
APJ as Promising Therapeutic Target of Peptide Analogues in Myocardial Infarction- and Hypertension-Induced Heart Failure

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. APJ and Its Endogenous Agonists
2.1. APJ
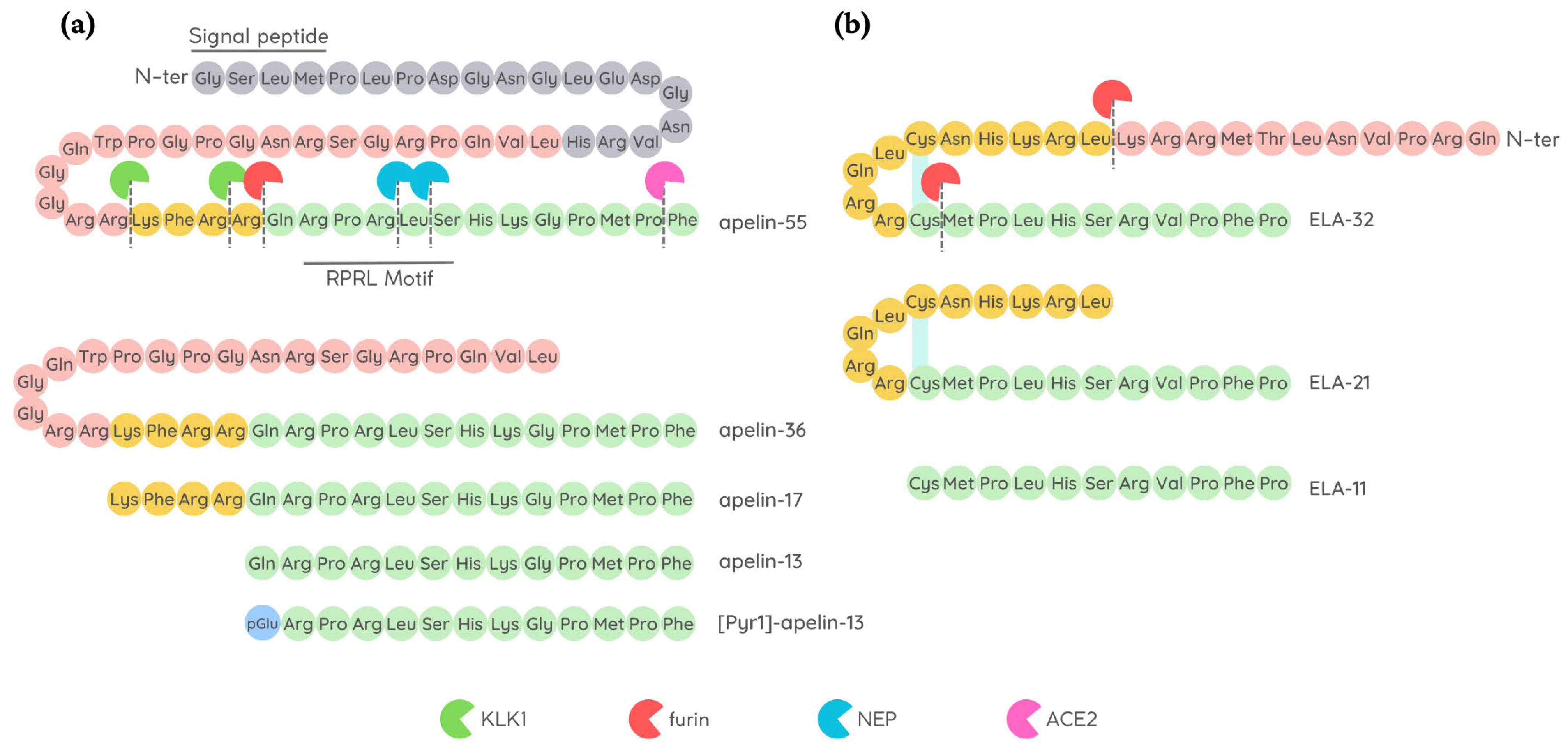
2.2. Apelin
2.3. ELABELA
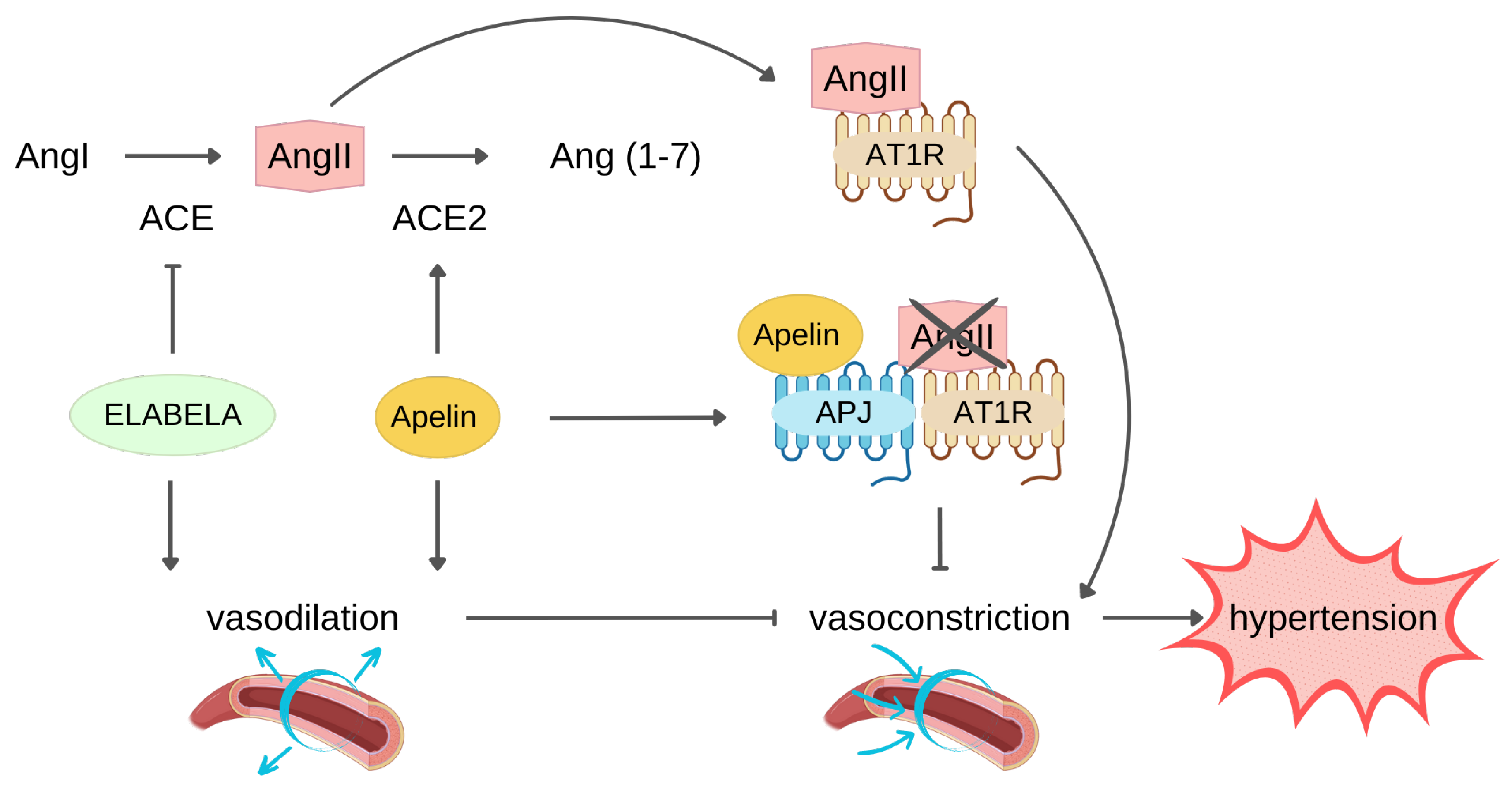
2.4. Physiological Ligand Activities in CV System
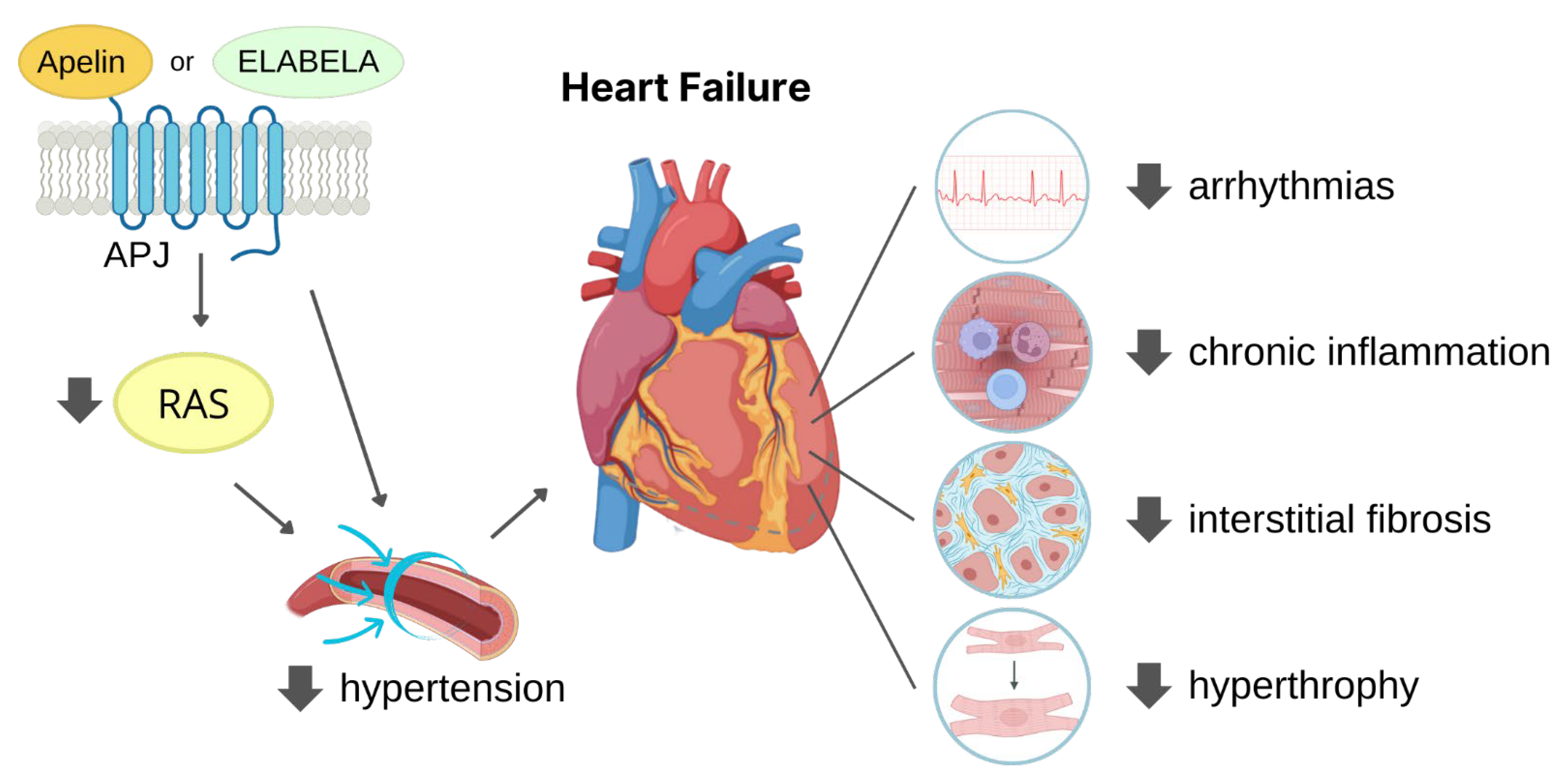
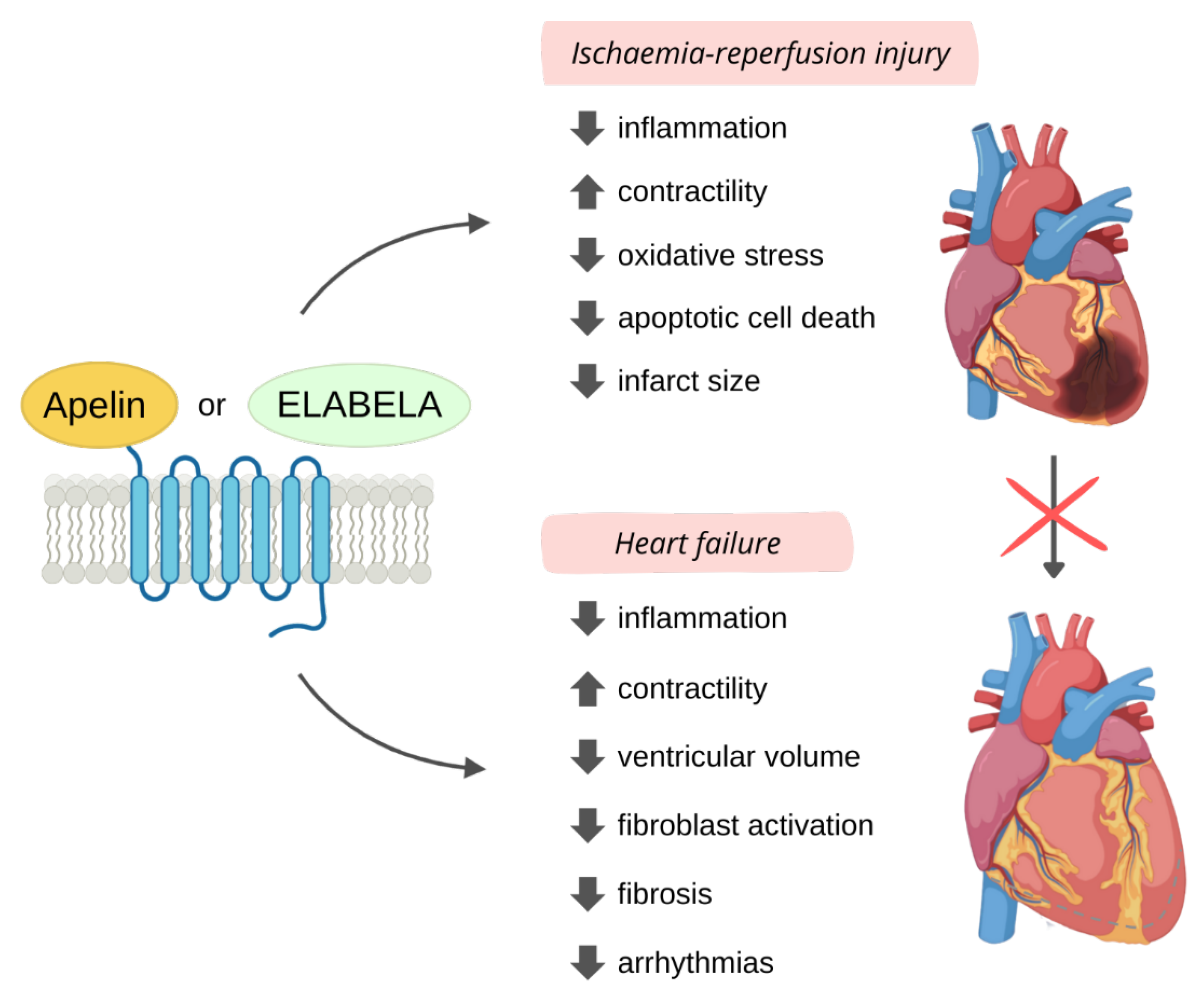
3. Cardioprotective Role of APJ Endogenous Ligands
3.1. Protection against Hypertension
3.2. Protection against MI
3.2.1. I/R
3.2.2. Adverse Remodelling
3.2.3. Vessel Formation
4. APJ Peptide Agonists
4.1. APJ Endogenous Ligand Main Chain Modifications
4.1.1. Modification of Apelin Cleavage Sites
4.1.2. Apelin Cyclization
4.1.3. Residue Modification of Apelin and ELA
4.2. Antibody-Based/Bound APJ Agonists
4.3. Side Effect and Delivery Issues
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Subramaniam, A.V.; Weston, S.A.; Killian, J.M.; Schulte, P.J.; Roger, V.L.; Redfield, M.M.; Blecker, S.B.; Dunlay, S.M. Development of Advanced Heart Failure: A Population-Based Study. Circ. Heart Fail. 2022, 15, e009218. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Fighting Cardiac Fibrosis with CAR T Cells. N. Engl. J. Med. 2022, 386, 1576–1578. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, B.F.; Heiber, M.; Chan, A.; Heng, H.H.; Tsui, L.C.; Kennedy, J.L.; Shi, X.; Petronis, A.; George, S.R.; Nguyen, T. A Human Gene That Shows Identity with the Gene Encoding the Angiotensin Receptor Is Located on Chromosome 11. Gene 1993, 136, 355–360. [Google Scholar] [CrossRef]
- Kleinz, M.J.; Skepper, J.N.; Davenport, A.P. Immunocytochemical Localisation of the Apelin Receptor, APJ, to Human Cardiomyocytes, Vascular Smooth Muscle and Endothelial Cells. Regul. Pept. 2005, 126, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tang, J.; Song, J.; Xie, M.; Liu, Y.; Dong, Z.; Liu, X.; Li, X.; Zhang, M.; Chen, Y.; et al. Elabela Alleviates Ferroptosis, Myocardial Remodeling, Fibrosis and Heart Dysfunction in Hypertensive Mice by Modulating the IL-6/STAT3/GPX4 Signaling. Free Radic. Biol. Med. 2022, 181, 130–142. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, X.; Zhou, X.; Cheng, B.; Li, G.; Bai, B. Temporal Expression of Apelin/Apelin Receptor in Ischemic Stroke and Its Therapeutic Potential. Front. Mol. Neurosci. 2017, 10, 1. [Google Scholar] [CrossRef]
- Dawid, M.; Mlyczyńska, E.; Jurek, M.; Respekta, N.; Pich, K.; Kurowska, P.; Gieras, W.; Milewicz, T.; Kotula-Balak, M.; Rak, A. Apelin, APJ, and ELABELA: Role in Placental Function, Pregnancy, and Foetal Development—An Overview. Cells 2021, 11, 99. [Google Scholar] [CrossRef]
- Chapman, N.A.; Dupré, D.J.; Rainey, J.K. The Apelin Receptor: Physiology, Pathology, Cell Signalling, and Ligand Modulation of a Peptide-Activated Class A GPCR. Biochem. Cell Biol. 2014, 92, 431–440. [Google Scholar] [CrossRef]
- Chen, J.; Chen, X.; Li, S.; Jiang, Y.; Mao, H.; Zhang, R.; Ji, B.; Yan, M.; Cai, X.; Wang, C. Individual Phosphorylation Sites at the C-Terminus of the Apelin Receptor Play Different Roles in Signal Transduction. Redox Biol. 2020, 36, 101629. [Google Scholar] [CrossRef]
- Scimia, M.C.; Hurtado, C.; Ray, S.; Metzler, S.; Wei, K.; Wang, J.; Woods, C.E.; Purcell, N.H.; Catalucci, D.; Akasaka, T.; et al. APJ Acts as a Dual Receptor in Cardiac Hypertrophy. Nature 2012, 488, 394–398. [Google Scholar] [CrossRef]
- D’Aniello, C.; Fiorenzano, A.; Iaconis, S.; Liguori, G.L.; Andolfi, G.; Cobellis, G.; Fico, A.; Minchiotti, G. The G-Protein-Coupled Receptor APJ Is Expressed in the Second Heart Field and Regulates Cerberus–Baf60c Axis in Embryonic Stem Cell Cardiomyogenesis. Cardiovasc. Res. 2013, 100, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Ho, L.; Ford, G.H.; Chen, H.I.; Goldstone, A.B.; Woo, Y.J.; Quertermous, T.; Reversade, B.; Red-Horse, K. Alternative Progenitor Cells Compensate to Rebuild the Coronary Vasculature in Elabela- and Apj-Deficient Hearts. Dev. Cell 2017, 42, 655–666.e3. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Xu, J.; Chen, L.; Li, L. Apelin/APJ Signaling in Hypoxia-Related Diseases. Clin. Chim. Acta 2015, 451, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Tatemoto, K.; Hosoya, M.; Habata, Y.; Fujii, R.; Kakegawa, T.; Zou, M.-X.; Kawamata, Y.; Fukusumi, S.; Hinuma, S.; Kitada, C.; et al. Isolation and Characterization of a Novel Endogenous Peptide Ligand for the Human APJ Receptor. Biochem. Biophys. Res. Commun. 1998, 251, 471–476. [Google Scholar] [CrossRef]
- Liu, J.; Liu, M.; Chen, L. Novel Pathogenesis: Regulation of Apoptosis by Apelin/APJ System. Acta Biochim. Biophys. Sin. 2017, 49, 471–478. [Google Scholar] [CrossRef]
- Kawamata, Y.; Habata, Y.; Fukusumi, S.; Hosoya, M.; Fujii, R.; Hinuma, S.; Nishizawa, N.; Kitada, C.; Onda, H.; Nishimura, O.; et al. Molecular Properties of Apelin: Tissue Distribution and Receptor Binding. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2001, 1538, 162–171. [Google Scholar] [CrossRef]
- De Falco, M.; De Luca, L.; Onori, N.; Cavallotti, I.; Artigiano, F.; Esposito, V.; De Luca, B.; Laforgia, V.; Groeger, A.M.; De Luca, A. Apelin Expression in Normal Human Tissues. In Vivo 2002, 16, 333–336. [Google Scholar]
- Shin, K.; Pandey, A.; Liu, X.-Q.; Anini, Y.; Rainey, J.K. Preferential Apelin-13 Production by the Proprotein Convertase PCSK3 Is Implicated in Obesity. FEBS Open Bio. 2013, 3, 328–333. [Google Scholar] [CrossRef]
- Zhang, Y.; Maitra, R.; Harris, D.L.; Dhungana, S.; Snyder, R.; Runyon, S.P. Identifying Structural Determinants of Potency for Analogs of Apelin-13: Integration of C-Terminal Truncation with Structure–Activity. Bioorg. Med. Chem. 2014, 22, 2992–2997. [Google Scholar] [CrossRef]
- Murza, A.; Belleville, K.; Longpré, J.-M.; Sarret, P.; Marsault, É. Stability and Degradation Patterns of Chemically Modified Analogs of Apelin-13 in Plasma and Cerebrospinal Fluid. Biopolymers 2014, 102, 297–303. [Google Scholar] [CrossRef]
- Zhen, E.Y.; Higgs, R.E.; Gutierrez, J.A. Pyroglutamyl Apelin-13 Identified as the Major Apelin Isoform in Human Plasma. Anal Biochem 2013, 442, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Kuc, R.E.; Brame, A.L.; Dyson, A.; Singer, M.; Glen, R.C.; Cheriyan, J.; Wilkinson, I.B.; Davenport, A.P.; Maguire, J.J. [Pyr1]Apelin-13(1–12) Is a Biologically Active ACE2 Metabolite of the Endogenous Cardiovascular Peptide [Pyr1]Apelin-13. Front. Neurosci. 2017, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Japp, A.G.; Newby, D.E. Unlocking the Therapeutic Potential of Apelin. Hypertension 2016, 68, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Brame, A.L.; Maguire, J.J.; Yang, P.; Dyson, A.; Torella, R.; Cheriyan, J.; Singer, M.; Glen, R.C.; Wilkinson, I.B.; Davenport, A.P. Design, Characterization, and First-In-Human Study of the Vascular Actions of a Novel Biased Apelin Receptor Agonist. Hypertension 2015, 65, 834–840. [Google Scholar] [CrossRef]
- Wang, W.; McKinnie, S.M.K.; Farhan, M.; Paul, M.; McDonald, T.; McLean, B.; Llorens-Cortes, C.; Hazra, S.; Murray, A.G.; Vederas, J.C.; et al. Angiotensin-Converting Enzyme 2 Metabolizes and Partially Inactivates Pyr-Apelin-13 and Apelin-17. Hypertension 2016, 68, 365–377. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of Biological Peptides by Human Angiotensin-Converting Enzyme-Related Carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef]
- McKinnie, S.M.K.; Fischer, C.; Tran, K.M.H.; Wang, W.; Mosquera, F.; Oudit, G.Y.; Vederas, J.C. The Metalloprotease Neprilysin Degrades and Inactivates Apelin Peptides. ChemBioChem 2016, 17, 1495–1498. [Google Scholar] [CrossRef]
- Zhong, J.-C.; Zhang, Z.-Z.; Wang, W.; McKinnie, S.M.K.; Vederas, J.C.; Oudit, G.Y. Targeting the Apelin Pathway as a Novel Therapeutic Approach for Cardiovascular Diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1942–1950. [Google Scholar] [CrossRef]
- De Hert, E.; Bracke, A.; Pintelon, I.; Janssens, E.; Lambeir, A.-M.; Van Der Veken, P.; De Meester, I. Prolyl Carboxypeptidase Mediates the C-Terminal Cleavage of (Pyr)-Apelin-13 in Human Umbilical Vein and Aortic Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 6698. [Google Scholar] [CrossRef]
- Fischer, C.; Lamer, T.; Wang, W.; McKinnie, S.M.K.; Iturrioz, X.; Llorens-Cortes, C.; Oudit, G.Y.; Vederas, J.C. Plasma Kallikrein Cleaves and Inactivates Apelin-17: Palmitoyl- and PEG-Extended Apelin-17 Analogs as Metabolically Stable Blood Pressure-Lowering Agents. Eur. J. Med. Chem. 2019, 166, 119–124. [Google Scholar] [CrossRef]
- Galanth, C.; Hus-Citharel, A.; Li, B.; Llorens-Cortès, C. Apelin in the Control of Body Fluid Homeostasis and Cardiovascular Functions. Curr. Pharm. Des. 2012, 18, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Read, C.; Kuc, R.E.; Buonincontri, G.; Southwood, M.; Torella, R.; Upton, P.D.; Crosby, A.; Sawiak, S.J.; Carpenter, T.A.; et al. Elabela/Toddler Is an Endogenous Agonist of the Apelin APJ Receptor in the Adult Cardiovascular System, and Exogenous Administration of the Peptide Compensates for the Downregulation of Its Expression in Pulmonary Arterial Hypertension. Circulation 2017, 135, 1160–1173. [Google Scholar] [CrossRef] [PubMed]
- Ronkainen, V.; Ronkainen, J.J.; Hänninen, S.L.; Leskinen, H.; Ruas, J.L.; Pereira, T.; Poellinger, L.; Vuolteenaho, O.; Tavi, P. Hypoxia Inducible Factor Regulates the Cardiac Expression and Secretion of Apelin. FASEB J. 2007, 21, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Y.; Chun, H.J.; Glassford, A.J.; Kundu, R.K.; Kutschka, I.; Ardigo, D.; Hendry, S.L.; Wagner, R.A.; Chen, M.M.; Ali, Z.A.; et al. In Vivo Genetic Profiling and Cellular Localization of Apelin Reveals a Hypoxia-Sensitive, Endothelial-Centered Pathway Activated in Ischemic Heart Failure. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H88–H98. [Google Scholar] [CrossRef] [PubMed]
- Guzelburc, O.; Demirtunc, R.; Altay, S.; Kemaloglu Oz, T.; Tayyareci, G. Plasma Apelin Level in Acute Myocardial Infarction and Its Relation with Prognosis: A Prospective Study. JRSM Cardiovasc. Dis. 2021, 10, 2048004020963970. [Google Scholar] [CrossRef]
- Pauli, A.; Norris, M.L.; Valen, E.; Chew, G.-L.; Gagnon, J.A.; Zimmerman, S.; Mitchell, A.; Ma, J.; Dubrulle, J.; Reyon, D.; et al. Toddler: An Embryonic Signal That Promotes Cell Movement via Apelin Receptors. Science 2014, 343, 1248636. [Google Scholar] [CrossRef]
- Chng, S.C.; Ho, L.; Tian, J.; Reversade, B. ELABELA: A Hormone Essential for Heart Development Signals via the Apelin Receptor. Dev. Cell 2013, 27, 672–680. [Google Scholar] [CrossRef]
- Murza, A.; Sainsily, X.; Coquerel, D.; Côté, J.; Marx, P.; Besserer-Offroy, É.; Longpré, J.-M.; Lainé, J.; Reversade, B.; Salvail, D.; et al. Discovery and Structure–Activity Relationship of a Bioactive Fragment of ELABELA That Modulates Vascular and Cardiac Functions. J. Med. Chem. 2016, 59, 2962–2972. [Google Scholar] [CrossRef]
- Couvineau, P.; Llorens-Cortes, C.; Iturrioz, X. Elabela/Toddler and Apelin Bind Differently to the Apelin Receptor. FASEB J. 2020, 34, 7989–8000. [Google Scholar] [CrossRef]
- Huang, S.K.; Shin, K.; Sarker, M.; Rainey, J.K. Apela Exhibits Isoform- and Headgroup-Dependent Modulation of Micelle Binding, Peptide Conformation and Dynamics. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2017, 1859, 767–778. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, D.; Wang, M.; Wang, Q.; Kouznetsova, J.; Yang, R.; Qian, K.; Wu, W.; Shuldiner, A.; Sztalryd, C.; et al. Elabela-Apelin Receptor Signaling Pathway Is Functional in Mammalian Systems. Sci. Rep. 2015, 5, 8170. [Google Scholar] [CrossRef] [PubMed]
- Perjés, Á.; Kilpiö, T.; Ulvila, J.; Magga, J.; Alakoski, T.; Szabó, Z.; Vainio, L.; Halmetoja, E.; Vuolteenaho, O.; Petäjä-Repo, U.; et al. Characterization of Apela, a Novel Endogenous Ligand of Apelin Receptor, in the Adult Heart. Basic Res. Cardiol. 2016, 111, 2. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Sato, T.; Imai, Y.; Yamaguchi, T. Apelin and Elabela/Toddler; Double Ligands for APJ/Apelin Receptor in Heart Development, Physiology, and Pathology. Peptides 2019, 111, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Coquerel, D.; Chagnon, F.; Sainsily, X.; Dumont, L.; Murza, A.; Côté, J.; Dumaine, R.; Sarret, P.; Marsault, É.; Salvail, D.; et al. ELABELA Improves Cardio-Renal Outcome in Fatal Experimental Septic Shock. Crit. Care Med. 2017, 45, e1139–e1148. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Zhang, L.; Imai, Y.; Arab, S.; Chen, M.; Maekawa, Y.; Leschnik, M.; Leibbrandt, A.; Markovic, M.; Schwaighofer, J.; et al. Impaired Heart Contractility in Apelin Gene–Deficient Mice Associated With Aging and Pressure Overload. Circ. Res. 2007, 101, e32–e42. [Google Scholar] [CrossRef]
- Perjés, Á.; Skoumal, R.; Tenhunen, O.; Kónyi, A.; Simon, M.; Horváth, I.G.; Kerkelä, R.; Ruskoaho, H.; Szokodi, I. Apelin Increases Cardiac Contractility via Protein Kinase Cε- and Extracellular Signal-Regulated Kinase-Dependent Mechanisms. PLoS ONE 2014, 9, e93473. [Google Scholar] [CrossRef]
- Seo, K.; Parikh, V.N.; Ashley, E.A. Stretch-Induced Biased Signaling in Angiotensin II Type 1 and Apelin Receptors for the Mediation of Cardiac Contractility and Hypertrophy. Front. Physiol. 2020, 11, 181. [Google Scholar] [CrossRef]
- Berry, M.F.; Pirolli, T.J.; Jayasankar, V.; Burdick, J.; Morine, K.J.; Gardner, T.J.; Woo, Y.J. Apelin Has In Vivo Inotropic Effects on Normal and Failing Hearts. Circulation 2004, 110 (Suppl. S1), II187–II193. [Google Scholar] [CrossRef]
- Folino, A.; Montarolo, P.G.; Samaja, M.; Rastaldo, R. Effects of Apelin on the Cardiovascular System. Heart Fail. Rev. 2015, 20, 505–518. [Google Scholar] [CrossRef]
- Peyronnet, R.; Bollensdorff, C.; Capel, R.A.; Rog-Zielinska, E.A.; Woods, C.E.; Charo, D.N.; Lookin, O.; Fajardo, G.; Ho, M.; Quertermous, T.; et al. Load-Dependent Effects of Apelin on Murine Cardiomyocytes. Prog. Biophys. Mol. Biol. 2017, 130, 333–343. [Google Scholar] [CrossRef]
- Wang, C.; Du, J.-F.; Wu, F.; Wang, H.-C. Apelin Decreases the SR Ca 2+ Content but Enhances the Amplitude of [Ca 2+ ] i Transient and Contractions during Twitches in Isolated Rat Cardiac Myocytes. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H2540–H2546. [Google Scholar] [CrossRef] [PubMed]
- Mughal, A.; Sun, C.; O’Rourke, S.T. Activation of Large Conductance, Calcium-Activated Potassium Channels by Nitric Oxide Mediates Apelin-Induced Relaxation of Isolated Rat Coronary Arteries. J. Pharmacol. Exp. Ther. 2018, 366, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Sahinturk, S.; Demirel, S.; Ozyener, F.; Isbil, N. [Pyr1]Apelin-13 Relaxes the Rat Thoracic Aorta via APJ, NO, AMPK, and Potassium Channels. Gen. Physiol. Biophys. 2021, 40, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Katugampola, S.D.; Maguire, J.J.; Matthewson, S.R.; Davenport, A.P. [(125)I]-(Pyr(1))Apelin-13 Is a Novel Radioligand for Localizing the APJ Orphan Receptor in Human and Rat Tissues with Evidence for a Vasoconstrictor Role in Man. Br. J. Pharmacol. 2001, 132, 1255–1260. [Google Scholar] [CrossRef]
- Rikitake, Y. The Apelin/APJ System in the Regulation of Vascular Tone: Friend or Foe? J. Biochem. 2021, 169, 383–386. [Google Scholar] [CrossRef]
- Sahinturk, S.; Demirel, S.; Ozyener, F.; Isbil, N. Vascular Functional Effect Mechanisms of Elabela in Rat Thoracic Aorta. Ann. Vasc. Surg. 2022, 84, 381–397. [Google Scholar] [CrossRef]
- Helker, C.S.; Eberlein, J.; Wilhelm, K.; Sugino, T.; Malchow, J.; Schuermann, A.; Baumeister, S.; Kwon, H.-B.; Maischein, H.-M.; Potente, M.; et al. Apelin Signaling Drives Vascular Endothelial Cells toward a Pro-Angiogenic State. eLife 2020, 9, e55589. [Google Scholar] [CrossRef]
- Wang, X.; Liang, G.; Guo, Q.; Cai, W.; Zhang, X.; Ni, J.; Tao, Y.; Niu, X.; Chen, S. ELABELA Improves Endothelial Cell Function via the ELA–APJ Axis by Activating the PI3K/Akt Signalling Pathway in HUVECs and EA.Hy926 Cells. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1953–1964. [Google Scholar] [CrossRef]
- Roberts, E.M.; Newson, M.J.F.; Pope, G.R.; Landgraf, R.; Lolait, S.J.; O’Carroll, A.-M. Abnormal Fluid Homeostasis in Apelin Receptor Knockout Mice. J. Endocrinol. 2009, 202, 453–462. [Google Scholar] [CrossRef]
- Hu, G.; Wang, Z.; Zhang, R.; Sun, W.; Chen, X. The Role of Apelin/Apelin Receptor in Energy Metabolism and Water Homeostasis: A Comprehensive Narrative Review. Front. Physiol. 2021, 12, 632886. [Google Scholar] [CrossRef]
- Xu, J.; Chen, L.; Jiang, Z.; Li, L. Biological Functions of Elabela, a Novel Endogenous Ligand of APJ Receptor. J. Cell Physiol. 2018, 233, 6472–6482. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, P.H.; Kalra, S. Selected Abstracts From Recent Publications in Cardiopulmonary Disease Prevention and Rehabilitation. J. Cardiopulm. Rehabil. Prev. 2016, 36, 140–144. [Google Scholar] [CrossRef]
- Vaughan, A.S.; Coronado, F.; Casper, M.; Loustalot, F.; Wright, J.S. County-Level Trends in Hypertension-Related Cardiovascular Disease Mortality—United States, 2000 to 2019. J. Am. Heart Assoc. 2022, 11, e024785. [Google Scholar] [CrossRef]
- Yildiz, M.; Oktay, A.A.; Stewart, M.H.; Milani, R.V.; Ventura, H.O.; Lavie, C.J. Left Ventricular Hypertrophy and Hypertension. Prog. Cardiovasc. Dis. 2020, 63, 10–21. [Google Scholar] [CrossRef]
- Arendse, L.B.; Danser AH, J.; Poglitsch, M.; Touyz, R.M.; Burnett, J.C.; Llorens-Cortes, C.; Ehlers, M.R.; Sturrock, E.D. Novel Therapeutic Approaches Targeting the Renin-Angiotensin System and Associated Peptides in Hypertension and Heart Failure. Pharmacol. Rev. 2019, 71, 539–570. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1–7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [PubMed]
- Sainsily, X.; Coquerel, D.; Giguère, H.; Dumont, L.; Tran, K.; Noll, C.; Ionescu, A.L.; Côté, J.; Longpré, J.-M.; Carpentier, A.; et al. Elabela Protects Spontaneously Hypertensive Rats From Hypertension and Cardiorenal Dysfunctions Exacerbated by Dietary High-Salt Intake. Front. Pharmacol. 2021, 12, 709467. [Google Scholar] [CrossRef]
- Wysocka, M.B.; Pietraszek-Gremplewicz, K.; Nowak, D. The Role of Apelin in Cardiovascular Diseases, Obesity and Cancer. Front. Physiol. 2018, 9, 557. [Google Scholar] [CrossRef]
- Sato, T.; Suzuki, T.; Watanabe, H.; Kadowaki, A.; Fukamizu, A.; Liu, P.P.; Kimura, A.; Ito, H.; Penninger, J.M.; Imai, Y.; et al. Apelin Is a Positive Regulator of ACE2 in Failing Hearts. J. Clin. Investig. 2013, 123, 5203–5211. [Google Scholar] [CrossRef]
- Zhang, Z.-Z.; Wang, W.; Jin, H.-Y.; Chen, X.; Cheng, Y.-W.; Xu, Y.-L.; Song, B.; Penninger, J.M.; Oudit, G.Y.; Zhong, J.-C. Apelin Is a Negative Regulator of Angiotensin II–Mediated Adverse Myocardial Remodeling and Dysfunction. Hypertension 2017, 70, 1165–1175. [Google Scholar] [CrossRef]
- Abbasloo, E.; Najafipour, H.; Vakili, A. Chronic Treatment with Apelin, Losartan and Their Combination Reduces Myocardial Infarct Size and Improves Cardiac Mechanical Function. Clin. Exp. Pharmacol. Physiol. 2020, 47, 393–402. [Google Scholar] [CrossRef]
- Tomek, J.; Bub, G. Hypertension-Induced Remodelling: On the Interactions of Cardiac Risk Factors. J. Physiol. 2017, 595, 4027–4036. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kadowaki, A.; Suzuki, T.; Ito, H.; Watanabe, H.; Imai, Y.; Kuba, K. Loss of Apelin Augments Angiotensin II-Induced Cardiac Dysfunction and Pathological Remodeling. Int. J. Mol. Sci. 2019, 20, 239. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Shen, M.; Fischer, C.; Basu, R.; Hazra, S.; Couvineau, P.; Paul, M.; Wang, F.; Toth, S.; Mix, D.S.; et al. Apelin Protects against Abdominal Aortic Aneurysm and the Therapeutic Role of Neutral Endopeptidase Resistant Apelin Analogs. Proc. Natl. Acad. Sci. USA 2019, 116, 13006–13015. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhang, Z.; Dong, Z.; Liu, X.; Liu, Y.; Li, X.; Xu, Y.; Guo, Y.; Wang, N.; Zhang, M.; et al. MicroRNA-122-5p Aggravates Angiotensin II-Mediated Myocardial Fibrosis and Dysfunction in Hypertensive Rats by Regulating the Elabela/Apelin-APJ and ACE2-GDF15-Porimin Signaling. J. Cardiovasc. Transl. Res. 2022, 15, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Zhang, L.; Cheng, X.; Wang, H.; Qin, W.; Zhou, X.; Tang, B. Apelin Inhibits Angiotensin II-Induced Atrial Fibrosis and Atrial Fibrillation via TGF-Β1/Smad2/α-SMA Pathway. Front. Physiol. 2020, 11, 583570. [Google Scholar] [CrossRef]
- Xu, C.; Wang, F.; Chen, Y.; Xie, S.; Sng, D.; Reversade, B.; Yang, T. ELABELA Antagonizes Intrarenal Renin-Angiotensin System to Lower Blood Pressure and Protects against Renal Injury. Am. J. Physiol. Renal Physiol. 2020, 318, F1122–F1135. [Google Scholar] [CrossRef]
- Sato, T.; Sato, C.; Kadowaki, A.; Watanabe, H.; Ho, L.; Ishida, J.; Yamaguchi, T.; Kimura, A.; Fukamizu, A.; Penninger, J.M.; et al. ELABELA-APJ Axis Protects from Pressure Overload Heart Failure and Angiotensin II-Induced Cardiac Damage. Cardiovasc. Res. 2017, 113, 760–769. [Google Scholar] [CrossRef]
- Hendrianus; Adiarto, S.; Prakoso, R.; Firdaus, I.; Indriani, S.; Rudiktyo, E.; Widyantoro, B.; Taofan, T.; Ambari, A.M.; Sukmawan, R. 10 A novel peptide elabela is associated with hypertension-related subclinical atherosclerosis. J. Hypertens. 2022, 40 (Suppl. S2), e3. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.; Ouyang, S.; He, J.; Yu, B.; Lin, X.; Zhang, Q.; Tao, J. Declined Circulating Elabela Levels in Patients with Essential Hypertension and Its Association with Impaired Vascular Function: A Preliminary Study. Clin. Exp. Hypertens. 2020, 42, 239–243. [Google Scholar] [CrossRef]
- Xie, H.; Luo, G.; Zheng, Y.; Hu, D.; Peng, F.; Xie, L. Lowered Circulating Apelin Is Significantly Associated with an Increased Risk for Hypertension: A Meta-Analysis. Clin. Exp. Hypertens. 2017, 39, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Baysal, S.S.; Pirat, B.; Okyay, K.; Bal, U.A.; Uluçam, M.Z.; Öztuna, D.; Müderrisoğlu, H. Treatment-Associated Change in Apelin Concentration in Patients with Hypertension and Its Relationship with Left Ventricular Diastolic Function. Anatol. J. Cardiol. 2017, 17, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhao, L.; Zhang, Y.; Zhong, J.; Yang, X. Declined ELABELA Plasma Levels in Hypertension Patients with Atrial Fibrillation: A Case Control Study. BMC Cardiovasc. Disord. 2021, 21, 390. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhao, L.; Martin, S.; Zhang, Y.; Dong, Y.; Zhong, J.-C.; Yang, X.-C. Lower Plasma Elabela Levels in Hypertensive Patients With Heart Failure Predict the Occurrence of Major Adverse Cardiac Events: A Preliminary Study. Front. Cardiovasc. Med. 2021, 8, 638468. [Google Scholar] [CrossRef]
- Yu, P.; Ma, S.; Dai, X.; Cao, F. Elabela Alleviates Myocardial Ischemia Reperfusion-Induced Apoptosis, Fibrosis and Mitochondrial Dysfunction through PI3K/AKT Signaling. Am. J. Transl. Res. 2020, 12, 4467–4477. [Google Scholar]
- Deng, J. Advanced Research on the Regulated Necrosis Mechanism in Myocardial Ischemia-Reperfusion Injury. Int. J. Cardiol. 2021, 334, 97–101. [Google Scholar] [CrossRef]
- Wang, W.; McKinnie, S.M.K.; Patel, V.B.; Haddad, G.; Wang, Z.; Zhabyeyev, P.; Das, S.K.; Basu, R.; McLean, B.; Kandalam, V.; et al. Loss of Apelin Exacerbates Myocardial Infarction Adverse Remodeling and Ischemia-Reperfusion Injury: Therapeutic Potential of Synthetic Apelin Analogues. J. Am. Heart Assoc. 2013, 2, e000249. [Google Scholar] [CrossRef]
- Rastaldo, R.; Cappello, S.; Folino, A.; Berta, G.N.; Sprio, A.E.; Losano, G.; Samaja, M.; Pagliaro, P. Apelin-13 Limits Infarct Size and Improves Cardiac Postischemic Mechanical Recovery Only If given after Ischemia. Am. J. Physiol.-Heart Circ. Physiol. 2011, 300, H2308–H2315. [Google Scholar] [CrossRef]
- Yang, S.; Li, H.; Tang, L.; Ge, G.; Ma, J.; Qiao, Z.; Liu, H.; Fang, W. Apelin-13 Protects the Heart against Ischemia-Reperfusion Injury through the RISK-GSK-3β-MPTP Pathway. Arch. Med. Sci. 2015, 11, 1065–1073. [Google Scholar] [CrossRef]
- Pisarenko, O.I.; Shulzhenko, V.S.; Studneva, I.M.; Serebryakova, L.I.; Pelogeykina, Y.A.; Veselova, O.M. Signaling Pathways of a Structural Analogue of Apelin-12 Involved in Myocardial Protection against Ischemia/Reperfusion Injury. Peptides 2015, 73, 67–76. [Google Scholar] [CrossRef]
- An, S.; Wang, X.; Shi, H.; Zhang, X.; Meng, H.; Li, W.; Chen, D.; Ge, J. Apelin Protects against Ischemia-Reperfusion Injury in Diabetic Myocardium via Inhibiting Apoptosis and Oxidative Stress through PI3K and P38-MAPK Signaling Pathways. Aging 2020, 12, 25120–25137. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.; Feng, M.; Xu, Z.; Cheng, Z.; Qian, L. ELA-11 Protects the Heart against Oxidative Stress Injury Induced Apoptosis through ERK/MAPK and PI3K/AKT Signaling Pathways. Front. Pharmacol. 2022, 13, 873614. [Google Scholar] [CrossRef] [PubMed]
- Rakhshan, K.; Sharifi, M.; Ramezani, F.; Azizi, Y.; Aboutaleb, N. ERK/HIF-1α/VEGF Pathway: A Molecular Target of ELABELA (ELA) Peptide for Attenuating Cardiac Ischemia–Reperfusion Injury in Rats by Promoting Angiogenesis. Mol. Biol. Rep. 2022, 49, 10509–10519. [Google Scholar] [CrossRef] [PubMed]
- Folino, A.; Accomasso, L.; Giachino, C.; Montarolo, P.G.; Losano, G.; Pagliaro, P.; Rastaldo, R. Apelin-Induced Cardioprotection against Ischaemia/Reperfusion Injury: Roles of Epidermal Growth Factor and Src. Acta Physiol. 2018, 222, e12924. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Lei, S.; Xia, Z.; Wu, Y.; Meng, Q.; Zhan, L.; Su, W.; Liu, H.; Xu, J.; Liu, Z.; et al. Selective Inhibition of PTEN Preserves Ischaemic Post-Conditioning Cardioprotection in STZ-Induced Type 1 Diabetic Rats: Role of the PI3K/Akt and JAK2/STAT3 Pathways. Clin. Sci. 2016, 130, 377–392. [Google Scholar] [CrossRef]
- Hasche, E.T.; Fernandes, C.; Ben Freedman, S.; Jeremy, R.W. Relation Between Ischemia Time, Infarct Size, and Left Ventricular Function in Humans. Circulation 1995, 92, 710–719. [Google Scholar] [CrossRef]
- Xu, W.; Yu, H.; Ma, R.; Ma, L.; Liu, Q.; Shan, H.; Wu, C.; Zhang, R.; Zhou, Y.; Shan, H. Apelin Protects against Myocardial Ischemic Injury by Inhibiting Dynamin-Related Protein 1. Oncotarget 2017, 8, 100034–100044. [Google Scholar] [CrossRef]
- Rakhshan, K.; Azizi, Y.; Naderi, N.; Ghardashi Afousi, A.; Aboutaleb, N. ELABELA (ELA) Peptide Exerts Cardioprotection Against Myocardial Infarction by Targeting Oxidative Stress and the Improvement of Heart Function. Int. J. Pept. Res. Ther. 2019, 25, 613–621. [Google Scholar] [CrossRef]
- Sánchez, E.C. Pathophysiology of Ischemia-Reperfusion Injury and Its Management with Hyperbaric Oxygen (HBO): A Review. J. Emerg. Crit. Care Med. 2019, 3, 22. [Google Scholar] [CrossRef]
- Boal, F.; Timotin, A.; Roumegoux, J.; Alfarano, C.; Calise, D.; Anesia, R.; Parini, A.; Valet, P.; Tronchere, H.; Kunduzova, O. Apelin-13 Administration Protects against Ischaemia/Reperfusion-Mediated Apoptosis through the FoxO1 Pathway in High-Fat Diet-Induced Obesity. Br. J. Pharmacol. 2016, 173, 1850–1863. [Google Scholar] [CrossRef]
- Zhang, M.-W.; Li, X.-T.; Zhang, Z.-Z.; Liu, Y.; Song, J.-W.; Liu, X.-M.; Chen, Y.-H.; Wang, N.; Guo, Y.; Liang, L.-R.; et al. Elabela Blunts Doxorubicin-Induced Oxidative Stress and Ferroptosis in Rat Aortic Adventitial Fibroblasts by Activating the KLF15/GPX4 Signaling. Cell Stress Chaperones 2022, 28, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Masoud, A.G.; Lin, J.; Azad, A.K.; Farhan, M.A.; Fischer, C.; Zhu, L.F.; Zhang, H.; Sis, B.; Kassiri, Z.; Moore, R.B.; et al. Apelin Directs Endothelial Cell Differentiation and Vascular Repair Following Immune-Mediated Injury. J. Clin. Investig. 2019, 130, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Chen, X.; Liu, X.; Xu, D.; Yang, H.; Zeng, C.; Long, H.; Zhou, C.; Wu, H.; Zheng, G.; et al. ELABELA Ameliorates Hypoxic/Ischemic-Induced Bone Mesenchymal Stem Cell Apoptosis via Alleviation of Mitochondrial Dysfunction and Activation of PI3K/AKT and ERK1/2 Pathways. Stem Cell Res. Ther. 2020, 11, 541. [Google Scholar] [CrossRef]
- Chen, G.; Liang, X.; Han, Q.; Mai, C.; Shi, L.; Shao, Z.; Hong, Y.; Lin, F.; Li, M.; Hu, B.; et al. Apelin-13 Pretreatment Promotes the Cardioprotective Effect of Mesenchymal Stem Cells against Myocardial Infarction by Improving Their Survival. Stem Cells Int. 2022, 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Guaricci, A.I.; Bulzis, G.; Pontone, G.; Scicchitano, P.; Carbonara, R.; Rabbat, M.; De Santis, D.; Ciccone, M.M. Current Interpretation of Myocardial Stunning. Trends Cardiovasc. Med. 2018, 28, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Sattler, S.M.; Skibsbye, L.; Linz, D.; Lubberding, A.F.; Tfelt-Hansen, J.; Jespersen, T. Ventricular Arrhythmias in First Acute Myocardial Infarction: Epidemiology, Mechanisms, and Interventions in Large Animal Models. Front. Cardiovasc. Med. 2019, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; He, Q.; Wu, C.; Chen, L.; Bi, F.; Zhou, Y.; Shan, H. Apelin Shorten QT Interval by Inhibiting Kir2.1/IK1 via a PI3K Way in Acute Myocardial Infarction. Biochem. Biophys. Res. Commun. 2019, 517, 272–277. [Google Scholar] [CrossRef]
- Farkasfalvi, K.; Stagg, M.A.; Coppen, S.R.; Siedlecka, U.; Lee, J.; Soppa, G.K.; Marczin, N.; Szokodi, I.; Yacoub, M.H.; Terracciano, C.M.N. Direct Effects of Apelin on Cardiomyocyte Contractility and Electrophysiology. Biochem. Biophys. Res. Commun. 2007, 357, 889–895. [Google Scholar] [CrossRef]
- Koval, S.; Iushko, K.; Starchenko, T. Relations of Apelin with Cardiac Remodeling in Patients with Hypertension and Type 2 Diabetes. Folia Med. 2018, 60, 117–123. [Google Scholar] [CrossRef]
- Xu, P.; Kong, L.; Tao, C.; Zhu, Y.; Cheng, J.; Li, W.; Shen, N.; Li, R.; Zhang, C.; Wang, L.; et al. Elabela-APJ Axis Attenuates Cerebral Ischemia/Reperfusion Injury by Inhibiting Neuronal Ferroptosis. Free Radic. Biol. Med. 2023, 196, 171–186. [Google Scholar] [CrossRef]
- Xia, F.; Chen, H.; Jin, Z.; Fu, Z. Apelin-13 Protects the Lungs from Ischemia-Reperfusion Injury by Attenuating Inflammatory and Oxidative Stress. Hum. Exp. Toxicol. 2021, 40, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, Y.; Zhou, Y.; Fei, B. Activation of Nrf2 Signaling by Apelin Attenuates Renal Ischemia Reperfusion Injury in Diabetic Rats. Diabetes Metab. Syndr. Obes. 2020, 13, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, L.; Wang, W.; Cheng, C.; Zhang, Y.; Zhou, Y.; Wang, C.; Miao, X.; Wang, J.; Wang, C.; et al. ELABELA and an ELABELA Fragment Protect against AKI. J. Am. Soc. Nephrol. 2017, 28, 2694–2707. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac Remodeling--Concepts and Clinical Implications: A Consensus Paper from an International Forum on Cardiac Remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef]
- Daubert, M.A.; White, J.A.; Al-Khalidi, H.R.; Velazquez, E.J.; Rao, S.V.; Crowley, A.L.; Zeymer, U.; Kasprzak, J.D.; Guetta, V.; Krucoff, M.W.; et al. Cardiac Remodeling after Large ST-Elevation Myocardial Infarction in the Current Therapeutic Era. Am. Heart J. 2020, 223, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, K.F.; Pothineni NV, K.; Rutland, J.; Ding, Z.; Mehta, J.L. Immunity, Inflammation, and Oxidative Stress in Heart Failure: Emerging Molecular Targets. Cardiovasc. Drugs Ther. 2017, 31, 593–608. [Google Scholar] [CrossRef]
- Leancă, S.A.; Crișu, D.; Petriș, A.O.; Afrăsânie, I.; Genes, A.; Costache, A.D.; Tesloianu, D.N.; Costache, I.I. Left Ventricular Remodeling after Myocardial Infarction: From Physiopathology to Treatment. Life 2022, 12, 1111. [Google Scholar] [CrossRef]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; de Boer, R.A. From Inflammation to Fibrosis—Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail Rep. 2017, 14, 235–250. [Google Scholar] [CrossRef]
- Piek, A.; de Boer, R.A.; Silljé, H.H.W. The Fibrosis-Cell Death Axis in Heart Failure. Heart Fail Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef]
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and Fibrosis. Circ. Res. 2020, 127, 427–447. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Myofibroblast Transdifferentiation: The Dark Force in Ocular Wound Healing and Fibrosis. Prog. Retin Eye Res. 2017, 60, 44–65. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, H.; Hanna, A.; Humeres, C.; Frangogiannis, N.G. Properties and Functions of Fibroblasts and Myofibroblasts in Myocardial Infarction. Cells 2022, 11, 1386. [Google Scholar] [CrossRef] [PubMed]
- Alex, L.; Frangogiannis, N.G. The Cellular Origin of Activated Fibroblasts in the Infarcted and Remodeling Myocardium. Circ. Res. 2018, 122, 540–542. [Google Scholar] [CrossRef]
- Thomas, T.P.; Grisanti, L.A. The Dynamic Interplay Between Cardiac Inflammation and Fibrosis. Front. Physiol. 2020, 11, 529075. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef]
- Tatin, F.; Renaud-Gabardos, E.; Godet, A.-C.; Hantelys, F.; Pujol, F.; Morfoisse, F.; Calise, D.; Viars, F.; Valet, P.; Masri, B.; et al. Apelin Modulates Pathological Remodeling of Lymphatic Endothelium after Myocardial Infarction. JCI Insight 2017, 2, 93887. [Google Scholar] [CrossRef]
- Czarzasta, K.; Wojno, O.; Zera, T.; Puchalska, L.; Dobruch, J.; Cudnoch-Jedrzejewska, A. The Influence of Post-Infarct Heart Failure and High Fat Diet on the Expression of Apelin APJ and Vasopressin V1a and V1b Receptors. Neuropeptides 2019, 78, 101975. [Google Scholar] [CrossRef]
- Zhong, S.; Guo, H.; Wang, H.; Xing, D.; Lu, T.; Yang, J.; Wang, C. Apelin-13 Alleviated Cardiac Fibrosis via Inhibiting the PI3K/Akt Pathway to Attenuate Oxidative Stress in Rats with Myocardial Infarction-Induced Heart Failure. Biosci. Rep. 2020, 40, BSR20200040. [Google Scholar] [CrossRef]
- Krasniqi, X.; Berisha, B.; Gashi, M.; Koçinaj, D.; Jashari, F.; Vincelj, J. Influence of Apelin-12 on Troponin Levels and the Rate of MACE in STEMI Patients. BMC Cardiovasc. Disord. 2017, 17, 195. [Google Scholar] [CrossRef]
- Dönmez, Y.; Acele, A. Increased Elabela Levels in the Acute ST Segment Elevation Myocardial Infarction Patients. Medicine 2019, 98, e17645. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, W.; Feng, F.; Xu, J.; Wu, F. Apelin-13 Protects against Myocardial Infarction-Induced Myocardial Fibrosis. Mol. Med. Rep. 2016, 13, 5262–5268. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.R.; Zornoff, L.A.M. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Severs, N.J. Gap Junction Remodeling in Heart Failure. J. Card Fail 2002, 8, S293–S299. [Google Scholar] [CrossRef] [PubMed]
- Vitale, E.; Rosso, R.; Lo Iacono, M.; Cristallini, C.; Giachino, C.; Rastaldo, R. Apelin-13 Increases Functional Connexin-43 through Autophagy Inhibition via AKT/MTOR Pathway in the Non-Myocytic Cell Population of the Heart. Int. J. Mol. Sci. 2022, 23, 13073. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-J.; De Jesus Morales, K.J.; Bheri, S.; Kassouf, B.P.; Davis, M.E. Bidirectional Relationship Between Cardiac Extracellular Matrix and Cardiac Cells in Ischemic Heart Disease. Stem Cells 2021, 39, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.-H.; Guo, C.-X.; Wang, H.-X.; Lu, L.-Q.; Wang, Y.-J.; Zhang, L.-K.; Du, F.-H.; Zeng, X.-J. Cardioprotective Effects of Adipokine Apelin on Myocardial Infarction. Heart Vessels 2014, 29, 679–689. [Google Scholar] [CrossRef] [PubMed]
- de Kam, P. Prediction of 6 Months Left Ventricular Dilatation after Myocardial Infarction in Relation to Cardiac Morbidity and Mortality. Application of a New Dilatation Model to GISSI-3 Data. Eur. Heart J. 2002, 23, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Macrae, R.G.C.; Colzani, M.T.; Williams, T.L.; Bayraktar, S.; Kuc, R.E.; Pullinger, A.L.; Bernard, W.G.; Robinson, E.L.; Davenport, E.E.; Maguire, J.J.; et al. Inducible Apelin Receptor Knockdown Reduces Differentiation Efficiency and Contractility of HESC-Derived Cardiomyocytes. Cardiovasc. Res. 2022, 119, 587–598. [Google Scholar] [CrossRef]
- Chong, K.S.; Gardner, R.S.; Morton, J.J.; Ashley, E.A.; McDonagh, T.A. Plasma Concentrations of the Novel Peptide Apelin Are Decreased in Patients with Chronic Heart Failure. Eur. J. Heart Fail 2006, 8, 355–360. [Google Scholar] [CrossRef]
- van den Borne SW, M.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial Remodeling after Infarction: The Role of Myofibroblasts. Nat. Rev. Cardiol. 2010, 7, 30–37. [Google Scholar] [CrossRef]
- Wu, X.; Reboll, M.R.; Korf-Klingebiel, M.; Wollert, K.C. Angiogenesis after Acute Myocardial Infarction. Cardiovasc. Res. 2021, 117, 1257–1273. [Google Scholar] [CrossRef] [PubMed]
- Kocijan, T.; Rehman, M.; Colliva, A.; Groppa, E.; Leban, M.; Vodret, S.; Volf, N.; Zucca, G.; Cappelletto, A.; Piperno, G.M.; et al. Genetic Lineage Tracing Reveals Poor Angiogenic Potential of Cardiac Endothelial Cells. Cardiovasc. Res. 2021, 117, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chen, L.; Li, L. Apelin/APJ System: A Novel Promising Therapy Target for Pathological Angiogenesis. Clin. Chim. Acta 2017, 466, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yan, J.; Pan, W.; Tang, M. Apelin/Elabela-APJ: A Novel Therapeutic Target in the Cardiovascular System. Ann. Transl. Med. 2020, 8, 243. [Google Scholar] [CrossRef]
- Cox, C.M.; D’Agostino, S.L.; Miller, M.K.; Heimark, R.L.; Krieg, P.A. Apelin, the Ligand for the Endothelial G-Protein-Coupled Receptor, APJ, Is a Potent Angiogenic Factor Required for Normal Vascular Development of the Frog Embryo. Dev. Biol. 2006, 296, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Helker, C.S.; Schuermann, A.; Pollmann, C.; Chng, S.C.; Kiefer, F.; Reversade, B.; Herzog, W. The Hormonal Peptide Elabela Guides Angioblasts to the Midline during Vasculogenesis. eLife 2015, 4, 6726. [Google Scholar] [CrossRef] [PubMed]
- Kidoya, H.; Naito, H.; Muramatsu, F.; Yamakawa, D.; Jia, W.; Ikawa, M.; Sonobe, T.; Tsuchimochi, H.; Shirai, M.; Adams, R.H.; et al. APJ Regulates Parallel Alignment of Arteries and Veins in the Skin. Dev. Cell 2015, 33, 247–259. [Google Scholar] [CrossRef]
- Kidoya, H.; Takakura, N. Biology of the Apelin-APJ Axis in Vascular Formation. J. Biochem. 2012, 152, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Hu, T.; He, L.; Huang, X.; Tian, X.; Zhang, H.; He, L.; Pu, W.; Zhang, L.; Sun, H.; et al. Genetic Targeting of Sprouting Angiogenesis Using Apln-CreER. Nat. Commun. 2015, 6, 6020. [Google Scholar] [CrossRef]
- Yavuz, F.; Kaplan, M. Associação Entre Os Níveis Séricos de Elabela e Oclusão Total Crônica Em Pacientes Com Angina Pectoris Estável. Arq. Bras. Cardiol. 2021, 117, 503–510. [Google Scholar] [CrossRef]
- Pan, Y.; Li, Q.; Yan, H.; Huang, J.; Wang, Z. Apela Improves Cardiac and Renal Function in Mice with Acute Myocardial Infarction. J. Cell Mol. Med. 2020, 24, 10382–10390. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Wang, X.; Shen, L.; Jiang, K.; Ding, X.; Cappetta, D.; Zhou, J.; Ge, J.; Zou, Y. Mechanical Stress Regulates Endothelial Progenitor Cell Angiogenesis Through VEGF Receptor Endocytosis. Int. Heart J. 2016, 57, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Eržen, B.; Šilar, M.; Šabovič, M. Stable Phase Post-MI Patients Have Elevated VEGF Levels Correlated with Inflammation Markers, but Not with Atherosclerotic Burden. BMC Cardiovasc. Disord. 2014, 14, 166. [Google Scholar] [CrossRef] [PubMed]
- Tempel, D.; de Boer, M.; van Deel, E.D.; Haasdijk, R.A.; Duncker, D.J.; Cheng, C.; Schulte-Merker, S.; Duckers, H.J. Apelin Enhances Cardiac Neovascularization After Myocardial Infarction by Recruiting Aplnr+ Circulating Cells. Circ. Res. 2012, 111, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Pan, Y.; Li, Q.; Li, J.; Wang, Z. Elabela Gene Therapy Promotes Angiogenesis after Myocardial Infarction. J. Cell Mol. Med. 2021, 25, 8537–8545. [Google Scholar] [CrossRef]
- Hou, X.; Zeng, H.; He, X.; Chen, J. Sirt3 Is Essential for Apelin-induced Angiogenesis in Post-myocardial Infarction of Diabetes. J. Cell Mol. Med. 2015, 19, 53–61. [Google Scholar] [CrossRef]
- Xue, S.; He, L.; Zhang, X.; Zhou, J.; Li, F.; Wang, X. Expression of Jagged1/Notch3 Signaling Pathway and Their Relationship with the Tumor Angiogenesis in TNBC. Arch. Med. Res. 2017, 48, 169–179. [Google Scholar] [CrossRef]
- Gomez, A.H.; Joshi, S.; Yang, Y.; Tune, J.D.; Zhao, M.-T.; Yang, H. Bioengineering Systems for Modulating Notch Signaling in Cardiovascular Development, Disease, and Regeneration. J. Cardiovasc. Dev. Dis. 2021, 8, 125. [Google Scholar] [CrossRef]
- Li, L.; Li, L.; Xie, F.; Zhang, Z.; Guo, Y.; Tang, G.; Lv, D.; Lu, Q.; Chen, L.; Li, J. Jagged-1/Notch3 Signaling Transduction Pathway Is Involved in Apelin-13-Induced Vascular Smooth Muscle Cells Proliferation. Acta Biochim. Biophys. Sin. 2013, 45, 875–881. [Google Scholar] [CrossRef]
- Hasan, S.S.; Tsaryk, R.; Lange, M.; Wisniewski, L.; Moore, J.C.; Lawson, N.D.; Wojciechowska, K.; Schnittler, H.; Siekmann, A.F. Endothelial Notch Signalling Limits Angiogenesis via Control of Artery Formation. Nat. Cell Biol. 2017, 19, 928–940. [Google Scholar] [CrossRef]
- Pitulescu, M.E.; Schmidt, I.; Giaimo, B.D.; Antoine, T.; Berkenfeld, F.; Ferrante, F.; Park, H.; Ehling, M.; Biljes, D.; Rocha, S.F.; et al. Dll4 and Notch Signalling Couples Sprouting Angiogenesis and Artery Formation. Nat. Cell Biol. 2017, 19, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zeng, H.; Chen, J.-X. Apelin-13 Increases Myocardial Progenitor Cells and Improves Repair Postmyocardial Infarction. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H605–H618. [Google Scholar] [CrossRef] [PubMed]
- Papangeli, I.; Kim, J.; Maier, I.; Park, S.; Lee, A.; Kang, Y.; Tanaka, K.; Khan, O.F.; Ju, H.; Kojima, Y.; et al. MicroRNA 139-5p Coordinates APLNR-CXCR4 Crosstalk during Vascular Maturation. Nat. Commun. 2016, 7, 11268. [Google Scholar] [CrossRef]
- Brakenhielm, E.; González, A.; Díez, J. Role of Cardiac Lymphatics in Myocardial Edema and Fibrosis. J. Am. Coll. Cardiol. 2020, 76, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-D.; Kang, Y.; Kim, J.; Papangeli, I.; Kang, H.; Wu, J.; Park, H.; Nadelmann, E.; Rockson, S.G.; Chun, H.J.; et al. Essential Role of Apelin Signaling During Lymphatic Development in Zebrafish. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct Target Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Murza, A.; Besserer-Offroy, É.; Côté, J.; Bérubé, P.; Longpré, J.-M.; Dumaine, R.; Lesur, O.; Auger-Messier, M.; Leduc, R.; Sarret, P.; et al. C-Terminal Modifications of Apelin-13 Significantly Change Ligand Binding, Receptor Signaling, and Hypotensive Action. J. Med. Chem. 2015, 58, 2431–2440. [Google Scholar] [CrossRef]
- Trân, K.; Murza, A.; Sainsily, X.; Coquerel, D.; Côté, J.; Belleville, K.; Haroune, L.; Longpré, J.-M.; Dumaine, R.; Salvail, D.; et al. A Systematic Exploration of Macrocyclization in Apelin-13: Impact on Binding, Signaling, Stability, and Cardiovascular Effects. J. Med. Chem. 2018, 61, 2266–2277. [Google Scholar] [CrossRef]
- Trân, K.; Van Den Hauwe, R.; Sainsily, X.; Couvineau, P.; Côté, J.; Simard, L.; Echevarria, M.; Murza, A.; Serre, A.; Théroux, L.; et al. Constraining the Side Chain of C-Terminal Amino Acids in Apelin-13 Greatly Increases Affinity, Modulates Signaling, and Improves the Pharmacokinetic Profile. J. Med. Chem. 2021, 64, 5345–5364. [Google Scholar] [CrossRef]
- Coquerel, D.; Delile, E.; Dumont, L.; Chagnon, F.; Murza, A.; Sainsily, X.; Salvail, D.; Sarret, P.; Marsault, E.; Auger-Messier, M.; et al. Gαi-Biased Apelin Analog Protects against Isoproterenol-Induced Myocardial Dysfunction in Rats. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H1646–H1656. [Google Scholar] [CrossRef]
- Juhl, C.; Els-Heindl, S.; Schönauer, R.; Redlich, G.; Haaf, E.; Wunder, F.; Riedl, B.; Burkhardt, N.; Beck-Sickinger, A.G.; Bierer, D. Development of Potent and Metabolically Stable APJ Ligands with High Therapeutic Potential. ChemMedChem 2016, 11, 2378–2384. [Google Scholar] [CrossRef] [PubMed]
- Théroux, L.; Van Den Hauwe, R.; Trân, K.; Fournier, J.; Desgagné, M.; Meneboo, N.; Lavallée, A.; Fröhlich, U.; Côté, J.; Hollanders, C.; et al. Signaling Modulation via Minimal C-Terminal Modifications of Apelin-13. ACS Pharmacol. Transl. Sci. 2023, 6, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Studneva, I.; Shulzhenko, V.; Veselova, O.; Pisarenko, O. Protective Effects of a Modified Apelin-12 and Dinitrosyl Iron Complexes in Experimental Cardioplegic Ischemia and Reperfusion. J. Physiol. Biochem. 2018, 74, 283–290. [Google Scholar] [CrossRef]
- Sidorova, M.; Studneva, I.; Bushuev, V.; Pal’keeva, M.; Molokoedov, A.; Veselova, O.; Ovchinnikov, M.; Pisarenko, O. [MeArg1, NLe10]-Apelin-12: Optimization of Solid-Phase Synthesis and Evaluation of Biological Properties in Vitro and in Vivo. Peptides 2020, 129, 170320. [Google Scholar] [CrossRef] [PubMed]
- Gerbier, R.; Alvear-Perez, R.; Margathe, J.; Flahault, A.; Couvineau, P.; Gao, J.; De Mota, N.; Dabire, H.; Li, B.; Ceraudo, E.; et al. Development of Original Metabolically Stable Apelin-17 Analogs with Diuretic and Cardiovascular Effects. FASEB J. 2017, 31, 687–700. [Google Scholar] [CrossRef]
- Flahault, A.; Keck, M.; Girault-Sotias, P.-E.; Esteoulle, L.; De Mota, N.; Bonnet, D.; Llorens-Cortes, C. LIT01-196, a Metabolically Stable Apelin-17 Analog, Normalizes Blood Pressure in Hypertensive DOCA-Salt Rats via a NO Synthase-Dependent Mechanism. Front. Pharmacol. 2021, 12, 715095. [Google Scholar] [CrossRef]
- McKinnie, S.M.K.; Wang, W.; Fischer, C.; McDonald, T.; Kalin, K.R.; Iturrioz, X.; Llorens-Cortes, C.; Oudit, G.Y.; Vederas, J.C. Synthetic Modification within the “RPRL” Region of Apelin Peptides: Impact on Cardiovascular Activity and Stability to Neprilysin and Plasma Degradation. J. Med. Chem. 2017, 60, 6408–6427. [Google Scholar] [CrossRef]
- Fernandez, K.X.; Fischer, C.; Vu, J.; Gheblawi, M.; Wang, W.; Gottschalk, S.; Iturrioz, X.; Llorens-Cortés, C.; Oudit, G.Y.; Vederas, J.C. Metabolically Stable Apelin-Analogues, Incorporating Cyclohexylalanine and Homoarginine, as Potent Apelin Receptor Activators. RSC Med. Chem. 2021, 12, 1402–1413. [Google Scholar] [CrossRef]
- Fischer, C.; Lamer, T.; Fernandez, K.; Gheblawi, M.; Wang, W.; Pascoe, C.; Lambkin, G.; Iturrioz, X.; Llorens-Cortes, C.; Oudit, G.Y.; et al. Optimizing PEG-Extended Apelin Analogues as Cardioprotective Drug Leads: Importance of the KFRR Motif and Aromatic Head Group for Improved Physiological Activity. J. Med. Chem. 2020, 63, 12073–12082. [Google Scholar] [CrossRef]
- Trân, K.; Murza, A.; Sainsily, X.; Delile, E.; Couvineau, P.; Côté, J.; Coquerel, D.; Peloquin, M.; Auger-Messier, M.; Bouvier, M.; et al. Structure–Activity Relationship and Bioactivity of Short Analogues of ELABELA as Agonists of the Apelin Receptor. J. Med. Chem. 2021, 64, 602–615. [Google Scholar] [CrossRef]
- Yang, P.; Read, C.; Kuc, R.E.; Nyimanu, D.; Williams, T.L.; Crosby, A.; Buonincontri, G.; Southwood, M.; Sawiak, S.J.; Glen, R.C.; et al. A Novel Cyclic Biased Agonist of the Apelin Receptor, MM07, Is Disease Modifying in the Rat Monocrotaline Model of Pulmonary Arterial Hypertension. Br. J. Pharmacol. 2019, 176, 1206–1221. [Google Scholar] [CrossRef]
- Murza, A.; Sainsily, X.; Côté, J.; Bruneau-Cossette, L.; Besserer-Offroy, É.; Longpré, J.-M.; Leduc, R.; Dumaine, R.; Lesur, O.; Auger-Messier, M.; et al. Structure–Activity Relationship of Novel Macrocyclic Biased Apelin Receptor Agonists. Org. Biomol. Chem. 2017, 15, 449–458. [Google Scholar] [CrossRef]
- Tran, K.; Sainsily, X.; Côté, J.; Coquerel, D.; Couvineau, P.; Saibi, S.; Haroune, L.; Besserer-Offroy, É.; Flynn-Robitaille, J.; Resua Rojas, M.; et al. Size-Reduced Macrocyclic Analogues of [Pyr]-Apelin-13 Showing Negative Gαi Bias Still Produce Prolonged Cardiac Effects. J. Med. Chem. 2022, 65, 531–551. [Google Scholar] [CrossRef]
- Marsault, E.; Peterson, M.L. Practical Medicinal Chemistry with Macrocycles; Marsault, E., Peterson, M.L., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ding, Y.; Song, X.; Ma, X.; Li, X.; Zhang, N.; Song, Y.; Sun, Y.; Shen, Y.; Zhong, W.; et al. Structure-Guided Discovery of a Single-Domain Antibody Agonist against Human Apelin Receptor. Sci. Adv. 2020, 6, eaax7379. [Google Scholar] [CrossRef] [PubMed]
- Read, C.; Yang, P.; Kuc, R.E.; Nyimanu, D.; Williams, T.L.; Glen, R.C.; Holt, L.J.; Arulanantham, H.; Smart, A.; Davenport, A.P.; et al. Apelin Peptides Linked to Anti-serum Albumin Domain Antibodies Retain Affinity in Vitro and Are Efficacious Receptor Agonists in Vivo. Basic Clin. Pharmacol. Toxicol. 2020, 126, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Yu, D.; Yang, R.; Zhao, Q.; Wang, J.; Zhang, H.; Qian, K.; Shi, Z.; Wang, W.; Brown, R.; et al. Recombinant Fc-Elabela Fusion Protein Has Extended Plasma Half-Life Andmitigates Post-Infarct Heart Dysfunction in Rats. Int. J. Cardiol. 2019, 292, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Holt, L.J.; Basran, A.; Jones, K.; Chorlton, J.; Jespers, L.S.; Brewis, N.D.; Tomlinson, I.M. Anti-Serum Albumin Domain Antibodies for Extending the Half-Lives of Short Lived Drugs. Protein Eng. Des. Sel. 2008, 21, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Kariya, T.; Ishikawa, K. Targeted Delivery of Therapeutic Agents to the Heart. Nat. Rev. Cardiol. 2021, 18, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/MTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef]
- Girault-Sotias, P.-E.; Gerbier, R.; Flahault, A.; de Mota, N.; Llorens-Cortes, C. Apelin and Vasopressin: The Yin and Yang of Water Balance. Front. Endocrinol. 2021, 12, 735515. [Google Scholar] [CrossRef] [PubMed]
- Jahangirian, H.; Ghasemian Lemraski, E.; Webster, T.J.; Rafiee-Moghaddam, R.; Abdollahi, Y. A Review of Drug Delivery Systems Based on Nanotechnology and Green Chemistry: Green Nanomedicine. Int. J. Nanomed. 2017, 12, 2957–2978. [Google Scholar] [CrossRef]
- Martinho, N.; Damgé, C.; Reis, C.P. Recent Advances in Drug Delivery Systems. J. Biomater. Nanobiotechnol. 2011, 2, 510–526. [Google Scholar] [CrossRef]
- Serpooshan, V.; Sivanesan, S.; Huang, X.; Mahmoudi, M.; Malkovskiy, A.V.; Zhao, M.; Inayathullah, M.; Wagh, D.; Zhang, X.J.; Metzler, S.; et al. [Pyr1]-Apelin-13 Delivery via Nano-Liposomal Encapsulation Attenuates Pressure Overload-Induced Cardiac Dysfunction. Biomaterials 2015, 37, 289–298. [Google Scholar] [CrossRef] [PubMed]
- de Villiers, M.M.; Aramwit, P.; Kwon, G.S. (Eds.) Nanotechnology in Drug Delivery; Springer New York: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Fang, J.; Koh, J.; Fang, Q.; Qiu, H.; Archang, M.M.; Hasani-Sadrabadi, M.M.; Miwa, H.; Zhong, X.; Sievers, R.; Gao, D.; et al. Injectable Drug-Releasing Microporous Annealed Particle Scaffolds for Treating Myocardial Infarction. Adv. Funct. Mater. 2020, 30, 2004307. [Google Scholar] [CrossRef] [PubMed]
- Almas, T.; Haider, R.; Malik, J.; Mehmood, A.; Alvi, A.; Naz, H.; Satti, D.I.; Zaidi, S.M.J.; AlSubai, A.K.; AlNajdi, S.; et al. Nanotechnology in Interventional Cardiology: A State-of-the-Art Review. IJC Heart Vasc. 2022, 43, 101149. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| APJ Ligand | Substituted Residues | Introduced Residues | Effects | Ref |
|---|---|---|---|---|
| [Pyr1]-apelin-13 | Leu5, Ser 6 | - | ↑ plasma protein stability | [167] |
| Phe13 | [(L-α-Me) Phe] | ↓ Pro12-Phe13 hydrolisis | ||
| Pro12-Phe13 | conformational constrained amino acids | ↑ binding affinity ↑ plasma protease stability | [168] | |
| Aia -Phe 1Nal-α,α-dibenzylglycine | ↑ Gα12 pathways ↑ affinity and resistance to ACE2 cleavage ↑ in vitro and in vivo pharmacokinetics | [169] | ||
| Phe13 | phenyl groups at the ortho-, meta-, and para-positions of Phe13 | slight effect on APJ binding affinity | ||
| (L-α-Me) Phe or p-benzoyl-L-Phe (Bpa) | ↑ APJ functional selectivity for Gαi proteins no effect on cardiac contractility in healthy rats ↑ binding affinity vs. [Pyr1]-apelin-13 ↑ resistance to ACE2 cleavage ↓ hypertrophy and fibrosis | [170] | ||
| N-Teminus (pGlu) | palmitic acid, (at Pro12) Aib, Nle | ↑ half-life (29 h in rat plasma) | [171] | |
| N-Terminus | Alkylated dipeptide | ↑ affinity in the low nM range | [172] | |
| extended and bulkier substituents | binding affinity close to [Pyr1]-apelin-13 ↓ signalling potency for the β-arrestin 2 pathway | |||
| C-Terminus | positive charge | binding affinity close to [Pyr1]-apelin-13 partial agonism for β-arrestin2 recruitment | ||
| 1NaI12 | amino-indoloazepinone-Orn | ↑affinity for APJ ↑ Gαi1 activation, ↑ stability ↓ hypotensive effects | ||
| apelin-12 | N-terminus Met10 | (NαMe) Arg Nle10 | ↑ proteolytic stability ↑ cardiac function ↓ membrane damage | [173,174] |
| apelin-17 | Classical substituted | P92 | ↑ plasma half-life sub-nanomolar affinity for APJ APJ internalization ↑ diuresis ↓ arterial blood pressure | [175] |
| LIT01-196 | fluoroaddiction N-terminal | |||
| ↓ blood pressure in normotensive and hypertensive rats | [176] | |||
| Arg, Leu | azapeptides | ↑ proteolytic stability vs. NEP | [177] | |
| l-hArg, l-Cha | - | ↑ proteolytic stability vs. NEP prolonged lowering blood pressure effect in mice | [178] | |
| N-terminus | palmitoylation PEGylation | ↑ plasma peptide half-life ↓ blood pressure in mice | [30] | |
| aromatic head groups carbonate analogues | ↑↑ proteolytic stability | [179] | ||
| ELA | Pro32 | Tyr (OBn), Bpa, or 1Nal | ↑ binding affinity vs. ELA | [180] |
| N-terminus | pyroglutamic acid (Pyr) residue to ELA(19-32) | ↓ arterial pressure ↓ inotropic effects on the heart ↑ half-life | [38] |
| Cyclized Peptide | Modified Residues | Cycle Position | Effects | Ref |
|---|---|---|---|---|
| MM07 | N-terminus | N-terminal cycle (MM07) | ↑ vasodilatation ↑ cardiac function vs. [Pyr1]-apelin-13 | [24] |
| ↑ cardiac function ↓ hypertrophy | [181] | |||
| Macrocycle analogues | G-P-M−P-F | [X-P-Nle-P-X] [B1-P-Nle-P-X] [B1-P-Nle-P-Xd] | ↑ plasma peptide half-life ↑ binding affinity vs. apelin-13 powerful modulators of cardiovascular system | [168] |
| C-terminal | C-thermal exocyclic residues endocyclic residues | ↑ G-protein activation on over-arresting signalling binding affinity close to [Pyr1]-apelin-13 | [182] | |
| Internal position | ||||
| β alanine spacer | Same APJ affinity of apelin-13 ↓ hypotensive effect similar to apelin-13 | |||
| apelin-13′s His8 position | Nγ-allyl-Nγ-nosyl-α,γ-diamino-butanoic acid Nπ-allyl-histidine linker | ↑ binding affinity ↑ potency | [183] |
| Agonist | Substituted Residues | Introduced Residues | Effects | Ref |
|---|---|---|---|---|
| JN241-9 | between E104 and S105 to JN241 | tyrosine | agonist activity for cAMP β-arrestin recruitment | [186] |
| MM202 (apelin-modified) | Met | Nle and Fluroaddition-C-terminal | high binding affinity vs [Pyr1]-apelin-13 = or ↑ potency than [Pyr1]-apelin-13 | [187] |
| MM202-AlbudAb | = or ↑ potency than [Pyr1]-apelin-13 | |||
| MM202 and AlbudAb fusion | ↑ cardiac function ↓ heart rate | |||
| Fc-ELA-21 | - | Fc and ELA fusion | ↑ angiogenesis ↓ apoptosis ↑ cardiomyocyte proliferation ↓ heart fibrosis | [188] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossin, D.; Vanni, R.; Lo Iacono, M.; Cristallini, C.; Giachino, C.; Rastaldo, R. APJ as Promising Therapeutic Target of Peptide Analogues in Myocardial Infarction- and Hypertension-Induced Heart Failure. Pharmaceutics 2023, 15, 1408. https://doi.org/10.3390/pharmaceutics15051408
Rossin D, Vanni R, Lo Iacono M, Cristallini C, Giachino C, Rastaldo R. APJ as Promising Therapeutic Target of Peptide Analogues in Myocardial Infarction- and Hypertension-Induced Heart Failure. Pharmaceutics. 2023; 15(5):1408. https://doi.org/10.3390/pharmaceutics15051408
Chicago/Turabian StyleRossin, Daniela, Roberto Vanni, Marco Lo Iacono, Caterina Cristallini, Claudia Giachino, and Raffaella Rastaldo. 2023. "APJ as Promising Therapeutic Target of Peptide Analogues in Myocardial Infarction- and Hypertension-Induced Heart Failure" Pharmaceutics 15, no. 5: 1408. https://doi.org/10.3390/pharmaceutics15051408
APA StyleRossin, D., Vanni, R., Lo Iacono, M., Cristallini, C., Giachino, C., & Rastaldo, R. (2023). APJ as Promising Therapeutic Target of Peptide Analogues in Myocardial Infarction- and Hypertension-Induced Heart Failure. Pharmaceutics, 15(5), 1408. https://doi.org/10.3390/pharmaceutics15051408