
Modulation of Brain Cholesterol Metabolism through CYP46A1 Overexpression for Rett Syndrome
 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. AAV Plasmid Design and Vector Production
2.3. Mecp2 Genotyping
2.4. Intravenous Injection
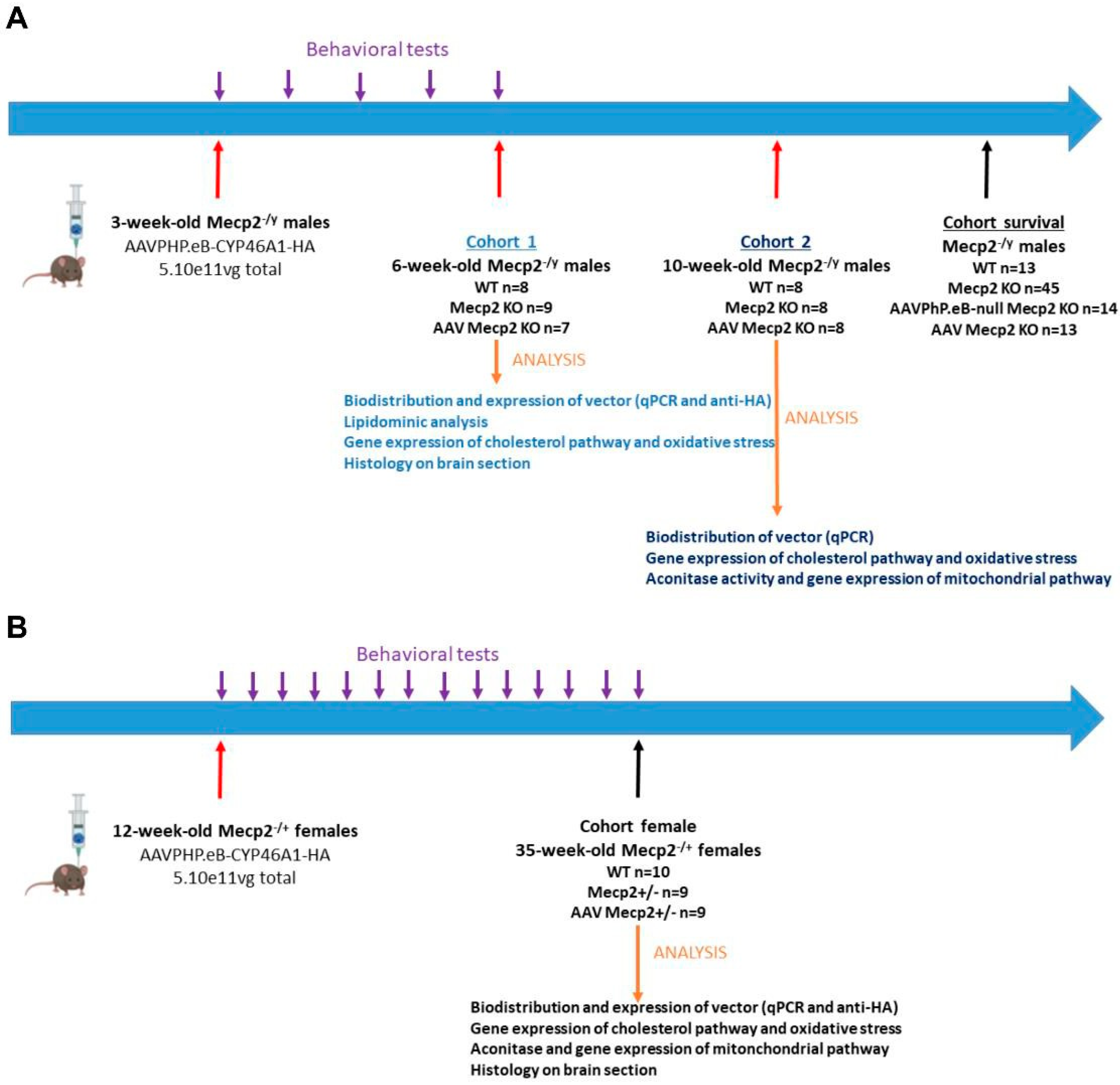
2.5. Experimental Design
2.5.1. Male Experiments (Refer to Table 1)
Cohort 1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | Age at Injection | WT | NT | AAV Treated | Age at Necropsy | |
|---|---|---|---|---|---|---|
| Cohort 1 | Males | 3 weeks | 8 | 9 | 7 | 6 weeks |
| Cohort 2 | Males | 3 weeks | 8 | 8 | 8 | 10 weeks |
| Cohort survival | Males | 3 weeks | 13 | 45 13 injected with AAV null | 14 | Variable depending on survival |
| Female cohort | Females | 12 weeks | 10 | 9 | 9 | 35 weeks |
Cohort 2
Cohort Survival
2.5.2. Female Experiment (Refer to Table 1)
2.6. Behavioral Tests
2.6.1. Weight Follow-Up
2.6.2. Clasping Test
2.6.3. Survival Evaluation
2.7. Tissue Collection
2.8. Biodistribution of Vector
2.9. Gene Expression
2.10. Cholesterol and Oxysterol Measurements
2.11. Mitochondrial Protein Enrichment and Aconitase Activity Test
2.12. Multiplex Immunofluorescence Staining
2.12.1. Development
2.12.2. Workflow
2.12.3. InForm® Analysis
2.13. Histological Analysis
2.13.1. Immunostaining
2.13.2. Image Acquisition
2.13.3. Quantification
2.14. Statistical Analysis
3. Results
3.1. Intravenous Administration of AAVPHP.eB-hCYP46A1-HA Vector Results in Widespread Distribution and Expression in CNS of Mecp2-Deficient Mice
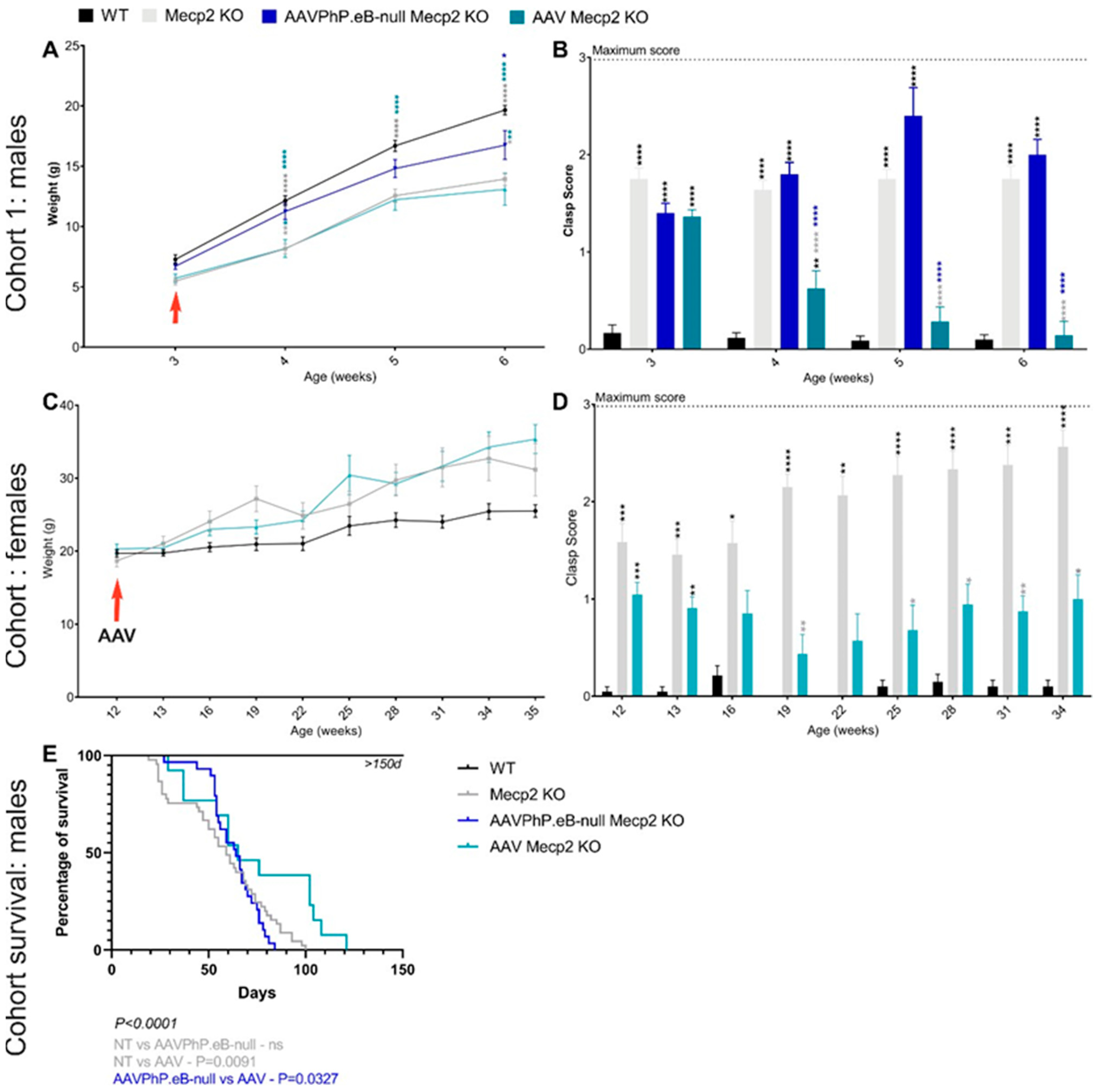
3.2. Overexpression of hCYP46A1 Led to Improvement of Behavioral Alterations as well as Life Expectancy in Mecp2-Deficient Male Mice
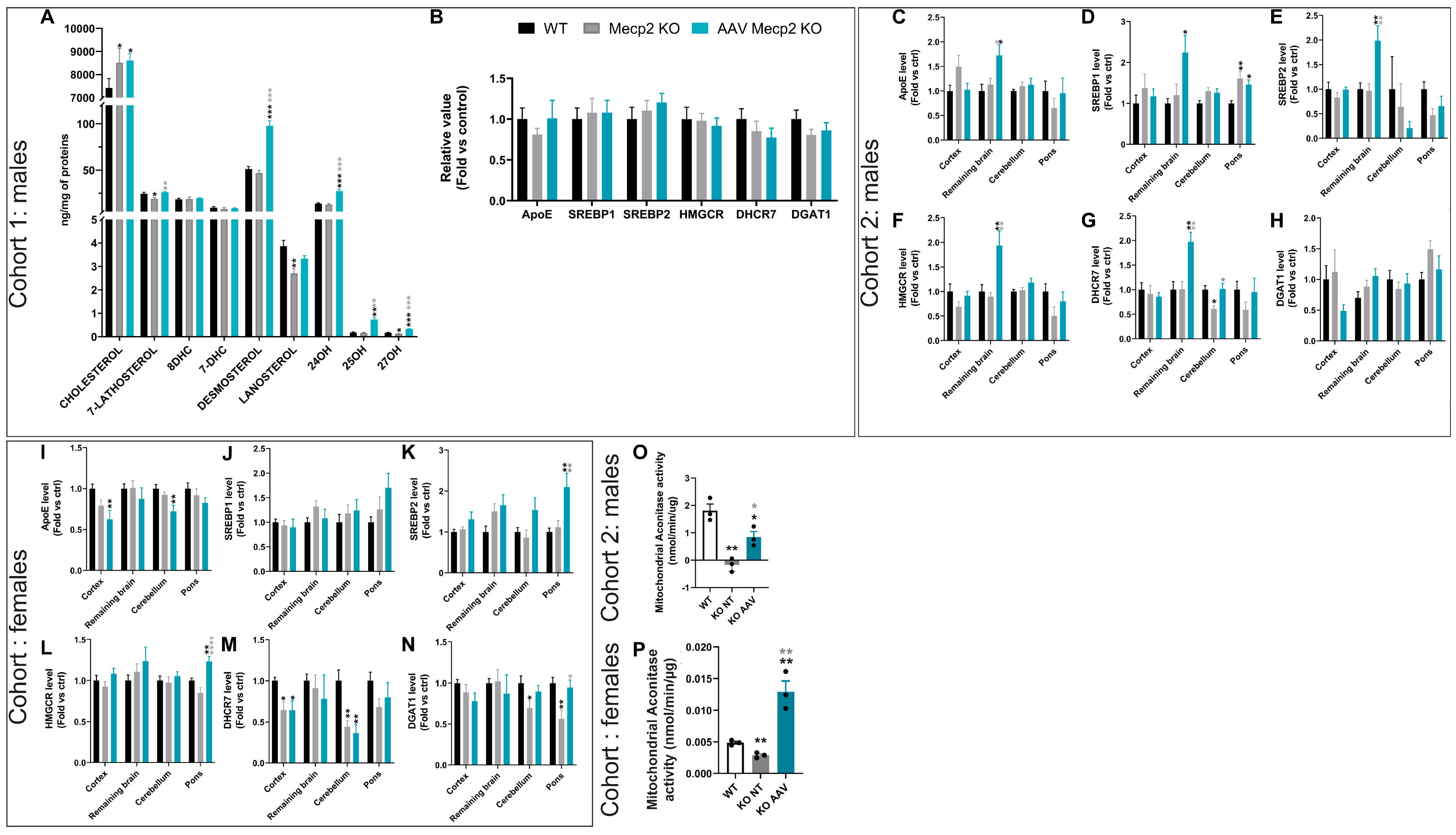
3.3. Overexpression of hCYP46A1 Led to Improvement/Activation of Mevalonate/Cholesterol Pathway and Mitochondrial Activity
3.4. Intravenous Administration of hCYP46A1 Did Not Lead to Severe Neuroinflammation in CNS or Neuronal Damage
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amir, R.E.; Van Den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett Syndrome Is Caused by Mutations in X-Linked MECP2, Encoding Methyl-CpG-Binding Protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Weaving, L.S.; Christodoulou, J.; Williamson, S.L.; Friend, K.L.; McKenzie, O.L.D.; Archer, H.; Evans, J.; Clarke, A.; Pelka, G.J.; Tam, P.P.L.; et al. Mutations of CDKL5 Cause a Severe Neurodevelopmental Disorder with Infantile Spasms and Mental Retardation. Am. J. Hum. Genet. 2004, 75, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Ariani, F.; Hayek, G.; Rondinella, D.; Artuso, R.; Mencarelli, M.A.; Spanhol-Rosseto, A.; Pollazzon, M.; Buoni, S.; Spiga, O.; Ricciardi, S.; et al. FOXG1 Is Responsible for the Congenital Variant of Rett Syndrome. Am. J. Hum. Genet. 2008, 83, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The Story of Rett Syndrome: From Clinic to Neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Ip, J.P.K.; Mellios, N.; Sur, M. Rett Syndrome: Insights into Genetic, Molecular and Circuit Mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M.; Goldstine, J.; Balmer, D.; Greco, C.M. Quantitative Localization of Heterogeneous Methyl-CpG-Binding Protein 2 (MeCP2) Expression Phenotypes in Normal and Rett Syndrome Brain by Laser Scanning Cytometry. Hum. Mol. Genet. 2001, 10, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett Syndrome: MeCP2 Levels Display Tissue- and Cell-Specific Differences and Correlate with Neuronal Maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Trofinetide: First Approval. Drugs 2023, 83, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Percy, A.K.; Benke, T.A.; Berry-Kravis, E.M.; Glaze, D.G.; Peters, S.U.; Jones, N.E.; Youakim, J.M. Design and Outcome Measures of LAVENDER, a Phase 3 Study of Trofinetide for Rett Syndrome. Contemp. Clin. Trials 2022, 114, 106704. [Google Scholar] [CrossRef]
- Fagiolini, M.; Patrizi, A.; LeBlanc, J.; Jin, L.W.; Maezawa, I.; Sinnett, S.; Gray, S.J.; Molholm, S.; Foxe, J.J.; Johnston, M.V.; et al. Intellectual and Developmental Disabilities Research Centers: A Multidisciplinary Approach to Understand the Pathogenesis of Methyl-CpG Binding Protein 2-Related Disorders. Neuroscience 2020, 445, 190–206. [Google Scholar] [CrossRef]
- Segatto, M.; Tonini, C.; Pfrieger, F.W.; Trezza, V.; Pallottini, V. Loss of Mevalonate/Cholesterol Homeostasis in the Brain: A Focus on Autism Spectrum Disorder and Rett Syndrome. Int. J. Mol. Sci. 2019, 20, 3317. [Google Scholar] [CrossRef] [PubMed]
- Segatto, M.; Trapani, L.; Di Tunno, I.; Sticozzi, C.; Valacchi, G.; Hayek, J.; Pallottini, V. Cholesterol Metabolism Is Altered in Rett Syndrome: A Study on Plasma and Primary Cultured Fibroblasts Derived from Patients. PLoS ONE 2014, 9, e104834. [Google Scholar] [CrossRef] [PubMed]
- Sticozzi, C.; Belmonte, G.; Pecorelli, A.; Cervellati, F.; Leoncini, S.; Signorini, C.; Ciccoli, L.; De Felice, C.; Hayek, J.; Valacchi, G. Scavenger Receptor B1 Post-Translational Modifications in Rett Syndrome. FEBS Lett. 2013, 587, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Zandl-lang, M.; Züllig, T.; Trötzmüller, M.; Naegelin, Y.; Abela, L.; Wilken, B.; Scholl-buergi, S.; Karall, D.; Kappos, L.; Köfeler, H.; et al. Changes in the Cerebrospinal Fluid and Plasma Lipidome in Patients with Rett Syndrome. Metabolites 2022, 12, 40291. [Google Scholar] [CrossRef] [PubMed]
- Buchovecky, C.M.; Turley, S.D.; Brown, H.M.; Kyle, S.M.; McDonald, J.G.; Liu, B.; Pieper, A.A.; Huang, W.; Katz, D.M.; Russell, D.W.; et al. A Suppressor Screen in Mecp2 Mutant Mice Implicates Cholesterol Metabolism in Rett Syndrome. Nat. Genet. 2013, 45, 1013–1020. [Google Scholar] [CrossRef]
- Lütjohann, D.; Lopez, A.M.; Chuang, J.C.; Kerksiek, A.; Turley, S.D. Identification of Correlative Shifts in Indices of Brain Cholesterol Metabolism in the C57BL6/Mecp2 Tm1.1Bird Mouse, a Model for Rett Syndrome. Lipids 2018, 53, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.M.; Chuang, J.C.; Posey, K.S.; Turley, S.D. Suppression of Brain Cholesterol Synthesis in Male Mecp2-Deficient Mice Is Age Dependent and Not Accompanied by a Concurrent Change in the Rate of Fatty Acid Synthesis. Brain Res. 2017, 1654, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.M.; Kasimov, M.R.; Zefirov, A.L. Brain Cholesterol Metabolism and Its Defects: Linkage to Neurodegenerative Diseases and Synaptic Dysfunction. Acta Nat. 2016, 8, 58–73. [Google Scholar] [CrossRef]
- Björkhem, I.; Meaney, S.; Fogelman, A.M. Brain Cholesterol: Long Secret Life behind a Barrier. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 806–815. [Google Scholar] [CrossRef]
- Björkhem, I.; Lütjohann, D.; Diczfalusy, U.; Ståhle, L.; Ahlborg, G.; Wahren, J. Cholesterol Homeostasis in Human Brain: Turnover of 24S- Hydroxycholesterol and Evidence for a Cerebral Origin of Most of This Oxysterol in the Circulation. J. Lipid Res. 1998, 39, 1594–1600. [Google Scholar] [CrossRef]
- Björkhem, I. Crossing the Barrier: Oxysterols as Cholesterol Transporters and Metabolic Modulators in the Brain. J. Intern. Med. 2006, 260, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W.; Halford, R.W.; Ramirez, D.M.O.; Shah, R.; Kotti, T. Cholesterol 24-Hydroxylase: An Enzyme of Cholesterol Turnover in the Brain. Annu. Rev. Biochem. 2009, 78, 1017–1040. [Google Scholar] [CrossRef]
- Puglielli, L.; Tanzi, R.E.; Kovacs, D.M. Alzheimer’s Disease: The Cholesterol Connection. Nat. Neurosci. 2003, 6, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B. Cholesterol and the Biology of Alzheimer’s Disease. Neuron 2004, 41, 7–10. [Google Scholar] [CrossRef]
- Valenza, M.; Rigamonti, D.; Goffredo, D.; Zuccato, C.; Fenu, S.; Jamot, L.; Strand, A.; Tarditi, A.; Woodman, B.; Racchi, M.; et al. Dysfunction of the Cholesterol Biosynthetic Pathway in Huntington’s Disease. J. Neurosci. 2005, 25, 9932–9939. [Google Scholar] [CrossRef]
- Valenza, M.; Leoni, V.; Karasinska, J.M.; Petricca, L.; Fan, J.; Carroll, J.; Pouladi, M.A.; Fossale, E.; Nguyen, H.P.; Riess, O.; et al. Cholesterol Defect Is Marked across Multiple Rodent Models of Huntington’s Disease and Is Manifest in Astrocytes. J. Neurosci. 2010, 30, 10844–10850. [Google Scholar] [CrossRef]
- Cheng, D.; Jenner, A.M.; Shui, G.; Cheong, W.F.; Mitchell, T.W.; Nealon, J.R.; Kim, W.S.; McCann, H.; Wenk, M.R.; Halliday, G.M.; et al. Lipid Pathway Alterations in Parkinson’s Disease Primary Visual Cortex. PLoS ONE 2011, 6, e17299. [Google Scholar] [CrossRef]
- Pikuleva, I. Targeting Cytochrome P450 46A1 and Brain Cholesterol 24-Hydroxylation to Treat Neurodegenerative Diseases. Explor. Neuroprotective Ther. 2021, 1, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Hudry, E.; Van Dam, D.; Kulik, W.; De Deyn, P.P.; Stet, F.S.; Ahouansou, O.; Benraiss, A.; Delacourte, A.; Bougnères, P.; Aubourg, P.; et al. Adeno-Associated Virus Gene Therapy with Cholesterol 24-Hydroxylase Reduces the Amyloid Pathology before or after the Onset of Amyloid Plaques in Mouse Models of Alzheimer’s Disease. Mol. Ther. 2010, 18, 44–53. [Google Scholar] [CrossRef]
- Boussicault, L.; Alves, S.; Lamazière, A.; Planques, A.; Heck, N.; Moumné, L.; Despres, G.; Bolte, S.; Hu, A.; Pagès, C.; et al. CYP46A1, the Rate-Limiting Enzyme for Cholesterol Degradation, Is Neuroprotective in Huntington’s Disease. Brain 2016, 139, 953–970. [Google Scholar] [CrossRef]
- Nagy, G.; Ackerman, S.L. Cholesterol Metabolism and Rett Syndrome Pathogenesis. Nat. Genet. 2013, 45, 965–967. [Google Scholar] [CrossRef] [PubMed]
- Kyle, S.M.; Saha, P.K.; Brown, H.M.; Chan, L.C.; Justice, M.J. MeCP2 Co-Ordinates Liver Lipid Metabolism with the NCoR1/HDAC3 Corepressor Complex. Hum. Mol. Genet. 2016, 25, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.L.; Sánchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for Efficient Noninvasive Gene Delivery to the Central and Peripheral Nervous Systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of Neurological Defects in a Mouse Model of Rett Syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Chevy, F.; Humbert, L.; Wolf, C. Sterol Profiling of Amniotic Fluid: A Routine Method for the Detection of Distal Cholesterol Synthesis Deficit. Prenat. Diagn. 2005, 25, 1000–1006. [Google Scholar] [CrossRef]
- Silvia Ricci, F.; Stanga, S.; Mezzanotte, M.; Marinaccio, C.; Somà, A.; Sottemano, S.; Conio, A.; Morana, G.; Spada, M.; Boido, M.; et al. Biochemical Characterization on Muscle Tissue of a Novel Biallelic ACO2 Mutation in an Infant with Progressive Encephalopathy. JIMD Rep. 2023, 65, 3–9. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A Mouse Mecp2-Null Mutation Causes Neurological Symptoms That Mimic Rett Syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Shulyakova, N.; Andreazza, A.C.; Mills, L.R.; Eubanks, J.H. Mitochondrial Dysfunction in the Pathogenesis of Rett Syndrome: Implications for Mitochondria-Targeted Therapies. Front. Cell. Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Dayton, R.D.; Grames, M.S.; Klein, R.L. More Expansive Gene Transfer to the Rat CNS: AAV PHP.EB Vector Dose–Response and Comparison to AAV PHP.B. Gene Ther. 2018, 25, 392–400. [Google Scholar] [CrossRef]
- Audouard, E.; Oger, V.; Meha, B.; Cartier, N.; Sevin, C.; Piguet, F. Complete Correction of Brain and Spinal Cord Pathology in Metachromatic Leukodystrophy Mice. Front. Mol. Neurosci. 2021, 14, 677895. [Google Scholar] [CrossRef]
- Audouard, E.; Khefif, N.; Mansat, C.; Nelcha, O.; Banchi, E.-G.; Lupiet, C.; Farabos, D.; Lamaziere, A.; Sevin, C.; Piguet, F. Dose Response Evaluation of Intravenous Gene Therapy in Symptomatic Mouse Model of Metachromatic Leukodystrophy. Mol. Ther. Methods Clin. Dev. 2023, 32, 101248. [Google Scholar] [CrossRef] [PubMed]
- Piguet, F.; de Montigny, C.; Vaucamps, N.; Reutenauer, L.; Eisenmann, A.; Puccio, H. Rapid and Complete Reversal of Sensory Ataxia by Gene Therapy in a Novel Model of Friedreich Ataxia. Mol. Ther. 2018, 26, 1940–1952. [Google Scholar] [CrossRef]
- Hammelrath, L.; Škokić, S.; Khmelinskii, A.; Hess, A.; van der Knaap, N.; Staring, M.; Lelieveldt, B.P.F.; Wiedermann, D.; Hoehn, M. Morphological Maturation of the Mouse Brain: An in Vivo MRI and Histology Investigation. Neuroimage 2016, 125, 144–152. [Google Scholar] [CrossRef]
- Xie, B.S.; Wang, X.; Pan, Y.H.; Jiang, G.; Feng, J.F.; Lin, Y. Apolipoprotein E, Low-Density Lipoprotein Receptor, and Immune Cells Control Blood-Brain Barrier Penetration by AAV-PHP.EB in Mice. Theranostics 2021, 11, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Knox, E.G.; Aburto, M.R.; Clarke, G.; Cryan, J.F.; O’driscoll, C.M. The Blood-Brain Barrier in Aging and Neurodegeneration. Mol. Psychiatry 2022, 27, 2659–2673. [Google Scholar] [CrossRef] [PubMed]
- Mezzanotte, M.; Ammirata, G.; Boido, M.; Stanga, S.; Roetto, A. Activation of the Hepcidin-Ferroportin1 Pathway in the Brain and Astrocytic-Neuronal Crosstalk to Counteract Iron Dyshomeostasis during Aging. Sci. Rep. 2022, 12, 1–11. [Google Scholar] [CrossRef]
- Gold, W.A.; Williamson, S.L.; Kaur, S.; Hargreaves, I.P.; Land, J.M.; Pelka, G.J.; Tam, P.P.L.; Christodoulou, J. Mitochondrial Dysfunction in the Skeletal Muscle of a Mouse Model of Rett Syndrome (RTT): Implications for the Disease Phenotype. Mitochondrion 2014, 15, 10–17. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Paterson, A.; Curtis, J.; Guy, J.; MacLeod, N.; Bird, A. Gene Expression Analysis Exposes Mitochondrial Abnormalities in a Mouse Model of Rett Syndrome. Mol. Cell. Biol. 2006, 26, 5033–5042. [Google Scholar] [CrossRef]
- Dotti, M.T.; Manneschi, L.; Malandrini, A.; De Stefano, N.; Caznerale, F.; Federico, A. Mitochondrial Dysfunction in Rett Syndrome. An Ultrastructural and Biochemical Study. Brain Dev. 1993, 15, 103–106. [Google Scholar] [CrossRef]
- Schon, E.A.; Manfredi, G. Neuronal Degeneration and Mitochondrial Dysfunction. J. Clin. Investig. 2003, 111, 303–312. [Google Scholar] [CrossRef]
- De Felice, C.; Ciccoli, L.; Leoncini, S.; Signorini, C.; Rossi, M.; Vannuccini, L.; Guazzi, G.; Latini, G.; Comporti, M.; Valacchi, G.; et al. Systemic Oxidative Stress in Classic Rett Syndrome. Free Radic. Biol. Med. 2009, 47, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Leoncini, S.; de Felice, C.; Signorini, C.; Pecorelli, A.; Durand, T.; Valacchi, G.; Ciccoli, L.; Hayek, J. Oxidative Stress in Rett Syndrome: Natural History, Genotype, and Variants. Redox Rep. 2011, 16, 145–153. [Google Scholar] [CrossRef]
- De Felice, C.; Della Ragione, F.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Ciccoli, L.; Scalabrì, F.; Marracino, F.; Madonna, M.; Belmonte, G.; et al. Oxidative Brain Damage in Mecp2-Mutant Murine Models of Rett Syndrome. Neurobiol. Dis. 2014, 68, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Swanberg, S.; Harvey, D.; LaSalle, J.M.; Jin, L.W. Rett Syndrome Astrocytes Are Abnormal and Spread MeCP2 Deficiency through Gap Junctions. J. Neurosci. 2009, 29, 5051–5061. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.O.; Zachariah, R.M.; Ezeonwuka, C.D.; Liyanage, V.R.B.; Rastegar, M. Brain Region-Specific Expression of MeCP2 Isoforms Correlates with DNA Methylation within Mecp2 Regulatory Elements. PLoS ONE 2014, 9, e90645. [Google Scholar] [CrossRef]
- Ballas, N.; Lioy, D.T.; Grunseich, C.; Mandel, G. Non-Cell Autonomous Influence of MeCP2-Deficient Glia on Neuronal Dendritic Morphology. Nat. Neurosci. 2009, 12, 311–317. [Google Scholar] [CrossRef]
- Williams, E.C.; Zhong, X.; Mohamed, A.; Li, R.; Liu, Y.; Dong, Q.; Ananiev, G.E.; Choongmok, J.C.; Lin, B.R.; Lu, J.; et al. Mutant Astrocytes Differentiated from Rett Syndrome Patients-Specific IPSCs Have Adverse Effects on Wild-Type Neurons. Hum. Mol. Genet. 2014, 23, 2968–2980. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.K.; Lioy, D.T.; Knopp, S.J.; Bissonnette, J.M. Conditional Depletion of Methyl-CpG-Binding Protein 2 in Astrocytes Depresses the Hypercapnic Ventilatory Response in Mice. J. Appl. Physiol. 2015, 119, 670–676. [Google Scholar] [CrossRef]
- Jin, X.R.; Chen, X.S.; Xiao, L. MeCP2 Deficiency in Neuroglia: New Progress in the Pathogenesis of Rett Syndrome. Front. Mol. Neurosci. 2017, 10, 316. [Google Scholar] [CrossRef]
- Lioy, D.T.; Garg, S.K.; Monaghan, C.E.; Raber, J.; Foust, K.D.; Kaspar, B.K.; Hirrlinger, P.G.; Kirchhoff, F.; Bissonnette, J.M.; Ballas, N.; et al. A Role for Glia in the Progression of Rett-Syndrome. Nature 2011, 475, 497–500. [Google Scholar] [CrossRef]
- Wu, W.; Gu, W.; Xu, X.; Shang, S.; Zhao, Z. Downregulation of CNPase in a MeCP2 Deficient Mouse Model of Rett Syndrome. Neurol. Res. 2012, 34, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Albizzati, E.; Florio, E.; Miramondi, F.; Sormonta, I.; Landsberger, N.; Frasca, A. Identification of Region-Specific Cytoskeletal and Molecular Alterations in Astrocytes of Mecp2 Deficient Animals. Front. Neurosci. 2022, 16, 823060. [Google Scholar] [CrossRef] [PubMed]
- Derecki, N.C.; Cronk, J.C.; Lu, Z.; Xu, E.; Abbott, S.B.G.; Guyenet, P.G.; Kipnis, J. Wild-Type Microglia Arrest Pathology in a Mouse Model of Rett Syndrome. Nature 2012, 484, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Jin, L.W. Rett Syndrome Microglia Damage Dendrites and Synapses by the Elevated Release of Glutamate. J. Neurosci. 2010, 30, 5346–5356. [Google Scholar] [CrossRef] [PubMed]
- Cronk, J.C.; Derecki, N.C.; Ji, E.; Xu, Y.; Lampano, A.E.; Smirnov, I.; Baker, W.; Norris, G.T.; Marin, I.; Coddington, N.; et al. Methyl-CpG Binding Protein 2 Regulates Microglia and Macrophage Gene Expression in Response to Inflammatory Stimuli. Immunity 2015, 42, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Achilly, N.P.; He, L.J.; Kim, O.A.; Ohmae, S.; Wojaczynski, G.J.; Lin, T.; Sillitoe, R.V.; Medina, J.F.; Zoghbi, H.Y. Deleting Mecp2 from the Cerebellum Rather than Its Neuronal Subtypes Causes a Delay in Motor Learning in Mice. Elife 2021, 10, 64833. [Google Scholar] [CrossRef] [PubMed]
- Oldfors, A.; Sourander, P.; Armstrong, D.L.; Percy, A.K.; Witt-Engerström, I.; Hagberg, B.A. Rett Syndrome: Cerebellar Pathology. Pediatr. Neurol. 1990, 6, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of Methyl-CpG Binding Protein-2 in CNS Neurons Results in a Rett-like Phenotype in Mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Pelka, G.J.; Watson, C.M.; Radziewic, T.; Hayward, M.; Lahooti, H.; Christodoulou, J.; Tam, P.P.L. Mecp2 Deficiency Is Associated with Learning and Cognitive Deficits and Altered Gene Activity in the Hippocampal Region of Mice. Brain 2006, 129, 887–898. [Google Scholar] [CrossRef]
- Pikuleva, I.A.; Cartier, N. Cholesterol Hydroxylating Cytochrome P450 46A1: From Mechanisms of Action to Clinical Applications. Front. Aging Neurosci. 2021, 13, 696778. [Google Scholar] [CrossRef]
- Vashi, N.; Justice, M.J. Treating Rett Syndrome: From Mouse Models to Human Therapies. Mamm. Genome 2019, 30, 90–110. [Google Scholar] [CrossRef] [PubMed]
- Nóbrega, C.; Mendonça, L.; Marcelo, A.; Lamazière, A.; Tomé, S.; Despres, G.; Matos, C.A.; Mechmet, F.; Langui, D.; den Dunnen, W.; et al. Restoring Brain Cholesterol Turnover Improves Autophagy and Has Therapeutic Potential in Mouse Models of Spinocerebellar Ataxia. Acta Neuropathol. 2019, 138, 837–858. [Google Scholar] [CrossRef] [PubMed]
- Panayotis, N.; Ehinger, Y.; Felix, M.S.; Roux, J.C. State-of-the-Art Therapies for Rett Syndrome. Dev. Med. Child Neurol. 2023, 65, 162–170. [Google Scholar] [CrossRef] [PubMed]





| Name | Primer 5′ -> 3′ |
|---|---|
| mADCK3 Forward | CCA CCT CTC CTA TGG GCA GA |
| mADCK3 Reverse | CCG GGC CTT TTC AAT GTC T |
| Actine Forward | TCC TGA GCG CAA GTA CTC TGT |
| Actine Reverse | CTG ATC CAC ATC TGC TGG AAG |
| Murine Cyp46A Forward | GGC TAA GAA GTA TGG TCC TGT TGT AAG A |
| Murine Cyp46A1 Reverse | GGT GGA CAT CAG GAA CTT CTT GAC T |
| Human Cyp46A1 Forward | CGA GTC CTG AGT CGG TTA AGA AGT T |
| Human Cyp46A1Reverse | AGT CTG GAG CGC ACG GTA CAT |
| ApoE Forward | GTC ACA TTG CTG ACA GGA TGC CTA |
| ApoE Reverse | GGG TTG GTT GCT TTG CCA CTC |
| Hmgcr Forward | CCC CAC ATT CAC TCT TGA CGC TCT |
| Hmgcr Reverse | GCT GGC GGA CGC CTG ACA T |
| Srebp1 Forward | GGT CCA GCA GGT CCC AGT TGT |
| Srebp1 Reverse | CTG CAG TCT TCA CGG TGG CTC |
| Srebp2 Forward | TGT TGA CGC AGA CAG CCA ATG |
| Dhcr7 Forward | AGACATTTGGGCCAAGACAC |
| Dhcr7 Reverse | AACCTGGCAGAAATCTGTGG |
| Dgat1 Forward | CCTCAGCCTTCTTCCATGAG |
| Dgat1 Reverse | ACTGGGGCATCGTAGTTGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Audouard, E.; Khefif, N.; Gillet-Legrand, B.; Nobilleau, F.; Bouazizi, O.; Stanga, S.; Despres, G.; Alves, S.; Lamazière, A.; Cartier, N.; et al. Modulation of Brain Cholesterol Metabolism through CYP46A1 Overexpression for Rett Syndrome. Pharmaceutics 2024, 16, 756. https://doi.org/10.3390/pharmaceutics16060756
Audouard E, Khefif N, Gillet-Legrand B, Nobilleau F, Bouazizi O, Stanga S, Despres G, Alves S, Lamazière A, Cartier N, et al. Modulation of Brain Cholesterol Metabolism through CYP46A1 Overexpression for Rett Syndrome. Pharmaceutics. 2024; 16(6):756. https://doi.org/10.3390/pharmaceutics16060756
Chicago/Turabian StyleAudouard, Emilie, Nicolas Khefif, Béatrix Gillet-Legrand, Fanny Nobilleau, Ouafa Bouazizi, Serena Stanga, Gaëtan Despres, Sandro Alves, Antonin Lamazière, Nathalie Cartier, and et al. 2024. "Modulation of Brain Cholesterol Metabolism through CYP46A1 Overexpression for Rett Syndrome" Pharmaceutics 16, no. 6: 756. https://doi.org/10.3390/pharmaceutics16060756
APA StyleAudouard, E., Khefif, N., Gillet-Legrand, B., Nobilleau, F., Bouazizi, O., Stanga, S., Despres, G., Alves, S., Lamazière, A., Cartier, N., & Piguet, F. (2024). Modulation of Brain Cholesterol Metabolism through CYP46A1 Overexpression for Rett Syndrome. Pharmaceutics, 16(6), 756. https://doi.org/10.3390/pharmaceutics16060756