
Delivery of DNA-Based Therapeutics for Treatment of Chronic Diseases

Abstract
1. Introduction
2. Adeno-Associated Virus-Based Gene Therapy
2.1. Attributes of Adeno-Associated Virus-Based Gene Therapy
2.2. AAV-Based Therapy in the Context of Chronic Disease
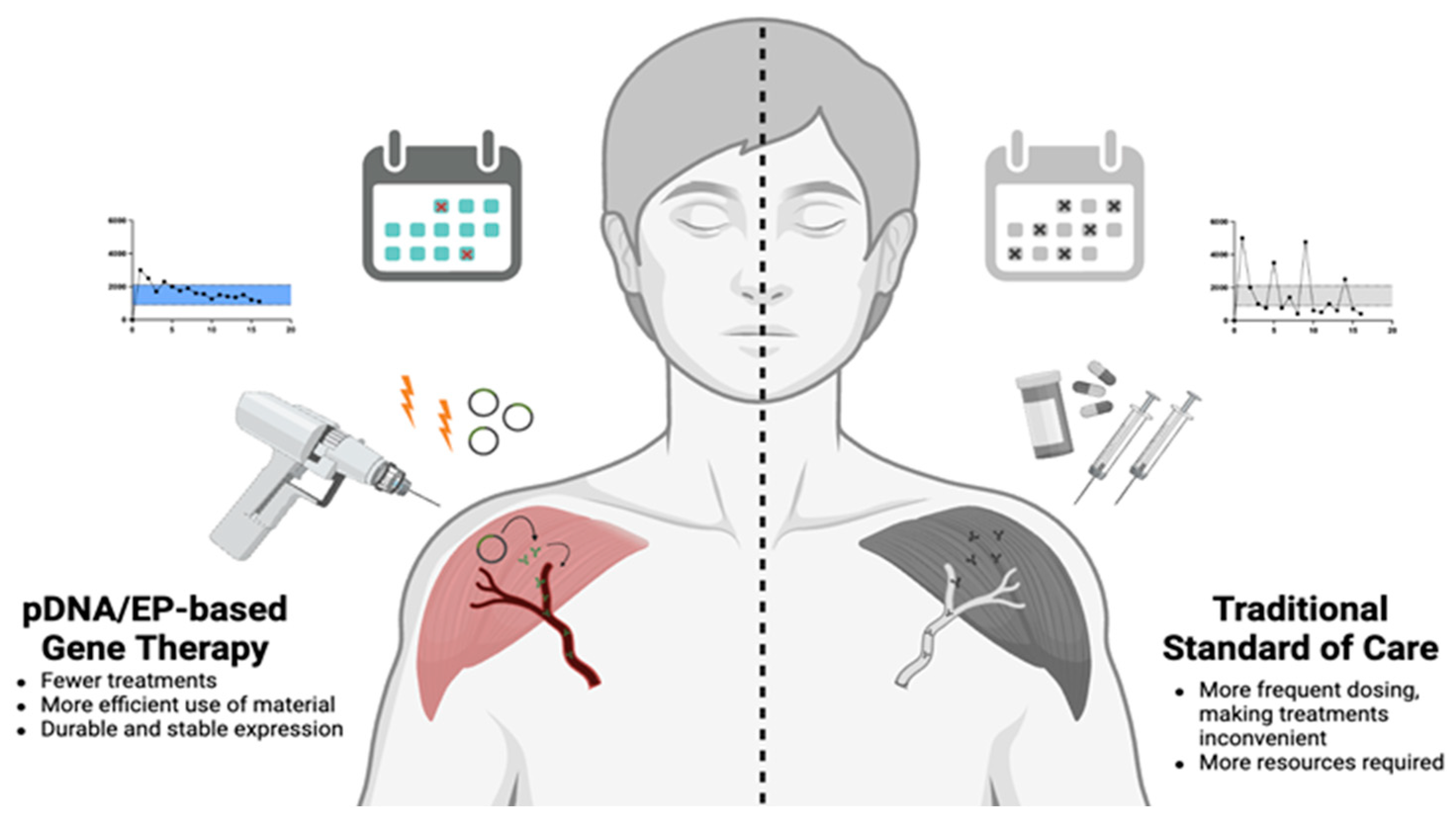
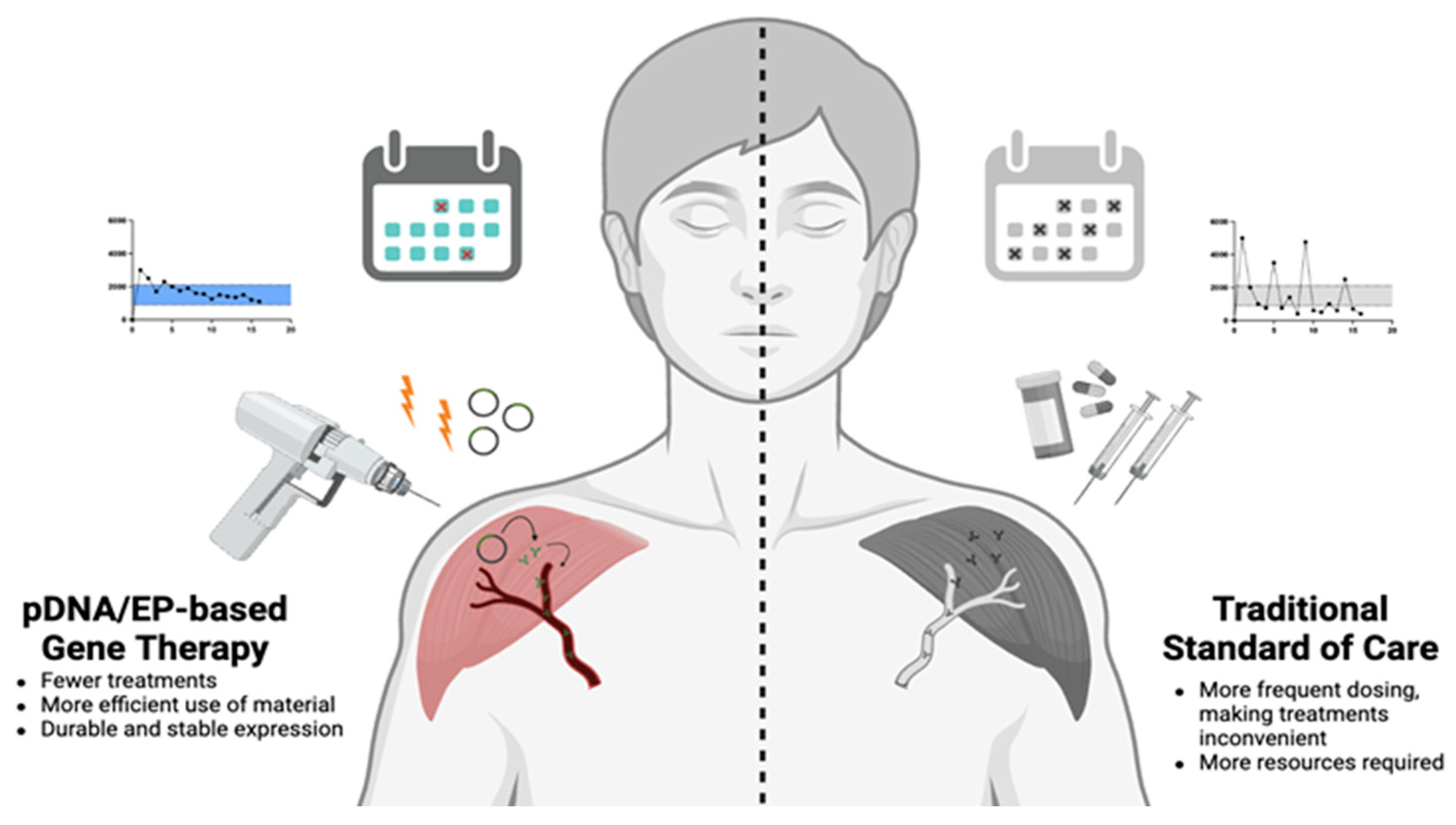
3. Use of pDNA in Gene Therapy
4. Electroporation as a Method for pDNA Delivery
4.1. Characteristics of Electroporation
4.2. Approaching Chronic Indications Using pDNA/EP
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bulaklak, K.; Gersbach, C.A. The once and future gene therapy. Nat. Commun. 2020, 11, 5820. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gao, G. State-of-the-Art Human Gene Therapy: Part I. Gene Delivery Technologies. Discov. Med. 2014, 18, 67–77. [Google Scholar] [PubMed]
- Ma, C.-C.; Wang, Z.-L.; Xu, T.; He, Z.-Y.; Wei, Y.-Q. The approved gene therapy drugs worldwide: From 1998 to 2019. Biotechnol. Adv. 2020, 40, 107502. [Google Scholar] [CrossRef] [PubMed]
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Afione, S.A.; Conrad, C.; McGrath, S.A.; Solow, R.; Oka, H.; Zeitlin, P.L.; Guggino, W.B.; Carter, B.J. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc. Natl. Acad. Sci. USA 1993, 90, 10613–10617. [Google Scholar] [CrossRef] [PubMed]
- Wagner, H.J.; Weber, W.; Fussenegger, M. Synthetic Biology: Emerging Concepts to Design and Advance Adeno-Associated Viral Vectors for Gene Therapy. Adv. Sci. 2021, 8, 2004018. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Viral Vectors in Gene Therapy: Where Do We Stand in 2023? Viruses 2023, 15, 698. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Hardee, C.L.; Arévalo-Soliz, L.M.; Hornstein, B.D.; Zechiedrich, L. Advances in Non-Viral DNA Vectors for Gene Therapy. Genes 2017, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Fynan, E.F.; Webster, R.G.; Fuller, D.H.; Haynes, J.R.; Santoro, J.C.; Robinson, H.L. DNA vaccines: Protective immunizations by parenteral, mucosal, and gene-gun inoculations. Proc. Natl. Acad. Sci. USA 1993, 90, 11478–11482. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, E.E.; DeGiulio, J.V.; Dean, D.A. Intracellular Trafficking of Plasmids for Gene Therapy: Mechanisms of Cytoplasmic Movement and Nuclear Import. Curr. Gene Ther. 2006, 6, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Widera, G.; Austin, M.; Rabussay, D.; Goldbeck, C.; Barnett, S.W.; Chen, M.; Leung, L.; Otten, G.R.; Thudium, K.; Selby, M.J.; et al. Increased DNA Vaccine Delivery and Immunogenicity by Electroporation In Vivo. J. Immunol. 2000, 164, 4635–4640. [Google Scholar] [CrossRef] [PubMed]
- Pagant, S.; Liberatore, R.A. In Vivo Electroporation of Plasmid DNA: A Promising Strategy for Rapid, Inexpensive, and Flexible Delivery of Anti-Viral Monoclonal Antibodies. Pharmaceutics 2021, 13, 1882. [Google Scholar] [CrossRef]
- Aihara, H.; Miyazaki, J. Gene transfer into muscle by electroporation in vivo. Nat. Biotechnol. 1998, 16, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen, I. Electropermeabilization of skeletal muscle enhances gene transfer in vivo. Gene Ther. 1999, 6, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Pupo, A.; Fernández, A.; Low, S.H.; François, A.; Suárez-Amarán, L.; Samulski, R.J. AAV vectors: The Rubik’s cube of human gene therapy. Mol. Ther. 2022, 30, 3515–3541. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.F.; Fraefel, C.; Seyffert, M. The Interplay between Adeno-Associated Virus and Its Helper Viruses. Viruses 2020, 12, 662. [Google Scholar] [CrossRef] [PubMed]
- Maurer, A.C.; Weitzman, M.D. Adeno-Associated Virus Genome Interactions Important for Vector Production and Transduction. Hum. Gene Ther. 2020, 31, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Schultz, B.R.; Chamberlain, J.S. Recombinant adeno-associated virus transduction and integration. Mol. Ther. 2008, 16, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Hermonat, P.L.; Muzyczka, N. Use of adeno-associated virus as a mammalian DNA cloning vector: Transduction of neomycin resistance into mammalian tissue culture cells. Proc. Natl. Acad. Sci. USA 1984, 81, 6466–6470. [Google Scholar] [CrossRef]
- Zhao, Z.; Anselmo, A.C.; Mitragotri, S. Viral vector-based gene therapies in the clinic. Bioeng. Transl. Med. 2022, 7, e10258. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef] [PubMed]
- Ylä-Herttuala, S. Endgame: Glybera Finally Recommended for Approval as the First Gene Therapy Drug in the European Union. Mol. Ther. 2012, 20, 1831–1832. [Google Scholar] [CrossRef] [PubMed]
- Senior, M. After Glybera’s withdrawal, what’s next for gene therapy? Nat. Biotechnol. 2017, 35, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Lusby, E.W.; Berns, K.I. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol. 1983, 45, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, H.; Colosi, P. Effect of Genome Size on AAV Vector Packaging. Mol. Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Dong, J.Y.; Fan, P.D.; Frizzell, R.A. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 1996, 7, 2101–2112. [Google Scholar] [CrossRef]
- Issa, S.S.; Shaimardanova, A.A.; Solovyeva, V.V.; Rizvanov, A.A. Various AAV Serotypes and Their Applications in Gene Therapy: An Overview. Cells 2023, 12, 785. [Google Scholar] [CrossRef]
- Muzyczka, N.; Warrington, K.H. Custom Adeno-Associated Virus Capsids: The Next Generation of Recombinant Vectors with Novel Tropism. Hum. Gene Ther. 2005, 16, 408–416. [Google Scholar] [CrossRef] [PubMed]
- White, A.F.; Mazur, M.; Sorscher, E.J.; Zinn, K.R.; Ponnazhagan, S. Genetic Modification of Adeno-Associated Viral Vector Type 2 Capsid Enhances Gene Transfer Efficiency in Polarized Human Airway Epithelial Cells. Hum. Gene Ther. 2008, 19, 1407–1414. [Google Scholar] [CrossRef]
- Huang, Q.; Chen, A.T.; Chan, K.Y.; Sorensen, H.; Barry, A.J.; Azari, B.; Zheng, Q.; Beddow, T.; Zhao, B.; Tobey, I.G.; et al. Targeting AAV vectors to the central nervous system by engineering capsid-receptor interactions that enable crossing of the blood-brain barrier. PLoS Biol. 2023, 21, e3002112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rossi, A.; Lange, L.; Meumann, N.; Koitzsch, U.; Christie, K.; Nesbit, M.A.; Moore, C.B.T.; Hacker, U.T.; Morgan, M.; et al. Capsid Engineering Overcomes Barriers toward Adeno-Associated Virus Vector-Mediated Transduction of Endothelial Cells. Hum. Gene Ther. 2019, 30, 1284–1296. [Google Scholar] [CrossRef] [PubMed]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV Serotypes 1–9 Mediated Gene Expression and Tropism in Mice After Systemic Injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Dhungel, B.P.; Winburn, I.; Pereira, C.d.F.; Huang, K.; Chhabra, A.; Rasko, J.E.J. Understanding AAV vector immunogenicity: From particle to patient. Theranostics 2024, 14, 1260–1288. [Google Scholar] [CrossRef] [PubMed]
- Klamroth, R.; Hayes, G.; Andreeva, T.; Gregg, K.; Suzuki, T.; Mitha, I.H.; Hardesty, B.; Shima, M.; Pollock, T.; Slev, P.; et al. Global Seroprevalence of Pre-existing Immunity Against AAV5 and Other AAV Serotypes in People with Hemophilia A. Hum. Gene Ther. 2022, 33, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.; Levy, D.I.; Petropoulos, C.J.; Bashirians, G.; Winburn, I.; Mahn, M.; Somanathan, S.; Cheng, S.H.; Byrne, B.J. Binding and neutralizing anti-AAV antibodies: Detection and implications for rAAV-mediated gene therapy. Mol. Ther. 2023, 31, 616–630. [Google Scholar] [CrossRef]
- Pipe, S.; Leebeek, F.W.G.; Ferreira, V.; Sawyer, E.K.; Pasi, J. Clinical Considerations for Capsid Choice in the Development of Liver-Targeted AAV-Based Gene Transfer. Mol. Ther. Methods Clin. Dev. 2019, 15, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Connolly, A.M.; Lehman, K.J.; Griffin, D.A.; Khan, S.Z.; Dharia, S.D.; Quintana-Gallardo, L.; Rodino-Klapac, L.R. Testing preexisting antibodies prior to AAV gene transfer therapy: Rationale, lessons and future considerations. Mol. Ther. Methods Clin. Dev. 2022, 25, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Immune responses to AAV in clinical trials. Curr. Gene Ther. 2007, 7, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Arjomandnejad, M.; Dasgupta, I.; Flotte, T.R.; Keeler, A.M. Immunogenicity of Recombinant Adeno-Associated Virus (AAV) Vectors for Gene Transfer. BioDrugs 2023, 37, 311–329. [Google Scholar] [CrossRef] [PubMed]
- Ertl, H.C.J. Immunogenicity and toxicity of AAV gene therapy. Front. Immunol. 2022, 13, 975803. [Google Scholar] [CrossRef] [PubMed]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.E.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shao, W. Innate Immune Response to Viral Vectors in Gene Therapy. Viruses 2023, 15, 1801. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Liu, S.; Ou, L. rAAV immunogenicity, toxicity, and durability in 255 clinical trials: A meta-analysis. Front. Immunol. 2022, 13, 1001263. [Google Scholar] [CrossRef] [PubMed]
- Kreppel, F.; Hagedorn, C. Capsid and Genome Modification Strategies to Reduce the Immunogenicity of Adenoviral Vectors. Int. J. Mol. Sci. 2021, 22, 2417. [Google Scholar] [CrossRef] [PubMed]
- Muhuri, M.; Maeda, Y.; Ma, H.; Ram, S.; Fitzgerald, K.A.; Tai, P.W.L.; Gao, G. Overcoming innate immune barriers that impede AAV gene therapy vectors. J. Clin. Investig. 2021, 131, e143780. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.K.; Samulski, R.J. Addressing high dose AAV toxicity—‘one and done’ or ‘slower and lower’? Expert. Opin. Biol. Ther. 2022, 22, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Urip, B.A.; Zhang, Z.; Ngan, C.C.; Feng, B. Evolving AAV-delivered therapeutics towards ultimate cures. J. Mol. Med. 2021, 99, 593–617. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Shitik, E.M.; Shalik, I.K.; Yudkin, D.V. AAV-based vector improvements unrelated to capsid protein modification. Front. Med. 2023, 10, 1106085. [Google Scholar] [CrossRef] [PubMed]
- Kolesnik, V.V.; Nurtdinov, R.F.; Oloruntimehin, E.S.; Karabelsky, A.V.; Malogolovkin, A.S. Optimization strategies and advances in the research and development of AAV-based gene therapy to deliver large transgenes. Clin. Transl. Med. 2024, 14, e1607. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.D.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2017, 8, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Domenger, C.; Grimm, D. Next-generation AAV vectors—Do not judge a virus (only) by its cover. Hum. Mol. Genet. 2019, 28, R3–R14. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Merchán, E.C.; De Pablo-Moreno, J.A.; Liras, A. Gene Therapy in Hemophilia: Recent Advances. Int. J. Mol. Sci. 2021, 22, 7647. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, L.; Staber, J.M.; Wright, J.F.; Valentino, L.A. The intersection of vector biology, gene therapy, and hemophilia. Res. Pract. Thromb. Haemost. 2021, 5, e12586. [Google Scholar] [CrossRef] [PubMed]
- Leebeek, F.W.G.; Miesbach, W. Gene therapy for hemophilia: A review on clinical benefit, limitations, and remaining issues. Blood 2021, 138, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Castaman, G.; Di Minno, G.; De Cristofaro, R.; Peyvandi, F. The Arrival of Gene Therapy for Patients with Hemophilia A. Int. J. Mol. Sci. 2022, 23, 10228. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento Doncel, S.; Díaz Mosquera, G.A.; Cortes, J.M.; Agudelo Rico, C.; Meza Cadavid, F.J.; Peláez, R.G. Haemophilia A: A Review of Clinical Manifestations, Treatment, Mutations, and the Development of Inhibitors. Hematol. Rep. 2023, 15, 130–150. [Google Scholar] [CrossRef] [PubMed]
- Berntorp, E.; Fischer, K.; Hart, D.P.; Mancuso, M.E.; Stephensen, D.; Shapiro, A.D.; Blanchette, V. Haemophilia. Nat. Rev. Dis. Primers 2021, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Castaman, G.; Matino, D. Hemophilia A and B: Molecular and clinical similarities and differences. Haematologica 2019, 104, 1702–1709. [Google Scholar] [CrossRef]
- Dolan, G.; Benson, G.; Duffy, A.; Hermans, C.; Jiménez-Yuste, V.; Lambert, T.; Ljung, R.; Morfini, M.; Zupančić Šalek, S. Haemophilia B: Where are we now and what does the future hold? Blood Rev. 2018, 32, 52–60. [Google Scholar] [CrossRef]
- Marchesini, E.; Morfini, M.; Valentino, L. Recent Advances in the Treatment of Hemophilia: A Review. Biol. Targets Ther. 2021, 15, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Makris, M. Prophylaxis in haemophilia should be life-long. Blood Transfus. 2012, 10, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Valoctocogene Roxaparvovec: First Approval. Drugs 2022, 82, 1505–1510. [Google Scholar] [CrossRef]
- Nathwani, A.C. Gene therapy for hemophilia. Hematol. Am. Soc. Hematol. Educ. Program. 2022, 2022, 569–578. [Google Scholar] [CrossRef]
- George, L.A.; Monahan, P.E.; Eyster, M.E.; Sullivan, S.K.; Ragni, M.V.; Croteau, S.E.; Rasko, J.E.J.; Recht, M.; Samelson-Jones, B.J.; MacDougall, A.; et al. Multiyear Factor VIII Expression after AAV Gene Transfer for Hemophilia A. N. Engl. J. Med. 2021, 385, 1961–1973. [Google Scholar] [CrossRef]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.J.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef]
- George, L.A.; Sullivan, S.K.; Rasko, J.E.J.; Giermasz, A.; Samelson-Jones, B.J.; Ducore, J.M.; Teitel, J.M.; McGuinn, C.E.; Runowski, A.R.; Wright, F.; et al. Efficacy and Safety in 15 Hemophilia B Patients Treated with the AAV Gene Therapy Vector Fidanacogene Elaparvovec and Followed for at Least 1 Year. Blood 2019, 134, 3347. [Google Scholar] [CrossRef]
- Frenzel, L.; Kavakli, K.; Klamroth, R.; Chiou, S.-S.; Shapiro, A.D.; Sun, P.; Fuiman, J.; McKay, J.; Fang, A.F.; Biondo, F.; et al. Characterizing a Cohort of Patients with Hemophilia B Treated with Fidanacogene Elaparvovec from the Phase 3 Benegene-2 Study Who Returned to Factor IX Prophylaxis. Blood 2023, 142, 2257. [Google Scholar] [CrossRef]
- Dev, S.; Kruse, R.L.; Hamilton, J.P.; Lutsenko, S. Wilson Disease: Update on Pathophysiology and Treatment. Front. Cell Dev. Biol. 2022, 10, 871877. [Google Scholar] [CrossRef] [PubMed]
- Shribman, S.; Poujois, A.; Bandmann, O.; Czlonkowska, A.; Warner, T.T. Wilson’s disease: Update on pathogenesis, biomarkers and treatments. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Greig, J.A.; Nordin, J.M.L.; Smith, M.K.; Ashley, S.N.; Draper, C.; Zhu, Y.; Bell, P.; Buza, E.L.; Wilson, J.M. A Gene Therapy Approach to Improve Copper Metabolism and Prevent Liver Damage in a Mouse Model of Wilson Disease. Hum. Gene Ther. Clin. Dev. 2019, 30, 29–39. [Google Scholar] [CrossRef]
- Murillo, O.; Luqui, D.M.; Gazquez, C.; Martinez-Espartosa, D.; Navarro-Blasco, I.; Monreal, J.I.; Guembe, L.; Moreno-Cermeño, A.; Corrales, F.J.; Prieto, J.; et al. Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy. J. Hepatol. 2016, 64, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Padula, A.; Petruzzelli, R.; Philbert, S.A.; Church, S.J.; Esposito, F.; Campione, S.; Monti, M.; Capolongo, F.; Perna, C.; Nusco, E.; et al. Full-length ATP7B reconstituted through protein trans-splicing corrects Wilson disease in mice. Mol. Ther. Methods Clin. Dev. 2022, 26, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, J.; Brunetti-Pierri, N.; Gissen, P. Liver-directed gene therapy for inherited metabolic diseases. J. Inherit. Metab. Dis. 2024, 47, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Baris, H.N.; Cohen, I.J.; Mistry, P.K. Gaucher disease: The metabolic defect, pathophysiology, phenotypes and natural history. Pediatr. Endocrinol. Rev. PER 2014, 12 (Suppl. 1), 72–81. [Google Scholar] [PubMed]
- Rosenbloom, B.E.; Weinreb, N.J. Gaucher disease: A comprehensive review. In Advances in Gaucher Disease: Basic and Clinical Perspectives; Future Medicine Ltd.: London, UK, 2013; pp. 26–49. [Google Scholar]
- Nalysnyk, L.; Rotella, P.; Simeone, J.C.; Hamed, A.; Weinreb, N. Gaucher disease epidemiology and natural history: A comprehensive review of the literature. Hematology 2017, 22, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Roshan Lal, T.; Sidransky, E. The Spectrum of Neurological Manifestations Associated with Gaucher Disease. Diseases 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Gary, S.E.; Ryan, E.; Steward, A.M.; Sidransky, E. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev. Endocrinol. Metab. 2018, 13, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.A.; Hassan, S.; Garcia, E.J.; Tayebi, N.; Sidransky, E. Exploring Genetic Modifiers of Gaucher Disease: The Next Horizon. Hum. Mutat. 2018, 39, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorth, M.; Narvekar, A. Non Viral Vectors in Gene Therapy—An Overview. J. Clin. Diagn. Res. 2015, 9, GE01–GE06. [Google Scholar] [CrossRef] [PubMed]
- Comerota, A.J.; Throm, R.C.; Miller, K.A.; Henry, T.; Chronos, N.; Laird, J.; Sequeira, R.; Kent, C.K.; Bacchetta, M.; Goldman, C.; et al. Naked plasmid DNA encoding fibroblast growth factor type 1 for the treatment of end-stage unreconstructible lower extremity ischemia: Preliminary results of a phase I trial. J. Vasc. Surg. 2002, 35, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Guthrie, K.A.; Liu, Y.; Coveler, A.L.; Higgins, D.M.; Childs, J.S.; Dang, Y.; Salazar, L.G. Safety and Outcomes of a Plasmid DNA Vaccine Encoding the ERBB2 Intracellular Domain in Patients with Advanced-Stage ERBB2-Positive Breast Cancer: A Phase 1 Nonrandomized Clinical Trial. JAMA Oncol. 2023, 9, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Khobragade, A.; Bhate, S.; Ramaiah, V.; Deshpande, S.; Giri, K.; Phophle, H.; Supe, P.; Godara, I.; Revanna, R.; Nagarkar, R.; et al. Efficacy, safety, and immunogenicity of the DNA SARS-CoV-2 vaccine (ZyCoV-D): The interim efficacy results of a phase 3, randomised, double-blind, placebo-controlled study in India. Lancet 2022, 399, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Schalk, J.A.C.; Mooi, F.R.; Berbers, G.A.M.; van Aerts, L.A.G.J.M.; Ovelgönne, H.; Kimman, T.G. Preclinical and Clinical Safety Studies on DNA Vaccines. Hum. Vaccines 2006, 2, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Donsante, A.; Miller, D.G.; Li, Y.; Vogler, C.; Brunt, E.M.; Russell, D.W.; Sands, M.S. AAV Vector Integration Sites in Mouse Hepatocellular Carcinoma. Science 2007, 317, 477. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Trivedi, N.S.; Carrillo-Carrasco, N.; Senac, J.S.; Wu, W.; Hoffmann, V.; Elkahloun, A.G.; Burgess, S.M.; et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Investig. 2015, 125, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Nakai, H.; Montini, E.; Fuess, S.; Storm, T.A.; Grompe, M.; Kay, M.A. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat. Genet. 2003, 34, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Lozier, J.N.; Csako, G.; Mondoro, T.H.; Krizek, D.M.; Metzger, M.E.; Costello, R.; Vostal, J.G.; Rick, M.E.; Donahue, R.E.; Morgan, R.A. Toxicity of a First-Generation Adenoviral Vector in Rhesus Macaques. Hum. Gene Ther. 2002, 13, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Sheets, R.L.; Stein, J.; Manetz, T.S.; Andrews, C.; Bailer, R.; Rathmann, J.; Gomez, P.L. Toxicological Safety Evaluation of DNA Plasmid Vaccines against HIV-1, Ebola, Severe Acute Respiratory Syndrome, or West Nile Virus Is Similar Despite Differing Plasmid Backbones or Gene-Inserts. Toxicol. Sci. 2006, 91, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Sheets, R.L.; Stein, J.; Manetz, T.S.; Duffy, C.; Nason, M.; Andrews, C.; Kong, W.-P.; Nabel, G.J.; Gomez, P.L. Biodistribution of DNA Plasmid Vaccines against HIV-1, Ebola, Severe Acute Respiratory Syndrome, or West Nile Virus Is Similar, without Integration, despite Differing Plasmid Backbones or Gene Inserts. Toxicol. Sci. 2006, 91, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Bah, M.A.; Weiner, D.B. In Vivo Delivery of Nucleic Acid-Encoded Monoclonal Antibodies. BioDrugs 2020, 34, 273–293. [Google Scholar] [CrossRef] [PubMed]
- Manam, S.; Ledwith, B.J.; Barnum, A.B.; Troilo, P.J.; Pauley, C.J.; Harper, L.B.; Griffiths, T.G., II; Niu, Z.; Denisova, L.; Follmer, T.T.; et al. Plasmid DNA Vaccines: Tissue Distribution and Effects of DNA Sequence, Adjuvants and Delivery Method on Integration into Host DNA. Intervirology 2001, 43, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.K.; Choi, S.M.; Choi, J.H.; Lee, D.S.; Kim, C.Y.; Ahn, B.O.; Kim, B.M.; Kim, W.B. Safety Evaluation of GX-12, a New HIV Therapeutic Vaccine: Investigation of Integration into the Host Genome and Expression in the Reproductive Organs. Intervirology 2003, 46, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Vilalta, A.; Mahajan, R.K.; Hartikka, J.; Rusalov, D.; Martin, T.; Bozoukova, V.; Leamy, V.; Hall, K.; Lalor, P.; Rolland, A.; et al. I. Poloxamer-Formulated Plasmid DNA-Based Human Cytomegalovirus Vaccine: Evaluation of Plasmid DNA Biodistribution/Persistence and Integration. Hum. Gene Ther. 2005, 16, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.R.; Pringle, I.A.; Hyde, S.C. Progress and Prospects: The design and production of plasmid vectors. Gene Ther. 2009, 16, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Baum, B.J. Evaluation of Promoters for Use in Tissue-Specific Gene Delivery. In Gene Therapy Protocols; Doux, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 205–219. [Google Scholar]
- van der Loo, J.C.M.; Wright, J.F. Progress and challenges in viral vector manufacturing. Hum. Mol. Genet. 2016, 25, R42–R52. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Dalby, P.A. Challenges in scaling up AAV-based gene therapy manufacturing. Trends Biotechnol. 2023, 41, 1268–1281. [Google Scholar] [CrossRef] [PubMed]
- Matange, K.; Tuck, J.M.; Keung, A.J. DNA stability: A central design consideration for DNA data storage systems. Nat. Commun. 2021, 12, 1358. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.B.; Harvey, B.K. Assaying the Stability and Inactivation of AAV Serotype 1 Vectors. Hum. Gene Ther. Methods 2017, 28, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Mairhofer, J.; Grabherr, R. Rational Vector Design for Efficient Non-viral Gene Delivery: Challenges Facing the Use of Plasmid DNA. Mol. Biotechnol. 2008, 39, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Carnes, A.E.; Hodgson, C.P. Plasmid DNA Vaccine vector design: Impact on efficacy, safety and upstream production. Biotechnol. Adv. 2009, 27, 353–370. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-Y.; Riu, E.; He, C.-Y.; Xu, H.; Kay, M.A. Silencing of Episomal Transgene Expression in Liver by Plasmid Bacterial Backbone DNA Is Independent of CpG Methylation. Mol. Ther. 2008, 16, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Darquet, A.-M.; Rangara, R.; Kreiss, P.; Schwartz, B.; Naimi, S.; Delaère, P.; Crouzet, J.; Scherman, D. Minicircle: An improved DNA molecule for in vitro and in vivo gene transfer. Gene Ther. 1999, 6, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.A.; He, C.-Y.; Chen, Z.-Y. A robust system for production of minicircle DNA vectors. Nat. Biotechnol. 2010, 28, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Barreira, M.; Kerridge, C.; Jorda, S.; Olofsson, D.; Neumann, A.; Horton, H.; Smith-Moore, S. Enzymatically amplified linear dbDNATM as a rapid and scalable solution to industrial lentiviral vector manufacturing. Gene Ther. 2023, 30, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Vandermeulen, G.; Marie, C.; Scherman, D.; Préat, V. New Generation of Plasmid Backbones Devoid of Antibiotic Resistance Marker for Gene Therapy Trials. Mol. Ther. 2011, 19, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- Dean, D.A. Nonviral gene transfer to skeletal, smooth, and cardiac muscle in living animals. Am. J. Physiol.-Cell Physiol. 2005, 289, C233–C245. [Google Scholar] [CrossRef] [PubMed]
- Young, J.L.; Dean, D.A. Nonviral gene transfer strategies for the vasculature. Microcirculation 2002, 9, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Mali, S. Delivery systems for gene therapy. Indian. J. Hum. Genet. 2013, 19, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.K.; Kim, S.W. Recent advances in the development of gene delivery systems. Biomater. Res. 2019, 23, 8. [Google Scholar] [CrossRef]
- Du, X.; Wang, J.; Zhou, Q.; Zhang, L.; Wang, S.; Zhang, Z.; Yao, C. Advanced physical techniques for gene delivery based on membrane perforation. Drug Deliv. 2018, 25, 1516–1525. [Google Scholar] [CrossRef]
- Thi, T.T.H.; Suys, E.J.A.; Lee, J.S.; Nguyen, D.H.; Park, K.D.; Truong, N.P. Lipid-Based Nanoparticles in the Clinic and Clinical Trials: From Cancer Nanomedicine to COVID-19 Vaccines. Vaccines 2021, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Jerzykiewicz, J.; Czogalla, A. Polyethyleneimine-Based Lipopolyplexes as Carriers in Anticancer Gene Therapies. Materials 2022, 15, 179. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.; Wu, S.; An, J.; Liang, Y.; Hou, L.; Zhang, Z. Self-responsive co-delivery system for remodeling tumor intracellular microenvironment to promote PTEN-mediated anti-tumor therapy. Nanoscale 2020, 12, 9392–9403. [Google Scholar] [CrossRef]
- Mir, L.M.; Bureau, M.F.; Gehl, J.; Rangara, R.; Rouy, D.; Caillaud, J.-M.; Delaere, P.; Branellec, D.; Schwartz, B.; Scherman, D. High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc. Natl. Acad. Sci. USA 1999, 96, 4262–4267. [Google Scholar] [CrossRef] [PubMed]
- Rosazza, C.; Meglic, S.H.; Zumbusch, A.; Rols, M.-P.; Miklavcic, D. Gene Electrotransfer: A Mechanistic Perspective. Curr. Gene Ther. 2016, 16, 98–129. [Google Scholar] [CrossRef]
- Todorova, B.; Adam, L.; Culina, S.; Boisgard, R.; Martinon, F.; Cosma, A.; Ustav, M.; Kortulewski, T.; Le Grand, R.; Chapon, C. Electroporation as a vaccine delivery system and a natural adjuvant to intradermal administration of plasmid DNA in macaques. Sci. Rep. 2017, 7, 4122. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Nguyen, B.; Lee, J.Y.; Browning, E.; Zhang, J.; Mukhopadhyay, A.; Gujar, R.; Salazar, J.; Hermiz, R.; Svenson, L.; et al. Intratumoral Electroporation of Plasmid Encoded IL12 and Membrane-Anchored Anti-CD3 Increases Systemic Tumor Immunity. Mol. Cancer Res. MCR 2022, 20, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Longino, N.V.; Miller, N.J.; Kulikauskas, R.; Iyer, J.G.; Ibrani, D.; Blom, A.; Byrd, D.R.; Parvathaneni, U.; Twitty, C.G.; et al. Intratumoral Delivery of Plasmid IL12 Via Electroporation Leads to Regression of Injected and Noninjected Tumors in Merkel Cell Carcinoma. Clin. Cancer Res. 2020, 26, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Greaney, S.K.; Algazi, A.P.; Tsai, K.K.; Takamura, K.T.; Chen, L.; Twitty, C.G.; Zhang, L.; Paciorek, A.; Pierce, R.H.; Le, M.H.; et al. Intratumoral Plasmid IL12 Electroporation Therapy in Patients with Advanced Melanoma Induces Systemic and Intratumoral T-cell Responses. Cancer Immunol. Res. 2020, 8, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Young, J.L.; Dean, D.A. Chapter Three—Electroporation-Mediated Gene Delivery. In Advances in Genetics; Huang, L., Liu, D., Wagner, E., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 49–88. [Google Scholar]
- Bhattacharya, S.; Silkunas, M.; Gudvangen, E.; Mangalanathan, U.; Pakhomova, O.N.; Pakhomov, A.G. Ca2+ dependence and kinetics of cell membrane repair after electropermeabilization. Biochim. Biophys. Acta BBA-Biomembr. 2022, 1864, 183823. [Google Scholar] [CrossRef] [PubMed]
- Silkunas, M.; Silkuniene, G.; Pakhomov, A.G. Real-time imaging of individual electropores proves their longevity in cells. Biochem. Biophys. Res. Commun. 2024, 695, 149408. [Google Scholar] [CrossRef] [PubMed]
- Wegener, J.; Keese, C.R.; Giaever, I. Recovery of Adherent Cells after In Situ Electroporation Monitored Electrically. BioTechniques 2002, 33, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Zhao, Y.; Lu, H.; Pang, W.; Xu, Y. In vivo plasmid DNA electroporation resulted in transfection of satellite cells and lasting transgene expression in regenerated muscle fibers. Biochem. Biophys. Res. Commun. 2005, 338, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Hollevoet, K.; Thomas, D.; Compernolle, G.; Vermeire, G.; De Smidt, E.; De Vleeschauwer, S.; Smith, T.R.F.; Fisher, P.D.; Dewilde, M.; Geukens, N.; et al. Clinically relevant dosing and pharmacokinetics of DNA-encoded antibody therapeutics in a sheep model. Front. Oncol. 2022, 12, 1017612. [Google Scholar] [CrossRef]
- Andrews, C.D.; Huang, Y.; Ho, D.D.; Liberatore, R.A. In vivo expressed biologics for infectious disease prophylaxis: Rapid delivery of DNA-based antiviral antibodies. Emerg. Microbes Infect. 2020, 9, 1523–1533. [Google Scholar] [CrossRef]
- Hollevoet, K.; De Vleeschauwer, S.; De Smidt, E.; Vermeire, G.; Geukens, N.; Declerck, P. Bridging the Clinical Gap for DNA-Based Antibody Therapy Through Translational Studies in Sheep. Hum. Gene Ther. 2019, 30, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- McNee, A.; Smith, T.R.F.; Holzer, B.; Clark, B.; Bessell, E.; Guibinga, G.; Brown, H.; Schultheis, K.; Fisher, P.; Ramos, S.; et al. Establishment of a Pig Influenza Challenge Model for Evaluation of Monoclonal Antibody Delivery Platforms. J. Immunol. 2020, 205, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Mau, T.; Amin, M.R.; Belafsky, P.C.; Best, S.R.; Friedman, A.D.; Klein, A.M.; Lott, D.G.; Paniello, R.C.; Pransky, S.M.; Saba, N.F.; et al. Interim Results of a Phase 1/2 Open-Label Study of INO-3107 for HPV-6 and/or HPV-11-Associated Recurrent Respiratory Papillomatosis. Laryngoscope 2023, 133, 3087–3093. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wise, M.C.; Choi, H.; Perales-Puchalt, A.; Patel, A.; Tello-Ruiz, E.; Chu, J.D.; Muthumani, K.; Weiner, D.B. Synthetic DNA delivery by electroporation promotes robust in vivo sulfation of broadly neutralizing anti-HIV immunoadhesin eCD4-Ig. eBioMedicine 2018, 35, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Edupuganti, S.; De Rosa, S.C.; Elizaga, M.; Lu, Y.; Han, X.; Huang, Y.; Swann, E.; Polakowski, L.; Kalams, S.A.; Keefer, M.; et al. Intramuscular and Intradermal Electroporation of HIV-1 PENNVAX-GP® DNA Vaccine and IL-12 Is Safe, Tolerable, Acceptable in Healthy Adults. Vaccines 2020, 8, 741. [Google Scholar] [CrossRef] [PubMed]
- Bettan, M.; Emmanuel, F.; Darteil, R.; Caillaud, J.-M.; Soubrier, F.; Delaere, P.; Branelec, D.; Mahfoudi, A.; Duverger, N.; Scherman, D. High-Level Protein Secretion into Blood Circulation after Electric Pulse-Mediated Gene Transfer into Skeletal Muscle. Mol. Ther. 2000, 2, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Fewell, J.G.; MacLaughlin, F.; Mehta, V.; Gondo, M.; Nicol, F.; Wilson, E.; Smith, L.C. Gene Therapy for the Treatment of Hemophilia B Using PINC-Formulated Plasmid Delivered to Muscle with Electroporation. Mol. Ther. 2001, 3, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Dale, D.C.; Bolyard, A.A. An update on the diagnosis and treatment of chronic idiopathic neutropenia. Curr. Opin. Hematol. 2017, 24, 46. [Google Scholar] [CrossRef]
- Rezaei, N.; Moazzami, K.; Aghamohammadi, A.; Klein, C. Neutropenia and Primary Immunodeficiency Diseases. Int. Rev. Immunol. 2009, 28, 335–366. [Google Scholar] [CrossRef] [PubMed]
- Spiekermann, K.; Roesler, J.; Emmendoerffer, A.; Elsner, J.; Welte, K. Functional features of neutrophils induced by G-CSF and GM-CSF treatment: Differential effects and clinical implications. Leukemia 1997, 11, 466–478. [Google Scholar] [CrossRef]
- Link, H. Current state and future opportunities in granulocyte colony-stimulating factor (G-CSF). Support. Care Cancer 2022, 30, 7067–7077. [Google Scholar] [CrossRef] [PubMed]
- Theyab, A.; Alsharif, K.F.; Alzahrani, K.J.; Oyouni, A.A.A.; Hawsawi, Y.M.; Algahtani, M.; Alghamdi, S.; Alshammary, A.F. New insight into strategies used to develop long-acting G-CSF biologics for neutropenia therapy. Front. Oncol. 2023, 12, 1026377. [Google Scholar] [CrossRef] [PubMed]
- Nikravesh, F.Y.; Shirkhani, S.; Bayat, E.; Talebkhan, Y.; Mirabzadeh, E.; Sabzalinejad, M.; Aliabadi, H.A.M.; Nematollahi, L.; Ardakani, Y.H.; Sardari, S. Extension of human GCSF serum half-life by the fusion of albumin binding domain. Sci. Rep. 2022, 12, 667. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Wassif, W.S. Adrenal insufficiency. J. Clin. Pathol. 2022, 75, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Barthel, A.; Benker, G.; Berens, K.; Diederich, S.; Manfras, B.; Gruber, M.; Kanczkowski, W.; Kline, G.; Kamvissi-Lorenz, V.; Hahner, S.; et al. An Update on Addison’s Disease. Exp. Clin. Endocrinol. Diabetes 2019, 127, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Draghia-Akli, R.; Ellis, K.M.; Hill, L.A.; Malone, P.B.; Fiorotto, M.L. High-efficiency growth hormone-releasing hormone plasmid vector administration into skeletal muscle mediated by electroporation in pigs. FASEB J. 2003, 3, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, M.-L.; Geukens, N.; Hollevoet, K.; Declerck, P.; Dewilde, M. Exploring the Fate of Antibody-Encoding pDNA after Intramuscular Electroporation in Mice. Pharmaceutics 2023, 15, 1160. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Danishmalik, S.N.; Hwang, H.; Sin, J.-I.; Oh, J.; Cho, Y.; Lee, H.; Jeong, M.; Kim, S.-H.; Hong, H.J. Gene therapy using plasmid DNA-encoded anti-HER2 antibody for cancers that overexpress HER2. Cancer Gene Ther. 2016, 23, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Parzych, E.M.; Du, J.; Ali, A.R.; Schultheis, K.; Frase, D.; Smith, T.R.F.; Cui, J.; Chokkalingam, N.; Tursi, N.J.; Andrade, V.M.; et al. DNA-delivered antibody cocktail exhibits improved pharmacokinetics and confers prophylactic protection against SARS-CoV-2. Nat. Commun. 2022, 13, 5886. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Ho, D.D.; Abdool Karim, Q. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet 2006, 368, 489–504. [Google Scholar] [CrossRef]
- Becerra, J.C.; Bildstein, L.S.; Gach, J.S. Recent Insights into the HIV/AIDS Pandemic. Microb. Cell 2016, 3, 451–475. [Google Scholar] [CrossRef] [PubMed]
- Kemnic, T.R.; Gulick, P.G. HIV Antiretroviral Therapy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Gandhi, R.T.; Bedimo, R.; Hoy, J.F.; Landovitz, R.J.; Smith, D.M.; Eaton, E.F.; Lehmann, C.; Springer, S.A.; Sax, P.E.; Thompson, M.A.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2022 Recommendations of the International Antiviral Society–USA Panel. JAMA 2023, 329, 63–84. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.H.R.; Henry, W.K.; Podzamczer, D.; Masiá, M.D.M.; Bettacchi, C.J.; Arasteh, K.; Jaeger, H.; Khuong-Josses, M.-A.; Montes-Ramírez, M.L.; Stellbrink, H.-J.; et al. Efficacy, Safety, and Durability of Long-Acting Cabotegravir and Rilpivirine in Adults with Human Immunodeficiency Virus Type 1 Infection: 5-Year Results from the LATTE-2 Study. Open Forum Infect. Dis. 2021, 8, ofab439. [Google Scholar] [CrossRef] [PubMed]
- Wise, M.C.; Xu, Z.; Tello-Ruiz, E.; Beck, C.; Trautz, A.; Patel, A.; Elliott, S.T.C.; Chokkalingam, N.; Kim, S.; Kerkau, M.G.; et al. In vivo delivery of synthetic DNA–encoded antibodies induces broad HIV-1–neutralizing activity. J. Clin. Investig. 2020, 130, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Greb, J.E.; Goldminz, A.M.; Elder, J.T.; Lebwohl, M.G.; Gladman, D.D.; Wu, J.J.; Mehta, N.N.; Finlay, A.Y.; Gottlieb, A.B. Psoriasis. Nat. Rev. Dis. Primers 2016, 2, 16082. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.W.; Read, C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA 2020, 323, 1945–1960. [Google Scholar] [CrossRef] [PubMed]
- Bellinato, F.; Gisondi, P.; Girolomoni, G. Latest Advances for the Treatment of Chronic Plaque Psoriasis with Biologics and Oral Small Molecules. Biologics 2021, 15, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes—State-of-the-art. Mol. Metab. 2021, 46, 101102. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.; Costello, R.A. Glucagon-like Peptide-1 Receptor Agonists. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Yao, H.; Zhang, A.; Li, D.; Wu, Y.; Wang, C.-Z.; Wan, J.-Y.; Yuan, C.-S. Comparative effectiveness of GLP-1 receptor agonists on glycaemic control, body weight, and lipid profile for type 2 diabetes: Systematic review and network meta-analysis. BMJ 2024, 384, e076410. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Feature | pDNA/EP Gene Therapy | AAV Gene Therapy |
|---|---|---|
| Durability | Months to years | Months to many years |
| Clinical safety of technology | *** | * |
| Redosable | **** | * |
| Large genetic payload | **** | ** |
| Opportunity | Systemic activity, Genetic disease, Chronic disease | Specific tissue target, Genetic disease, Chronic disease |
| Large scale manufacturing | **** | * |
| Freedom from cold chain | **** | * |
| Natural AAV Serotype Tissue Tropisms | |
|---|---|
| Tissue Type | AAV Serotype |
| Skeletal Muscle | AAV1, AAV2, AAV6, AAV7, AAV8, AAV9, AAV12 |
| Cardio-myocytes | AAV1, AAV4, AAV6, AAV7, AAV8, AAV9 |
| Endothelial Vascular Smooth Muscles | AAV1, AAV5, AAV7 |
| Inner Ear Cells | AAV3 |
| Retinal Cells | AAV1, AAV2, AAV4, AAV5, AAV7, AAV8, AAV9, AAV10 |
| CNS | AAV1, AAV2, AAV4, AAV5, AAV7, AAV9, AAV10, AAV11 |
| Airway Epithelia | AAV4, AAV5, AAV6, AAV9, AAV10, AAV12 |
| Hepatocyes | AAV2, AAV3, AAV5, AAV7, AAV8, AAV9, AAV10, AAV11 |
| Salivary Glands | AAV12 |
| Pancreatic Cells | AAV8, AAV9, AAV10 |
| Small Intestine Cells | AAV10, AAV11 |
| Colon Cells | AAV10 |
| Lymph Nodes | AAV10, AAV11 |
| Leydig Cells | AAV9 |
| Adrenal Glands | AAV10, AAV11 |
| Renal Tissue | AAV2, AAV4, AAV8, AAV9, AAV10, AAV11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sussman, C.; Liberatore, R.A.; Drozdz, M.M. Delivery of DNA-Based Therapeutics for Treatment of Chronic Diseases. Pharmaceutics 2024, 16, 535. https://doi.org/10.3390/pharmaceutics16040535
Sussman C, Liberatore RA, Drozdz MM. Delivery of DNA-Based Therapeutics for Treatment of Chronic Diseases. Pharmaceutics. 2024; 16(4):535. https://doi.org/10.3390/pharmaceutics16040535
Chicago/Turabian StyleSussman, Carleigh, Rachel A. Liberatore, and Marek M. Drozdz. 2024. "Delivery of DNA-Based Therapeutics for Treatment of Chronic Diseases" Pharmaceutics 16, no. 4: 535. https://doi.org/10.3390/pharmaceutics16040535
APA StyleSussman, C., Liberatore, R. A., & Drozdz, M. M. (2024). Delivery of DNA-Based Therapeutics for Treatment of Chronic Diseases. Pharmaceutics, 16(4), 535. https://doi.org/10.3390/pharmaceutics16040535