell gene therapy for X-linked adrenoleukodystrophy. Methods Enzymol. 2012, 507, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.; Aubourg, P.; Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Dalcq, M.; Feigenbaum, V.; Aubourg, P. The neurobiology of X-linked adrenoleukodystrophy, a demyelinating peroxisomal disorder. Trends Neurosci. 1999, 22, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Moser, H.W.; Mahmood, A.; Raymond, G.V. X-linked adrenoleukodystrophy. Nat. Clin. Pract. Neurol. 2007, 3, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Aubourg, P.; Blanche, S.; Jambaque, I.; Rocchiccioli, F.; Kalifa, G.; Naud-Saudreau, C.; Rolland, M.O.; Debre, M.; Chaussain, J.L.; Griscelli, C.; et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N. Engl. J. Med. 1990, 322, 1860–1866. [Google Scholar] [CrossRef]
- Shapiro, E.; Krivit, W.; Lockman, L.; Jambaque, I.; Peters, C.; Cowan, M.; Harris, R.; Blanche, S.; Bordigoni, P.; Loes, D.; et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet 2000, 356, 713–718. [Google Scholar] [CrossRef]
- Moser, H.W.; Raymond, G.V.; Lu, S.E.; Muenz, L.R.; Moser, A.B.; Xu, J.; Jones, R.O.; Loes, D.J.; Melhem, E.R.; Dubey, P.; et al. Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo’s oil. Arch. Neurol. 2005, 62, 1073–1080. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Kartha, R.V.; Brundage, R.C.; Cloyd, J.; Basu, C.; Carlin, B.P.; Jones, R.O.; Moser, A.B.; Fatemi, A.; Raymond, G.V. A model-based approach to assess the exposure-response relationship of Lorenzo’s oil in adrenoleukodystrophy. Br. J. Clin. Pharmacol. 2016, 81, 1058–1066. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef]
- Greenfield, J.G. A Form of Progressive Cerebral Sclerosis in Infants Associated with Primary Degeneration of the Interfascicular Glia. J. Neurol. Psychopathol. 1933, 13, 289–302. [Google Scholar] [CrossRef]
- Austin, J.H. Metachromatic form of diffuse cerebral sclerosis. III. Significance of sulfatide and other lipid abnormalities in white matter and kidney. Neurology 1960, 10, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.; McAfee, D.; Armstrong, D.; O’Rourke, M.; Shearer, L.; Bachhawat, B. Abnormal sulphatase activities in two human diseases (metachromatic leucodystrophy and gargoylism). Biochem. J. 1964, 93, 15C–17C. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, C.; Brown, J.A.; Shows, T.B. Lysosomal arylsulfatase deficiencies in humans: Chromosome assignments for arylsulfatase A and B. Proc. Natl. Acad. Sci. USA 1979, 76, 1957–1961. [Google Scholar] [CrossRef] [PubMed]
- Shaimardanova, A.A.; Chulpanova, D.S.; Solovyeva, V.V.; Mullagulova, A.I.; Kitaeva, K.V.; Allegrucci, C.; Rizvanov, A.A. Metachromatic Leukodystrophy: Diagnosis, Modeling, and Treatment Approaches. Front. Med. 2020, 7, 576221. [Google Scholar] [CrossRef] [PubMed]
- Barth, M.L.; Fensom, A.; Harris, A. The arylsulphatase A gene and molecular genetics of metachromatic leucodystrophy. J. Med. Genet. 1994, 31, 663–666. [Google Scholar] [CrossRef]
- Cesani, M.; Lorioli, L.; Grossi, S.; Amico, G.; Fumagalli, F.; Spiga, I.; Filocamo, M.; Biffi, A. Mutation Update of ARSA and PSAP Genes Causing Metachromatic Leukodystrophy. Hum. Mutat. 2016, 37, 16–27. [Google Scholar] [CrossRef]
- Gieselmann, V. Metachromatic leukodystrophy: Genetics, pathogenesis and therapeutic options. Acta Paediatr. 2008, 97, 15–21. [Google Scholar] [CrossRef]
- Bayever, E.; Ladisch, S.; Philippart, M.; Brill, N.; Nuwer, M.; Sparkes, R.S.; Feig, S.A. Bone-marrow transplantation for metachromatic leucodystrophy. Lancet 1985, 2, 471–473. [Google Scholar] [CrossRef]
- Boucher, A.A.; Miller, W.; Shanley, R.; Ziegler, R.; Lund, T.; Raymond, G.; Orchard, P.J. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: The largest single-institution cohort report. Orphanet J. Rare Dis. 2015, 10, 94. [Google Scholar] [CrossRef]
- Martin, H.R.; Poe, M.D.; Provenzale, J.M.; Kurtzberg, J.; Mendizabal, A.; Escolar, M.L. Neurodevelopmental outcomes of umbilical cord blood transplantation in metachromatic leukodystrophy. Biol. Blood Marrow Transplant. 2013, 19, 616–624. [Google Scholar] [CrossRef]
- Kapaun, P.; Dittmann, R.W.; Granitzny, B.; Eickhoff, W.; Wulbrand, H.; Neumaier-Probst, E.; Zander, A.; Kohlschuetter, A. Slow progression of juvenile metachromatic leukodystrophy 6 years after bone marrow transplantation. J. Child Neurol. 1999, 14, 222–228. [Google Scholar] [CrossRef]
- Gorg, M.; Wilck, W.; Granitzny, B.; Suerken, A.; Lukacs, Z.; Ding, X.; Schulte-Markwort, M.; Kohlschutter, A. Stabilization of juvenile metachromatic leukodystrophy after bone marrow transplantation: A 13-year follow-up. J. Child Neurol. 2007, 22, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Al-Saady, M.; Beerepoot, S.; Plug, B.C.; Breur, M.; Galabova, H.; Pouwels, P.J.W.; Boelens, J.J.; Lindemans, C.; van Hasselt, P.M.; Matzner, U.; et al. Neurodegenerative disease after hematopoietic stem cell transplantation in metachromatic leukodystrophy. Ann. Clin. Transl. Neurol. 2023, 10, 1146–1159. [Google Scholar] [CrossRef] [PubMed]
- Kopen, G.C.; Prockop, D.J.; Phinney, D.G. Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proc. Natl. Acad. Sci. USA 1999, 96, 10711–10716. [Google Scholar] [CrossRef] [PubMed]
- Azizi, S.A.; Stokes, D.; Augelli, B.J.; DiGirolamo, C.; Prockop, D.J. Engraftment and migration of human bone marrow stromal cells implanted in the brains of albino rats--similarities to astrocyte grafts. Proc. Natl. Acad. Sci. USA 1998, 95, 3908–3913. [Google Scholar] [CrossRef]
- Koc, O.N.; Peters, C.; Aubourg, P.; Raghavan, S.; Dyhouse, S.; DeGasperi, R.; Kolodny, E.H.; Yoseph, Y.B.; Gerson, S.L.; Lazarus, H.M.; et al. Bone marrow-derived mesenchymal stem cells remain host-derived despite successful hematopoietic engraftment after allogeneic transplantation in patients with lysosomal and peroxisomal storage diseases. Exp. Hematol. 1999, 27, 1675–1681. [Google Scholar] [CrossRef]
- Koc, O.N.; Day, J.; Nieder, M.; Gerson, S.L.; Lazarus, H.M.; Krivit, W. Allogeneic mesenchymal stem cell infusion for treatment of metachromatic leukodystrophy (MLD) and Hurler syndrome (MPS-IH). Bone Marrow Transplant 2002, 30, 215–222. [Google Scholar] [CrossRef]
- Cabanillas Stanchi, K.M.; Bohringer, J.; Strolin, M.; Groeschel, S.; Lenglinger, K.; Treuner, C.; Kehrer, C.; Laugwitz, L.; Bevot, A.; Kaiser, N.; et al. Hematopoietic Stem Cell Transplantation with Mesenchymal Stromal Cells in Children with Metachromatic Leukodystrophy. Stem Cells Dev. 2022, 31, 163–175. [Google Scholar] [CrossRef]
- Zychlinski, D.; Schambach, A.; Modlich, U.; Maetzig, T.; Meyer, J.; Grassman, E.; Mishra, A.; Baum, C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol. Ther. 2008, 16, 718–725. [Google Scholar] [CrossRef]
- Schwarzer, A.; Talbot, S.R.; Selich, A.; Morgan, M.; Schott, J.W.; Dittrich-Breiholz, O.; Bastone, A.L.; Weigel, B.; Ha, T.C.; Dziadek, V.; et al. Predicting genotoxicity of viral vectors for stem cell gene therapy using gene expression-based machine learning. Mol. Ther. 2021, 29, 3383–3397. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Calbi, V.; Natali Sora, M.G.; Sessa, M.; Baldoli, C.; Rancoita, P.M.V.; Ciotti, F.; Sarzana, M.; Fraschini, M.; Zambon, A.A.; et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: Long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 2022, 399, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- EMA. Libmeldy Autologous CD34+ Cells Encoding ARSA Gene. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/libmeldy (accessed on 14 September 2023).
- Krabbe, K. A new familial infantile form of diffuse brain-sclerosis. Brain 1916, 39, 74–114. [Google Scholar] [CrossRef]
- Hagberg, B.; Sourander, P.; Svennerholm, L. Diagnosis of Krabbe’s infantile leucodystrophy. J. Neurol. Neurosurg. Psychiatry 1963, 26, 195–198. [Google Scholar] [CrossRef]
- Kass, A. Acute diffuse infantile sclerosis of the brain (Krabbe’s disease); a report of two cases in sibs. Acta Paediatr. 1953, 42, 70–76. [Google Scholar] [CrossRef]
- Hagberg, B. Clinical aspects of globoid cell and metachromatic leukodystrophies. Birth Defects Orig. Artic. Ser. 1971, 7, 103–112. [Google Scholar]
- Bradbury, A.M.; Bongarzone, E.R.; Sands, M.S. Krabbe disease: New hope for an old disease. Neurosci. Lett. 2021, 752, 135841. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Rafi, M.A.; de Gala, G.; Wenger, D.A. Cloning and expression of cDNA encoding human galactocerebrosidase, the enzyme deficient in globoid cell leukodystrophy. Hum. Mol. Genet. 1993, 2, 1841–1845. [Google Scholar] [CrossRef]
- Luzi, P.; Rafi, M.A.; Wenger, D.A. Structure and organization of the human galactocerebrosidase (GALC) gene. Genomics 1995, 26, 407–409. [Google Scholar] [CrossRef]
- Krivit, W.; Shapiro, E.G.; Peters, C.; Wagner, J.E.; Cornu, G.; Kurtzberg, J.; Wenger, D.A.; Kolodny, E.H.; Vanier, M.T.; Loes, D.J.; et al. Hematopoietic stem-cell transplantation in globoid-cell leukodystrophy. N. Engl. J. Med. 1998, 338, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Escolar, M.L.; Poe, M.D.; Provenzale, J.M.; Richards, K.C.; Allison, J.; Wood, S.; Wenger, D.A.; Pietryga, D.; Wall, D.; Champagne, M.; et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N. Engl. J. Med. 2005, 352, 2069–2081. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Li, Y.; Nikolaishvili-Feinberg, N.; Scesa, G.; Bi, Y.; Pan, D.; Moore, D.; Bongarzone, E.R.; Sands, M.S.; Miller, R.; et al. Hematopoietic Stem cell transplantation and lentiviral vector-based gene therapy for Krabbe’s disease: Present convictions and future prospects. J. Neurosci. Res. 2016, 94, 1152–1168. [Google Scholar] [CrossRef] [PubMed]
- Spratley, S.J.; Deane, J.E. New therapeutic approaches for Krabbe disease: The potential of pharmacological chaperones. J. Neurosci. Res. 2016, 94, 1203–1219. [Google Scholar] [CrossRef]
- Visigalli, I.; Ungari, S.; Martino, S.; Park, H.; Cesani, M.; Gentner, B.; Sergi Sergi, L.; Orlacchio, A.; Naldini, L.; Biffi, A. The galactocerebrosidase enzyme contributes to the maintenance of a functional hematopoietic stem cell niche. Blood 2010, 116, 1857–1866. [Google Scholar] [CrossRef]
- Gentner, B.; Visigalli, I.; Hiramatsu, H.; Lechman, E.; Ungari, S.; Giustacchini, A.; Schira, G.; Amendola, M.; Quattrini, A.; Martino, S.; et al. Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci. Transl. Med. 2010, 2, 58ra84. [Google Scholar] [CrossRef]
- Ungari, S.; Montepeloso, A.; Morena, F.; Cocchiarella, F.; Recchia, A.; Martino, S.; Gentner, B.; Naldini, L.; Biffi, A. Design of a regulated lentiviral vector for hematopoietic stem cell gene therapy of globoid cell leukodystrophy. Mol. Ther. Methods Clin. Dev. 2015, 2, 15038. [Google Scholar] [CrossRef]
- Bushman, F.D. Retroviral Insertional Mutagenesis in Humans: Evidence for Four Genetic Mechanisms Promoting Expansion of Cell Clones. Mol. Ther. 2020, 28, 352–356. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef]
- Goyal, S.; Tisdale, J.; Schmidt, M.; Kanter, J.; Jaroscak, J.; Whitney, D.; Bitter, H.; Gregory, P.D.; Parsons, G.; Foos, M.; et al. Acute Myeloid Leukemia Case after Gene Therapy for Sickle Cell Disease. N. Engl. J. Med. 2022, 386, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, R.; Bernardo, M.E. Haematopoietic stem cell gene therapy in inborn errors of metabolism. Br. J. Haematol. 2022, 198, 227–243. [Google Scholar] [CrossRef]
- Sheridan, C. Bluebird’s CALD gene therapy poised for approval. Nat. Biotechnol. 2022, 40, 985. [Google Scholar] [CrossRef]
- Williams, D.A.; Bledsoe, J.R.; Duncan, C.N.; Eichler, F.S.; Grzywacz, B.; Gupta, A.O.; Lund, T.; Orchard, P.J.; Slauson, S.; Whitney, D.; et al. Myelodysplastic syndromes after eli-cel gene therapy for cerebral adrenoleukodystrophy (CALD). In Proceedings of the ASGCT 25th Annual Meeting, Washington, DC, USA, 16–19 May 2022. [Google Scholar]
- FDA. Biologics License Application Approval for Elivaldogene Autotemcel. Available online: https://www.fda.gov/media/161665/download (accessed on 14 September 2023).
- Keam, S.J. Elivaldogene Autotemcel: First Approval. Mol. Diagn. Ther. 2021, 25, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Şeker, M.; Erol, Ö.; Pervin, B.; Wagemaker, G.; van Til, N.; Aerts-Kaya, F. Non-Myelotoxic Agents as a Preparatory Regimen for Hematopoietic Stem Cell Gene Therapy. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Saha, A.; Blazar, B.R. Antibody based conditioning for allogeneic hematopoietic stem cell transplantation. Front. Immunol. 2022, 13, 1031334. [Google Scholar] [CrossRef]
- EMA. Skysona Withdrawal of the Marketing Authorisation in the European Union. Available online: https://www.ema.europa.eu/en/documents/public-statement/public-statement-skysona-withdrawal-marketing-authorisation-european-union_.pdf (accessed on 9 October 2023).
- Hampson, G.; Towse, A.; Pearson, S.D.; Dreitlein, W.B.; Henshall, C. Gene therapy: Evidence, value and affordability in the US health care system. J. Comp. Eff. Res. 2018, 7, 15–28. [Google Scholar] [CrossRef]
- Kohn, D.B.; Booth, C.; Shaw, K.L.; Xu-Bayford, J.; Garabedian, E.; Trevisan, V.; Carbonaro-Sarracino, D.A.; Soni, K.; Terrazas, D.; Snell, K.; et al. Autologous Ex Vivo Lentiviral Gene Therapy for Adenosine Deaminase Deficiency. N. Engl. J. Med. 2021, 384, 2002–2013. [Google Scholar] [CrossRef]
- Richter, M.; Saydaminova, K.; Yumul, R.; Krishnan, R.; Liu, J.; Nagy, E.E.; Singh, M.; Izsvak, Z.; Cattaneo, R.; Uckert, W.; et al. In vivo transduction of primitive mobilized hematopoietic stem cells after intravenous injection of integrating adenovirus vectors. Blood 2016, 128, 2206–2217. [Google Scholar] [CrossRef]
- Wang, H.; Georgakopoulou, A.; Zhang, W.; Kim, J.; Gil, S.; Ehrhardt, A.; Lieber, A. HDAd6/35++—A new helper-dependent adenovirus vector platform for in vivo transduction of hematopoietic stem cells. Mol. Ther. Methods Clin. Dev. 2023, 29, 213–226. [Google Scholar] [CrossRef]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.L.; Sanchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen, S.N.; Lock, J.L.; Schoderboeck, L.; Abraham, W.C.; Hughes, S.M. CNS Transduction Benefits of AAV-PHP.eB over AAV9 Are Dependent on Administration Route and Mouse Strain. Mol. Ther. Methods Clin. Dev. 2020, 19, 447–458. [Google Scholar] [CrossRef]
- Chuapoco, M.; Flytzanis, N.; Goeden, N.; Octeau, J.; Roxas, K.; Chan, K.; Scherrer, J.; Winchester, J.; Blackburn, R.; Campos, L.; et al. Intravenous functional gene transfer throughout the brain of non-human primates using AAV. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Chuapoco, M.R.; Flytzanis, N.C.; Goeden, N.; Christopher Octeau, J.; Roxas, K.M.; Chan, K.Y.; Scherrer, J.; Winchester, J.; Blackburn, R.J.; Campos, L.J.; et al. Adeno-associated viral vectors for functional intravenous gene transfer throughout the non-human primate brain. Nat. Nanotechnol. 2023. [Google Scholar] [CrossRef]
- Huang, Q.; Chen, A.T.; Chan, K.Y.; Sorensen, H.; Barry, A.J.; Azari, B.; Zheng, Q.; Beddow, T.; Zhao, B.; Tobey, I.G.; et al. Targeting AAV vectors to the central nervous system by engineering capsid-receptor interactions that enable crossing of the blood-brain barrier. PLoS Biol. 2023, 21, e3002112. [Google Scholar] [CrossRef]
- Nonnenmacher, M.; Wang, W.; Child, M.A.; Ren, X.Q.; Huang, C.; Ren, A.Z.; Tocci, J.; Chen, Q.; Bittner, K.; Tyson, K.; et al. Rapid evolution of blood-brain-barrier-penetrating AAV capsids by RNA-driven biopanning. Mol. Ther. Methods Clin. Dev. 2021, 20, 366–378. [Google Scholar] [CrossRef]
- Gray, S.J.; Matagne, V.; Bachaboina, L.; Yadav, S.; Ojeda, S.R.; Samulski, R.J. Preclinical differences of intravascular AAV9 delivery to neurons and glia: A comparative study of adult mice and nonhuman primates. Mol. Ther. 2011, 19, 1058–1069. [Google Scholar] [CrossRef]
- Bey, K.; Ciron, C.; Dubreil, L.; Deniaud, J.; Ledevin, M.; Cristini, J.; Blouin, V.; Aubourg, P.; Colle, M.A. Efficient CNS targeting in adult mice by intrathecal infusion of single-stranded AAV9-GFP for gene therapy of neurological disorders. Gene Ther. 2017, 24, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Bey, K.; Deniaud, J.; Dubreil, L.; Joussemet, B.; Cristini, J.; Ciron, C.; Hordeaux, J.; Le Boulc’h, M.; Marche, K.; Maquigneau, M.; et al. Intra-CSF AAV9 and AAVrh10 Administration in Nonhuman Primates: Promising Routes and Vectors for Which Neurological Diseases? Mol. Ther. Methods Clin. Dev. 2020, 17, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Bugiani, M.; Abbink, T.E.M.; Edridge, A.W.D.; van der Hoek, L.; Hillen, A.E.J.; van Til, N.P.; Hu, A.N.G.V.; Breur, M.; Aiach, K.; Drevot, P.; et al. Focal lesions following intracerebral gene therapy for mucopolysaccharidosis IIIA. Ann. Clin. Transl. Neurol. 2023, 10, 904–917. [Google Scholar] [CrossRef]
- Francis, J.S.; Markov, V.; Wojtas, I.D.; Gray, S.; McCown, T.; Samulski, R.J.; Figueroa, M.; Leone, P. Preclinical biodistribution, tropism, and efficacy of oligotropic AAV/Olig001 in a mouse model of congenital white matter disease. Mol. Ther. Methods Clin. Dev. 2021, 20, 520–534. [Google Scholar] [CrossRef]
- Leone, P.; Shera, D.; McPhee, S.W.; Francis, J.S.; Kolodny, E.H.; Bilaniuk, L.T.; Wang, D.J.; Assadi, M.; Goldfarb, O.; Goldman, H.W.; et al. Long-term follow-up after gene therapy for canavan disease. Sci. Transl. Med. 2012, 4, 165ra163. [Google Scholar] [CrossRef]
- Corti, M.; Byrne, B.J.; Gessler, D.J.; Thompson, G.; Norman, S.; Lammers, J.; Coleman, K.E.; Liberati, C.; Elder, M.E.; Escolar, M.L.; et al. Adeno-associated virus-mediated gene therapy in a patient with Canavan disease using dual routes of administration and immune modulation. Mol. Ther. Methods Clin. Dev. 2023, 30, 303–314. [Google Scholar] [CrossRef]
- Uhlenberg, B.; Schuelke, M.; Ruschendorf, F.; Ruf, N.; Kaindl, A.M.; Henneke, M.; Thiele, H.; Stoltenburg-Didinger, G.; Aksu, F.; Topaloglu, H.; et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am. J. Hum. Genet 2004, 75, 251–260. [Google Scholar] [CrossRef]
- Menichella, D.M.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Connexins are critical for normal myelination in the CNS. J. Neurosci. 2003, 23, 5963–5973. [Google Scholar] [CrossRef]
- Georgiou, E.; Sidiropoulou, K.; Richter, J.; Papaneophytou, C.; Sargiannidou, I.; Kagiava, A.; von Jonquieres, G.; Christodoulou, C.; Klugmann, M.; Kleopa, K.A. Gene therapy targeting oligodendrocytes provides therapeutic benefit in a leukodystrophy model. Brain 2017, 140, 599–616. [Google Scholar] [CrossRef]
- Dooves, S.; Bugiani, M.; Postma, N.L.; Polder, E.; Land, N.; Horan, S.T.; van Deijk, A.L.; van de Kreeke, A.; Jacobs, G.; Vuong, C.; et al. Astrocytes are central in the pathomechanisms of vanishing white matter. J. Clin. Investig. 2016, 126, 1512–1524. [Google Scholar] [CrossRef]
- Sanchez, A.; Garcia-Lareu, B.; Puig, M.; Prat, E.; Ruberte, J.; Chillon, M.; Nunes, V.; Estevez, R.; Bosch, A. Cerebellar Astrocyte Transduction as Gene Therapy for Megalencephalic Leukoencephalopathy. Neurotherapeutics 2020, 17, 2041–2053. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Miyake, K.; Sakai, A.; Yamamoto, M.; Suzuki, H.; Shimada, T. Treatment of adult metachromatic leukodystrophy model mice using intrathecal administration of type 9 AAV vector encoding arylsulfatase A. Sci. Rep. 2021, 11, 20513. [Google Scholar] [CrossRef] [PubMed]
- Hordeaux, J.; Jeffrey, B.A.; Jian, J.; Choudhury, G.R.; Michalson, K.; Mitchell, T.W.; Buza, E.L.; Chichester, J.; Dyer, C.; Bagel, J.; et al. Efficacy and Safety of a Krabbe Disease Gene Therapy. Hum. Gene Ther. 2022, 33, 499–517. [Google Scholar] [CrossRef] [PubMed]
- Neri, I.; Ramazzotti, G.; Mongiorgi, S.; Rusciano, I.; Bugiani, M.; Conti, L.; Cousin, M.; Giorgio, E.; Padiath, Q.S.; Vaula, G.; et al. Understanding the Ultra-Rare Disease Autosomal Dominant Leukodystrophy: An Updated Review on Morpho-Functional Alterations Found in Experimental Models. Mol. Neurobiol. 2023, 60, 6362–6372. [Google Scholar] [CrossRef]
- Cucchiarini, M.; Ren, X.L.; Perides, G.; Terwilliger, E.F. Selective gene expression in brain microglia mediated via adeno-associated virus type 2 and type 5 vectors. Gene Ther. 2003, 10, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Hosoi, N.; Matsuzaki, Y.; Fukai, Y.; Hiraga, A.; Nakai, J.; Nitta, K.; Shinohara, Y.; Konno, A.; Hirai, H. Development of microglia-targeting adeno-associated viral vectors as tools to study microglial behavior in vivo. Commun. Biol. 2022, 5, 1224. [Google Scholar] [CrossRef]
- Samaranch, L.; Salegio, E.A.; San Sebastian, W.; Kells, A.P.; Bringas, J.R.; Forsayeth, J.; Bankiewicz, K.S. Strong cortical and spinal cord transduction after AAV7 and AAV9 delivery into the cerebrospinal fluid of nonhuman primates. Hum. Gene Ther. 2013, 24, 526–532. [Google Scholar] [CrossRef]
- Yang, B.; Li, S.; Wang, H.; Guo, Y.; Gessler, D.J.; Cao, C.; Su, Q.; Kramer, J.; Zhong, L.; Ahmed, S.S.; et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol. Ther. 2014, 22, 1299–1309. [Google Scholar] [CrossRef]
- Hordeaux, J.; Yuan, Y.; Clark, P.M.; Wang, Q.; Martino, R.A.; Sims, J.J.; Bell, P.; Raymond, A.; Stanford, W.L.; Wilson, J.M. The GPI-Linked Protein LY6A Drives AAV-PHP.B Transport across the Blood-Brain Barrier. Mol. Ther. 2019, 27, 912–921. [Google Scholar] [CrossRef]
- Huang, Q.; Chan, K.Y.; Tobey, I.G.; Chan, Y.A.; Poterba, T.; Boutros, C.L.; Balazs, A.B.; Daneman, R.; Bloom, J.M.; Seed, C.; et al. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS ONE 2019, 14, e0225206. [Google Scholar] [CrossRef]
- Han, J.; Sarlus, H.; Wszolek, Z.K.; Karrenbauer, V.D.; Harris, R.A. Microglial replacement therapy: A potential therapeutic strategy for incurable CSF1R-related leukoencephalopathy. Acta Neuropathol. Commun. 2020, 8, 217. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Takahashi, K. Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J. Neuroimmunol. 2007, 184, 92–99. [Google Scholar] [CrossRef]
- Picache, J.A.; Zheng, W.; Chen, C.Z. Therapeutic Strategies For Tay-Sachs Disease. Front. Pharmacol. 2022, 13, 906647. [Google Scholar] [CrossRef] [PubMed]
- Golebiowski, D.; van der Bom, I.M.J.; Kwon, C.S.; Miller, A.D.; Petrosky, K.; Bradbury, A.M.; Maitland, S.; Kuhn, A.L.; Bishop, N.; Curran, E.; et al. Direct Intracranial Injection of AAVrh8 Encoding Monkey beta-N-Acetylhexosaminidase Causes Neurotoxicity in the Primate Brain. Hum. Gene Ther. 2017, 28, 510–522. [Google Scholar] [CrossRef]
- Norflus, F.; Tifft, C.J.; McDonald, M.P.; Goldstein, G.; Crawley, J.N.; Hoffmann, A.; Sandhoff, K.; Suzuki, K.; Proia, R.L. Bone marrow transplantation prolongs life span and ameliorates neurologic manifestations in Sandhoff disease mice. J. Clin. Investig. 1998, 101, 1881–1888. [Google Scholar] [CrossRef]
- Wada, R.; Tifft, C.J.; Proia, R.L. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. USA 2000, 97, 10954–10959. [Google Scholar] [CrossRef]
- Sala, D.; Ornaghi, F.; Morena, F.; Argentati, C.; Valsecchi, M.; Alberizzi, V.; Di Guardo, R.; Bolino, A.; Aureli, M.; Martino, S.; et al. Therapeutic advantages of combined gene/cell therapy strategies in a murine model of GM2 gangliosidosis. Mol. Ther. Methods Clin. Dev. 2022, 25, 170–189. [Google Scholar] [CrossRef]
- Gleitz, H.F.; Liao, A.Y.; Cook, J.R.; Rowlston, S.F.; Forte, G.M.; D’Souza, Z.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 2018, 10, e8730. [Google Scholar] [CrossRef]
- Dogan, Y.; Barese, C.N.; Schindler, J.W.; Yoon, J.K.; Unnisa, Z.; Guda, S.; Jacobs, M.E.; Oborski, C.; Maiwald, T.; Clarke, D.L.; et al. Screening chimeric GAA variants in preclinical study results in hematopoietic stem cell gene therapy candidate vectors for Pompe disease. Mol. Ther. Methods Clin. Dev. 2022, 27, 464–487. [Google Scholar] [CrossRef] [PubMed]
- Unnisa, Z.; Yoon, J.K.; Schindler, J.W.; Mason, C.; van Til, N.P. Gene Therapy Developments for Pompe Disease. Biomedicines 2022, 10, 302. [Google Scholar] [CrossRef]
- Yoshimitsu, M.; Higuchi, K.; Ramsubir, S.; Nonaka, T.; Rasaiah, V.I.; Siatskas, C.; Liang, S.B.; Murray, G.J.; Brady, R.O.; Medin, J.A. Efficient correction of Fabry mice and patient cells mediated by lentiviral transduction of hematopoietic stem/progenitor cells. Gene Ther. 2007, 14, 256–265. [Google Scholar] [CrossRef] [PubMed]
- van Til, N.P.; Stok, M.; Aerts Kaya, F.S.; de Waard, M.C.; Farahbakhshian, E.; Visser, T.P.; Kroos, M.A.; Jacobs, E.H.; Willart, M.A.; van der Wegen, P.; et al. Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood 2010, 115, 5329–5337. [Google Scholar] [CrossRef] [PubMed]
- Stok, M.; de Boer, H.; Huston, M.W.; Jacobs, E.H.; Roovers, O.; Visser, T.P.; Jahr, H.; Duncker, D.J.; van Deel, E.D.; Reuser, A.J.J.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Corrects Murine Pompe Disease. Mol. Ther. Methods Clin. Dev. 2020, 17, 1014–1025. [Google Scholar] [CrossRef]
- Puzzo, F.; Colella, P.; Biferi, M.G.; Bali, D.; Paulk, N.K.; Vidal, P.; Collaud, F.; Simon-Sola, M.; Charles, S.; Hardet, R.; et al. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid alpha-glucosidase. Sci. Transl. Med. 2017, 99, eaam6375. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Huston, M.W.; Pagant, S.; Gan, L.; St Martin, S.; Sproul, S.; Richards, D.; Ballaron, S.; Hettini, K.; Ledeboer, A.; et al. AAV2/6 Gene Therapy in a Murine Model of Fabry Disease Results in Supraphysiological Enzyme Activity and Effective Substrate Reduction. Mol. Ther. Methods Clin. Dev. 2020, 18, 607–619. [Google Scholar] [CrossRef]
- Jeyakumar, J.M.; Kia, A.; Tam, L.C.S.; McIntosh, J.; Spiewak, J.; Mills, K.; Heywood, W.; Chisari, E.; Castaldo, N.; Verhoef, D.; et al. Preclinical evaluation of FLT190, a liver-directed AAV gene therapy for Fabry disease. Gene Ther. 2023, 30, 487–502. [Google Scholar] [CrossRef]
- Hillen, A.E.J.; Hruzova, M.; Rothgangl, T.; Breur, M.; Bugiani, M.; van der Knaap, M.S.; Schwank, G.; Heine, V.M. In vivo targeting of a variant causing vanishing white matter using CRISPR/Cas9. Mol. Ther. Methods Clin. Dev. 2022, 25, 17–25. [Google Scholar] [CrossRef]
- Bloomer, H.; Smith, R.H.; Hakami, W.; Larochelle, A. Genome editing in human hematopoietic stem and progenitor cells via CRISPR-Cas9-mediated homology-independent targeted integration. Mol. Ther. 2021, 29, 1611–1624. [Google Scholar] [CrossRef]
- Tran, N.T.; Graf, R.; Wulf-Goldenberg, A.; Stecklum, M.; Strauss, G.; Kuhn, R.; Kocks, C.; Rajewsky, K.; Chu, V.T. CRISPR-Cas9-Mediated ELANE Mutation Correction in Hematopoietic Stem and Progenitor Cells to Treat Severe Congenital Neutropenia. Mol. Ther. 2020, 28, 2621–2634. [Google Scholar] [CrossRef]
- Antony, J.S.; Daniel-Moreno, A.; Lamsfus-Calle, A.; Raju, J.; Kaftancioglu, M.; Urena-Bailen, G.; Rottenberger, J.; Hou, Y.; Santhanakumaran, V.; Lee, J.H.; et al. A Mutation-Agnostic Hematopoietic Stem Cell Gene Therapy for Metachromatic Leukodystrophy. CRISPR J. 2022, 5, 66–79. [Google Scholar] [CrossRef]
- Dorset, S.R.; Bak, R.O. The p53 challenge of hematopoietic stem cell gene editing. Mol. Ther. Methods Clin. Dev. 2023, 30, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.H.; Chiang, H.; Rees, H.A.; Edge, A.S.B.; Liu, D.R. In vivo base editing of post-mitotic sensory cells. Nat. Commun. 2018, 9, 2184. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Neugebauer, M.E.; Hsu, A.; Arbab, M.; Krasnow, N.A.; McElroy, A.N.; Pandey, S.; Doman, J.L.; Huang, T.P.; Raguram, A.; Banskota, S.; et al. Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nat. Biotechnol. 2023, 41, 673–685. [Google Scholar] [CrossRef]
- Lim, C.K.W.; Gapinske, M.; Brooks, A.K.; Woods, W.S.; Powell, J.E.; Zeballos, C.M.; Winter, J.; Perez-Pinera, P.; Gaj, T. Treatment of a Mouse Model of ALS by In Vivo Base Editing. Mol. Ther. 2020, 28, 1177–1189. [Google Scholar] [CrossRef]
- Levy, J.M.; Yeh, W.H.; Pendse, N.; Davis, J.R.; Hennessey, E.; Butcher, R.; Koblan, L.W.; Comander, J.; Liu, Q.; Liu, D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020, 4, 97–110. [Google Scholar] [CrossRef]
- Saito, M.; Xu, P.; Faure, G.; Maguire, S.; Kannan, S.; Altae-Tran, H.; Vo, S.; Desimone, A.; Macrae, R.K.; Zhang, F. Fanzor is a eukaryotic programmable RNA-guided endonuclease. Nature 2023, 620, 660–668. [Google Scholar] [CrossRef]
- Hakim, C.H.; Kumar, S.R.P.; Perez-Lopez, D.O.; Wasala, N.B.; Zhang, D.; Yue, Y.; Teixeira, J.; Pan, X.; Zhang, K.; Million, E.D.; et al. Cas9-specific immune responses compromise local and systemic AAV CRISPR therapy in multiple dystrophic canine models. Nat. Commun. 2021, 12, 6769. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Franklin, B.; Koob, J.; Kellner, M.J.; Ladha, A.; Joung, J.; Kirchgatterer, P.; Cox, D.B.T.; Zhang, F. A cytosine deaminase for programmable single-base RNA editing. Science 2019, 365, 382–386. [Google Scholar] [CrossRef] [PubMed]

| Clinical Trial | Title/Year | Country | Vector/ Transgene | NTC No |
|---|---|---|---|---|
| Ex vivo gene therapy | ||||
| Phase I | Gene Therapy for Gaucher’s and Fabry Disease Using Viruses and Blood-Forming Cells (1988–2022) | USA | RV-aGLA | NCT00001234 |
| Phase I active | Autologous Stem Cell Transplantation of Cells Engineered to Express Alpha-Galactosidase A in Patients With Fabry Disease (2016–2024) | Canada | LV-aGLA | NCT02800070 |
| Phase I/II terminated | Open Label, Study Of Efficacy and Safety Of AVR-RD-01 for Treatment-Naive Subjects With Classic Fabry Disease (2018–2022) | USA | LV-hGLA (AVR-RD-01) | NCT03454893 |
| Follow-up terminated | Long-Term Follow-up Study of Subjects With Fabry Disease Who Received Lentiviral Gene Therapy in Study AVRO-RD-01-201 (2019–2023) | Australia | LV-hGLA (AVRO-RD-01-201) | NCT04999059 |
| In vivo gene therapy | ||||
| Phase I/II completed | Safety Study of Recombinant Adeno-Associated Virus Acid Alpha-Glucosidase to Treat Pompe Disease (2010–2015) | USA | rAAV1-CMV-hGAA | NCT00976352 |
| Phase I/II completed | Re-administration of Intramuscular AAV9 in Patients With Late-Onset Pompe Disease (AAV9-GAA_IM) (2017–2021) | USA | rAAV9-DES-hGAA | NCT02240407 |
| Phase I/II | AAV2/8-LSPhGAA (ACTUS-101) in Late-Onset Pompe Disease (2018–2026) | USA | AAV2/8-LSP-hGAA (ACTUS-101) | NCT03533673 |
| Phase I/II terminated | A Fabry Disease Gene Therapy Study (MARVEL1) (2019–2023) | USA | AAV (FLT190) | NCT04040049 |
| Phase I/II recruiting | Dose-Ranging Study of ST-920, an AAV2/6 Human Alpha Galactosidase A Gene Therapy in Subjects With Fabry Disease (STAAR) (2019–2024) | USA | AAV2/6—hGLA (ST-920) | NCT04046224 |
| Phase I/II | A Gene Transfer Study for Late-Onset Pompe Disease (RESOLUTE) (2020–2027) | USA | AAV-rh74-GAA (SPK-3006) | NCT04093349 |
| Phase I/II recruiting | Gene Transfer Study in Patients With Late Onset Pompe Disease (FORTIS) (2020–2029) | USA | AAV8-GAA (AT845) | NCT04174105 |
| Follow-up active | A Long Term Follow-Up Study of Fabry Disease Subjects Treated With FLT190 (2020–2030) | Germany, UK | AAV (FLT190) | NCT04455230 |
| Phase I/II recruiting | An Open-label, Phase 1/2 Trial of Gene Therapy 4D-310 in Adults With Fabry Disease (2020–2027) | USA | AAV (4D-310) | NCT04519749 |
| Follow-up active | Long-Term Follow-up of Subjects Who Were Treated With ST-920 (2021–2029) | USA | AAV2/6—hGLA (ST-920) | NCT05039866 |
| Phase I/II recruiting | Clinical Exploration of Adeno-associated Virus (AAV) Expressing Human Acid Alpha-Glucosidase (GAA) Gene Therapy for Patients With Infantile-onset Pompe Disease (2022–2025) | China | AAV9-hGAA (GC301) | NCT05567627 |
| Phase I/II active | 4D-310 in Adults With Fabry Disease and Cardiac Involvement (2022–2028) | Australia | AAV(4D-310) | NCT05629559 |
| Phase I/II recruiting | Evaluation of the Safety and Efficacy of Infantile-onset Pompe Disease Gene Therapy Drug (2023–2024) | China | AAV9-hGAA (GC301) | NCT05793307 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aerts-Kaya, F.; van Til, N.P. Gene and Cellular Therapies for Leukodystrophies. Pharmaceutics 2023, 15, 2522. https://doi.org/10.3390/pharmaceutics15112522
Aerts-Kaya F, van Til NP. Gene and Cellular Therapies for Leukodystrophies. Pharmaceutics. 2023; 15(11):2522. https://doi.org/10.3390/pharmaceutics15112522
Chicago/Turabian StyleAerts-Kaya, Fatima, and Niek P. van Til. 2023. "Gene and Cellular Therapies for Leukodystrophies" Pharmaceutics 15, no. 11: 2522. https://doi.org/10.3390/pharmaceutics15112522
APA StyleAerts-Kaya, F., & van Til, N. P. (2023). Gene and Cellular Therapies for Leukodystrophies. Pharmaceutics, 15(11), 2522. https://doi.org/10.3390/pharmaceutics15112522