


A Novel Class of Human ADAM8 Inhibitory Antibodies for Treatment of Triple-Negative Breast Cancer

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. ADP Generation
2.3. ELISA
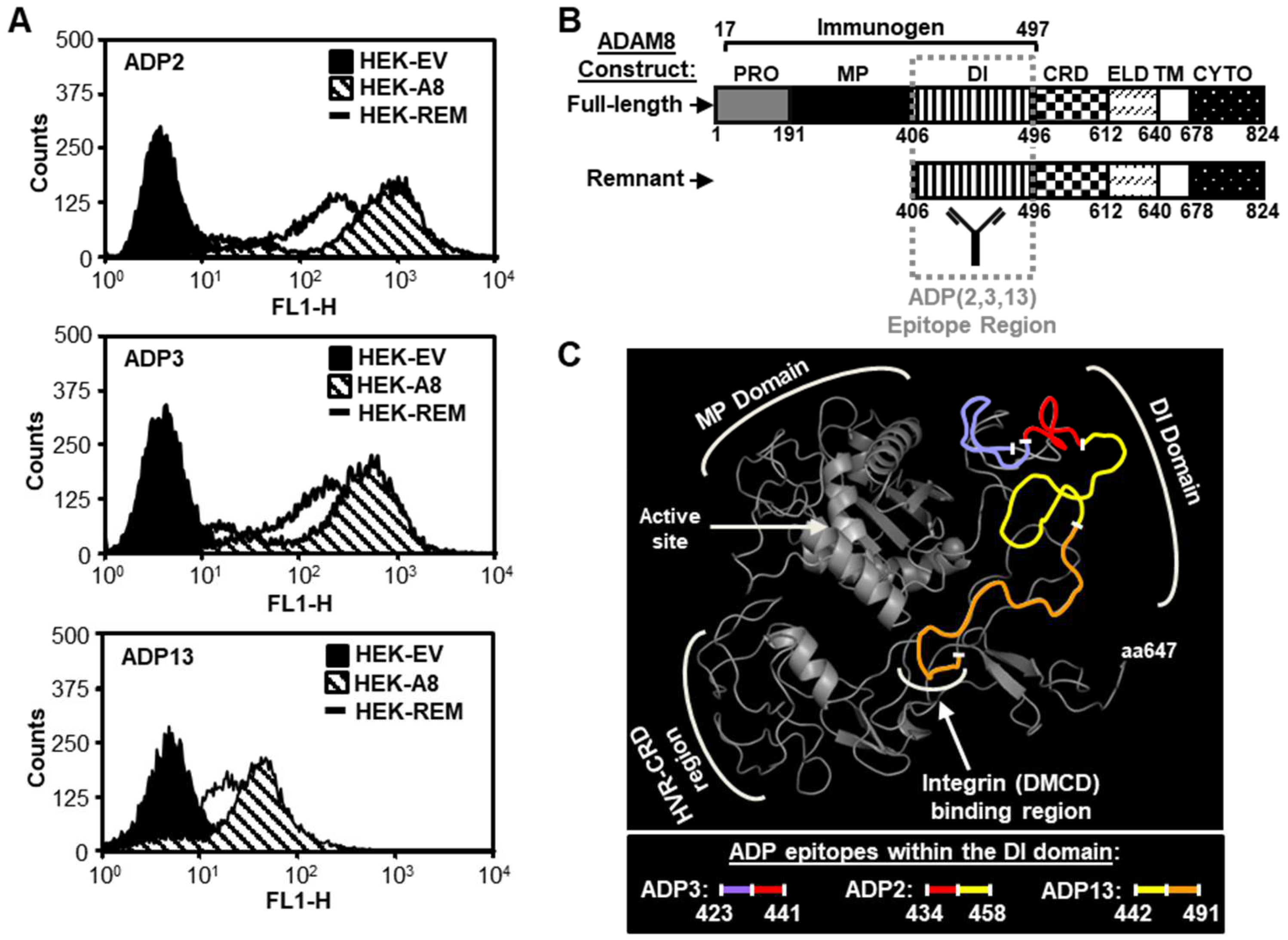
2.4. Flow Cytometry
2.5. Biacore Surface Plasmon Resonance
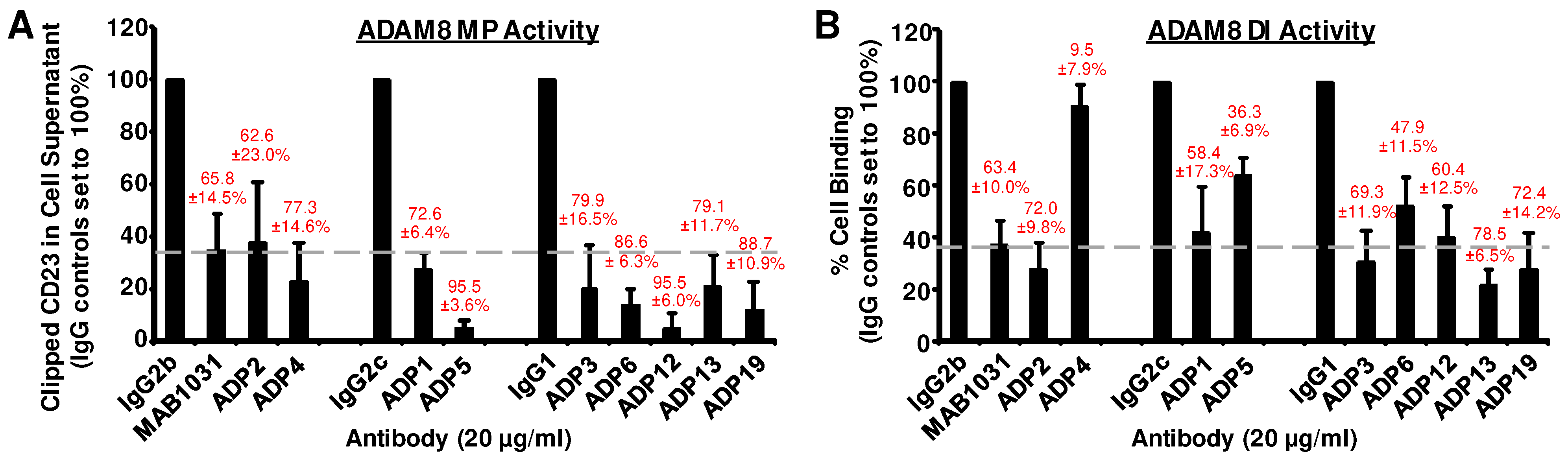
2.6. ADAM8 Functional Assays
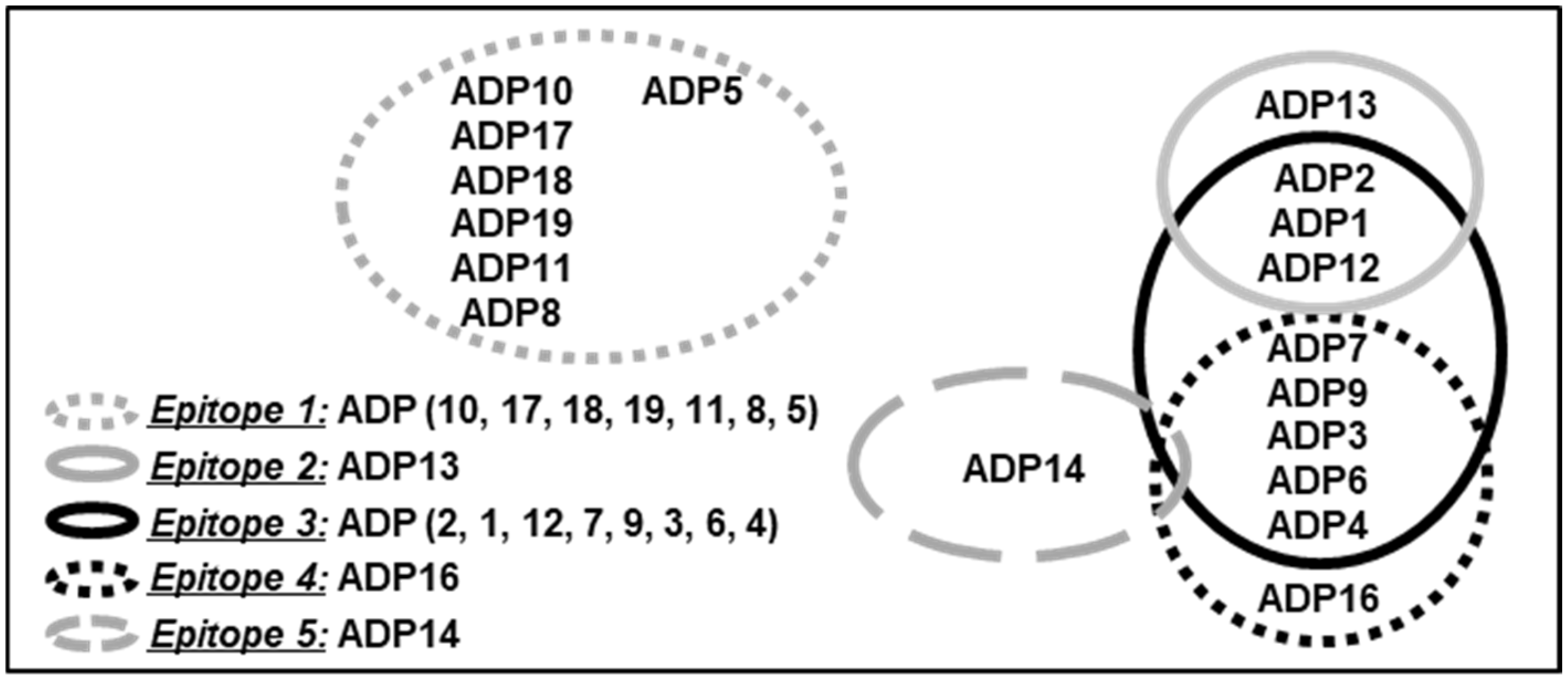
2.7. Epitope Binning
2.8. Hydrogen/Deuterium Exchange–Mass Spectrometry (HDX-MS)
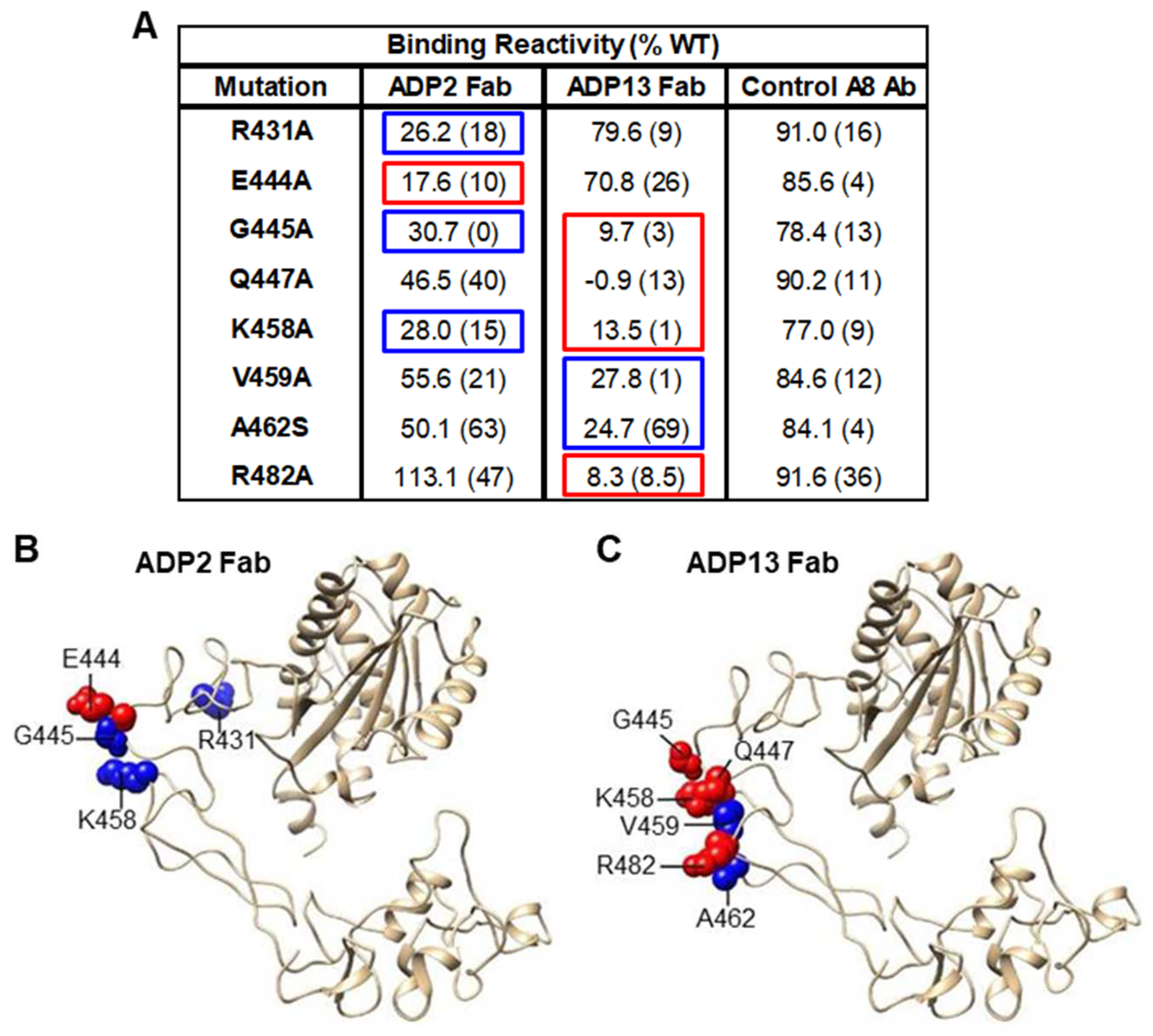
2.9. Mutagenesis Mapping
2.10. Orthotopic TNBC Mouse Models
2.10.1. Primary Tumor Growth
2.10.2. Tumor Regrowth
2.10.3. Metastasis and Outcome
2.11. Statistical Analyses
3. Results
3.1. Development of a Panel of ADAM8 MP and DI Inhibitory mAbs
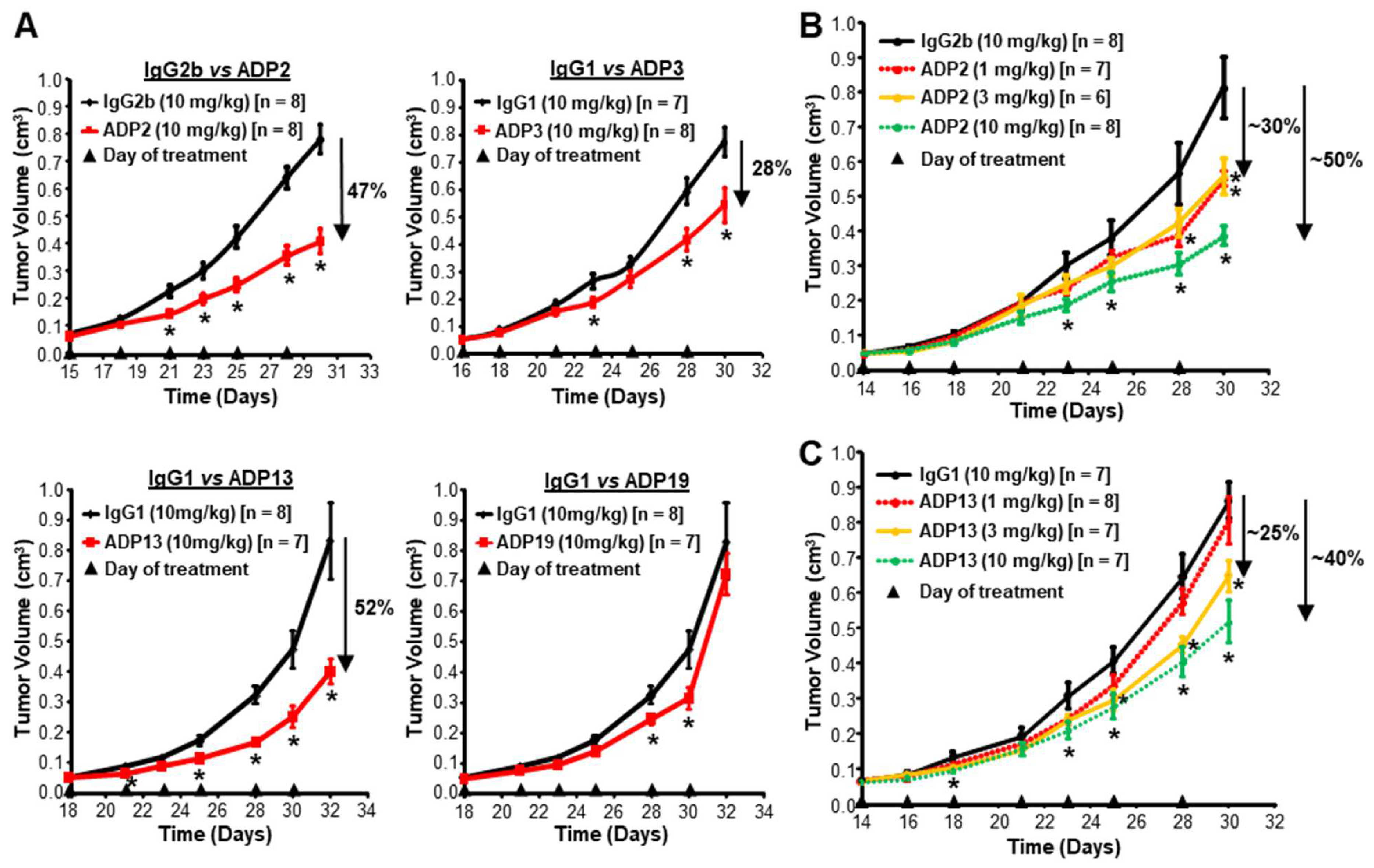
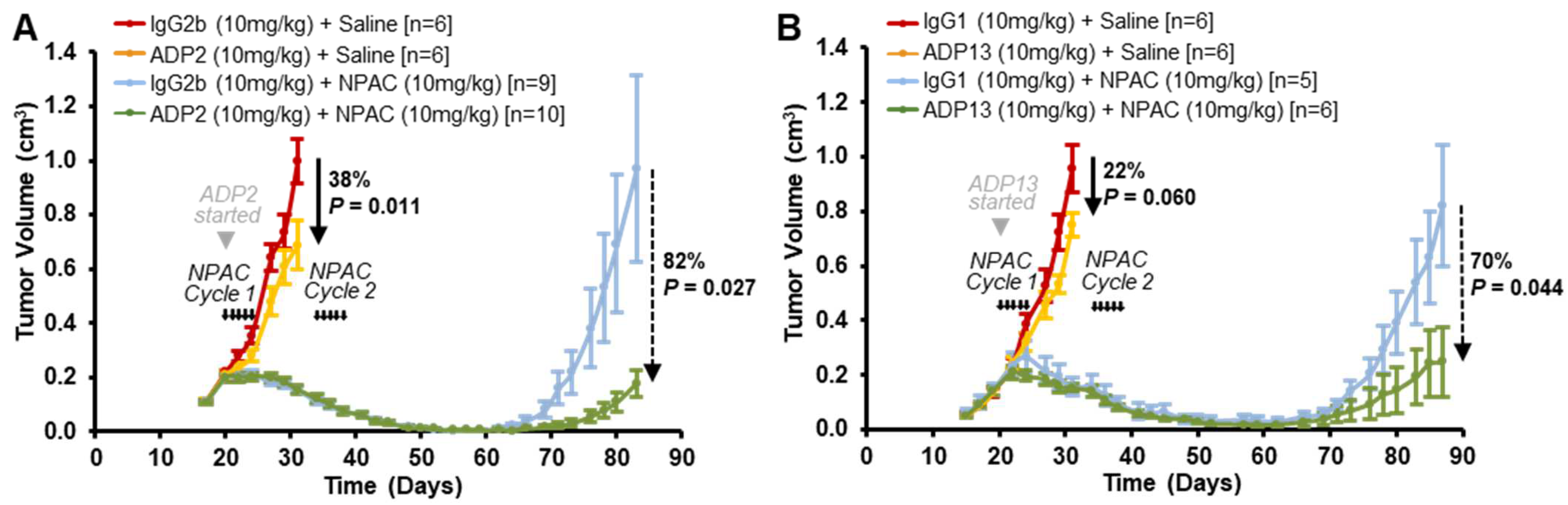
3.2. ADP2, ADP13, and to a Lesser Extent ADP3 Inhibit Primary TNBC Tumor Growth
3.3. Interactions of ADP2, ADP3, and ADP13 with ADAM8 Map to the DI
3.4. ADP2 and ADP13 Inhibit TNBC Tumor Regrowth
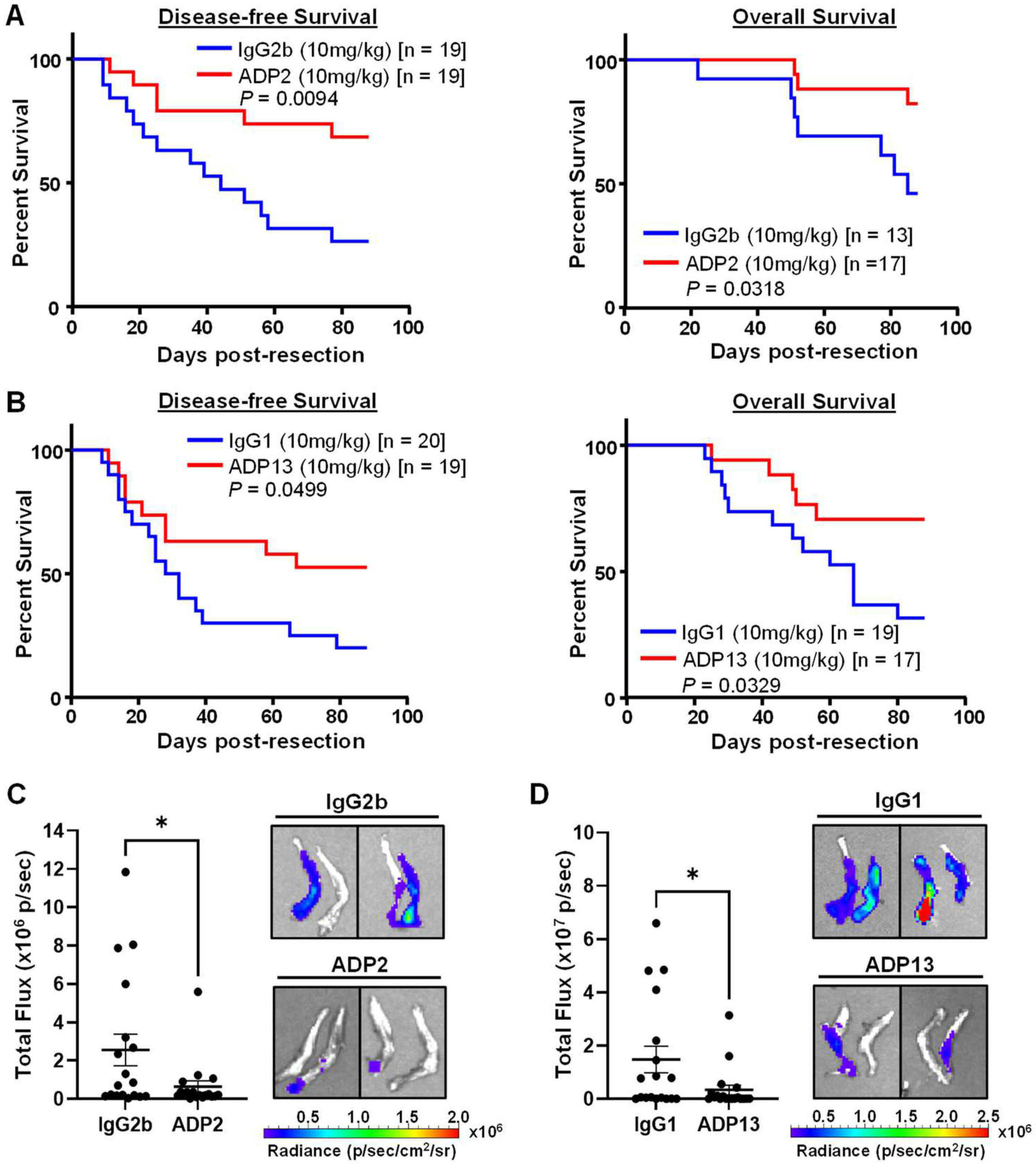
3.5. ADP2 or ADP13 Treatment Improves TNBC Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kerr, A.J.; Dodwell, D.; McGale, P.; Holt, F.; Duane, F.; Mannu, G.; Darby, S.C.; Taylor, C.W. Adjuvant and neoadjuvant breast cancer treatments: A systematic review of their effects on mortality. Cancer Treat. Rev. 2022, 105, 102375. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Niwinska, A.; Murawska, M.; Pienkowski, T. Analysis of pattern, time and risk factors influencing recurrence in triple-negative breast cancer patients. Med. Oncol. 2013, 30, 388. [Google Scholar] [CrossRef] [PubMed]
- Khadela, A.; Soni, S.; Shah, A.C.; Pandya, A.J.; Megha, K.; Kothari, N.; Cb, A. Unveiling the antibody-drug conjugates portfolio in battling Triple-negative breast cancer: Therapeutic trends and Future horizon. Med. Oncol. 2022, 40, 25. [Google Scholar] [CrossRef] [PubMed]
- Morganti, S.; Tolaney, S.M. Role of Immunotherapy in Early- and Late-Stage Triple-Negative Breast Cancer. Hematol. Oncol. Clin. N. Am. 2023, 37, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, M.; Mineva, N.D.; Polmear, M.; Conrad, C.; Srinivasan, S.; Loussouarn, D.; Barille-Nion, S.; Georgakoudi, I.; Dagg, A.; McDermott, E.W.; et al. ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis. EMBO Mol. Med. 2014, 6, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Pianetti, S.; Miller, K.D.; Chen, H.H.; Althouse, S.; Cao, S.; Michael, S.J.; Sonenshein, G.E.; Mineva, N.D. ADAM8 is expressed widely in breast cancer and predicts poor outcome in hormone receptor positive, HER-2 negative patients. Cancer Cell Int. 2023, 23, 165. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.; Polmear, M.; Mineva, N.D.; Romagnoli, M.; Sonenshein, G.E.; Georgakoudi, I. Endogenous light scattering as an optical signature of circulating tumor cell clusters. Biomed. Opt. Express 2016, 7, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, K.; Higuchi, Y.; Kataoka, M.; Matsuura, K.; Yamamoto, S. CD156 (human ADAM8): Expression, primary amino acid sequence, and gene location. Genomics 1997, 41, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Schlomann, U.; Wildeboer, D.; Webster, A.; Antropova, O.; Zeuschner, D.; Knight, C.G.; Docherty, A.J.; Lambert, M.; Skelton, L.; Jockusch, H.; et al. The metalloprotease disintegrin ADAM8. Processing by autocatalysis is required for proteolytic activity and cell adhesion. J. Biol. Chem. 2002, 277, 48210–48219. [Google Scholar] [CrossRef]
- Das, S.G.; Romagnoli, M.; Mineva, N.D.; Barille-Nion, S.; Jezequel, P.; Campone, M.; Sonenshein, G.E. miR-720 is a downstream target of an ADAM8-induced ERK signaling cascade that promotes the migratory and invasive phenotype of triple-negative breast cancer cells. Breast Cancer Res. 2016, 18, 40. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.; Shieh, H.S.; Day, J.E.; Caspers, N.; Chrencik, J.E.; Williams, J.M.; Pegg, L.E.; Pauley, A.M.; Moon, A.F.; Krahn, J.M.; et al. Structure of human ADAM-8 catalytic domain complexed with batimastat. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Schlomann, U.; Dorzweiler, K.; Nuti, E.; Tuccinardi, T.; Rossello, A.; Bartsch, J.W. Metalloprotease inhibitor profiles of human ADAM8 in vitro and in cell-based assays. Biol. Chem. 2019, 400, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Schlomann, U.; Koller, G.; Conrad, C.; Ferdous, T.; Golfi, P.; Garcia, A.M.; Hofling, S.; Parsons, M.; Costa, P.; Soper, R.; et al. ADAM8 as a drug target in pancreatic cancer. Nat. Commun. 2015, 6, 6175. [Google Scholar] [CrossRef] [PubMed]
- Yim, V.; Noisier, A.F.M.; Hung, K.Y.; Bartsch, J.W.; Schlomann, U.; Brimble, M.A. Synthesis and biological evaluation of analogues of the potent ADAM8 inhibitor cyclo(RLsKDK) for the treatment of inflammatory diseases and cancer metastasis. Bioorg. Med. Chem. 2016, 24, 4032–4037. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.; Lu, G.; Kajiya, H.; Garcia-Palacios, V.; Kurihara, N.; Anderson, J.; Patrene, K.; Sheppard, D.; Blair, H.C.; Windle, J.J.; et al. Alpha9beta1: A novel osteoclast integrin that regulates osteoclast formation and function. J. Bone Miner. Res. 2006, 21, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Pascal, B.D.; Willis, S.; Lauer, J.L.; Landgraf, R.R.; West, G.M.; Marciano, D.; Novick, S.; Goswami, D.; Chalmers, M.J.; Griffin, P.R. HDX workbench: Software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 2012, 23, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Davidson, E.; Doranz, B.J. A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology 2014, 143, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Volk, L.D.; Flister, M.J.; Bivens, C.M.; Stutzman, A.; Desai, N.; Trieu, V.; Ran, S. Nab-paclitaxel efficacy in the orthotopic model of human breast cancer is significantly enhanced by concurrent anti-vascular endothelial growth factor A therapy. Neoplasia 2008, 10, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Igarashi, T.; Mori, H.; Araki, S. Crystal structures of VAP1 reveal ADAMs’ MDC domain architecture and its unique C-shaped scaffold. EMBO J. 2006, 25, 2388–2396. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Krasnojon, D.; Cheporov, S.; Makhson, A.N.; Manikhas, G.M.; Clawson, A.; Bhar, P. Significantly longer progression-free survival with nab-paclitaxel compared with docetaxel as first-line therapy for metastatic breast cancer. J. Clin. Oncol. 2009, 27, 3611–3619. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O’Shaughnessy, J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J. Clin. Oncol. 2005, 23, 7794–7803. [Google Scholar] [CrossRef]
- Dorman, G.; Cseh, S.; Hajdu, I.; Barna, L.; Konya, D.; Kupai, K.; Kovacs, L.; Ferdinandy, P. Matrix metalloproteinase inhibitors: A critical appraisal of design principles and proposed therapeutic utility. Drugs 2010, 70, 949–964. [Google Scholar] [CrossRef]
- Marra, A.; Trapani, D.; Viale, G.; Criscitiello, C.; Curigliano, G. Practical classification of triple-negative breast cancer: Intratumoral heterogeneity, mechanisms of drug resistance, and novel therapies. NPJ Breast Cancer 2020, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 386, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Rehim, D.M.; Osman, N.A. Expression of a disintegrin and metalloprotease 8 and endostatin in human osteosarcoma: Implication in tumor progression and prognosis. J. Egypt. Natl. Cancer Inst. 2015, 27, 1–9. [Google Scholar] [CrossRef][Green Version]
- Huang, J.; Bai, Y.; Huo, L.; Xiao, J.; Fan, X.; Yang, Z.; Chen, H.; Yang, Z. Upregulation of a disintegrin and metalloprotease 8 is associated with progression and prognosis of patients with gastric cancer. Transl. Res. 2015, 166, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Daigo, Y.; Yasui, W.; Inai, K.; Nishimura, H.; Tsuchiya, E.; Kohno, N.; Nakamura, Y. ADAM8 as a novel serological and histochemical marker for lung cancer. Clin. Cancer Res. 2004, 10, 8363–8370. [Google Scholar] [CrossRef] [PubMed]
- Valkovskaya, N.; Kayed, H.; Felix, K.; Hartmann, D.; Giese, N.A.; Osinsky, S.P.; Friess, H.; Kleeff, J. ADAM8 expression is associated with increased invasiveness and reduced patient survival in pancreatic cancer. J. Cell. Mol. Med. 2007, 11, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Bai, Y.; Huo, L.; Chen, H.; Huang, J.; Li, J.; Fan, X.; Yang, Z.; Wang, L.; Wang, J. Expression of A disintegrin and metalloprotease 8 is associated with cell growth and poor survival in colorectal cancer. BMC Cancer 2014, 14, 568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tan, Y.F.; Jiang, C.; Zhang, K.; Zha, T.Z.; Zhang, M. High ADAM8 expression is associated with poor prognosis in patients with hepatocellular carcinoma. Pathol. Oncol. Res. 2013, 19, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, V.; Brunner, M.; Heiduschka, G.; Schneider, S.; Seemann, R.; Erovic, B.; Thurnher, D. ADAM8 in squamous cell carcinoma of the head and neck: A retrospective study. BMC Cancer 2012, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, J.W.; Wildeboer, D.; Koller, G.; Naus, S.; Rittger, A.; Moss, M.L.; Minai, Y.; Jockusch, H. Tumor necrosis factor-alpha (TNF-alpha) regulates shedding of TNF-alpha receptor 1 by the metalloprotease-disintegrin ADAM8: Evidence for a protease-regulated feedback loop in neuroprotection. J. Neurosci. 2010, 30, 12210–12218. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.; Hutchinson, G.; Nebenius-Oosthuizen, D.; Smith, A.J.; Bartsch, J.W.; Horiuchi, K.; Rittger, A.; Manova, K.; Docherty, A.J.; Blobel, C.P. Metalloprotease-disintegrin ADAM8: Expression analysis and targeted deletion in mice. Dev. Dyn. 2005, 232, 221–231. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding to Native ADAM8 in FACS (MFI) | ||||||
|---|---|---|---|---|---|---|
| Cell Line | HEK-A8 | HEK-EV | ||||
| Ab (µg/mL) | 10 | 1 | 0.1 | 10 | 1 | 0.1 |
| ADP1 | 185.7 | 171.2 | 46.8 | 3.1 | 3.1 | 3.1 |
| ADP2 | 158.1 | 156.5 | 58.4 | 2.6 | 2.7 | 2.9 |
| ADP3 | 86.9 | 96.5 | 51.8 | 2.8 | 2.7 | 2.7 |
| ADP4 | 160.8 | 140.0 | 47.4 | 2.6 | 2.7 | 2.7 |
| ADP5 | 149.0 | 105.9 | 26.8 | 2.7 | 2.8 | 3.1 |
| ADP6 | 62.4 | 65.4 | 28.2 | 2.7 | 2.7 | 2.9 |
| ADP7 | 68.1 | 63.1 | 25.8 | 2.7 | 2.9 | 2.8 |
| ADP8 | 48.6 | 53.1 | 17.0 | 2.6 | 2.6 | 2.6 |
| ADP9 | 62.1 | 61.1 | 22.1 | 3.2 | 2.9 | 2.7 |
| ADP10 | 67.2 | 58.2 | 18.7 | 3.2 | 2.8 | 2.7 |
| ADP11 | 56.6 | 64.2 | 24.1 | 3.1 | 2.7 | 2.6 |
| ADP12 | 106.7 | 68.7 | 17.4 | 3.1 | 3.1 | 3.0 |
| ADP13 | 62.2 | 21.1 | 7.1 | 2.6 | 2.7 | 2.6 |
| ADP14 | 58.6 | 17.9 | 5.9 | 3.3 | 3.2 | 3.0 |
| ADP16 | 147.3 | 89.6 | 24.6 | 2.6 | 2.9 | 2.9 |
| ADP17 | 53.6 | 63.8 | 25.5 | 2.8 | 2.7 | 2.8 |
| ADP18 | 53.7 | 61.0 | 23.5 | 2.8 | 2.8 | 2.7 |
| ADP19 | 59.0 | 52.9 | 17.5 | 2.6 | 2.6 | 3.0 |
| Binding to rHuADAM8 | ||||
|---|---|---|---|---|
| ELISA | Biacore | |||
| Ab | EC50 (nM) | ka (1/Ms) | kd (1/s) | KD (M) |
| ADP1 | 0.032 | 7.04 × 104 | 3.15 × 10−4 | 4.47 × 10−9 |
| ADP2 | 0.029 | 8.95 × 104 | 2.99 × 10−4 | 3.34 × 10−9 |
| ADP3 | 0.049 | 3.47 × 104 | 6.34 × 10−4 | 1.83 × 10−8 |
| ADP4 | 0.041 | 3.52 × 104 | 2.55 × 10−2 | 7.23 × 10−8 |
| ADP5 | 0.059 | 9.51 × 103 | 9.27 × 10−5 | 9.75 × 10−9 |
| ADP6 | 0.059 | 2.62 × 105 | 4.07 × 10−3 | 1.55 × 10−8 |
| ADP7 | 0.091 | 1.68 × 104 | 8.89 × 10−4 | 5.29 × 10−8 |
| ADP8 | 0.097 | 3.14 × 103 | 1.48 × 10−4 | 4.72 × 10−8 |
| ADP9 | 0.053 | 2.78 × 104 | 1.37 × 10−3 | 4.92 × 10−8 |
| ADP10 | 0.091 | 2.45 × 103 | 7.52 × 10−5 | 3.07 × 10−8 |
| ADP11 | 0.097 | 5.07 × 103 | 1.09 × 10−4 | 2.14 × 10−8 |
| ADP12 | 0.061 | 4.97 × 104 | 4.14 × 10−4 | 8.33 × 10−9 |
| ADP13 | 0.054 | 3.54 × 104 | 4.60 × 10−5 | 1.30 × 10−9 |
| ADP14 | 0.531 | 2.32 × 104 | 6.12 × 10−4 | 2.64 × 10−8 |
| ADP16 | 0.053 | 7.27 × 104 | 5.12 × 10−3 | 7.05 × 10−8 |
| ADP17 | 0.049 | 6.78 × 103 | 1.26 × 10−4 | 1.86 × 10−8 |
| ADP18 | 0.024 | 8.05 × 103 | 1.23 × 10−4 | 1.53 × 10−8 |
| ADP19 | 0.085 | 1.60 × 104 | 1.45 × 10−4 | 9.11 × 10−9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mineva, N.D.; Pianetti, S.; Das, S.G.; Srinivasan, S.; Billiald, N.M.; Sonenshein, G.E. A Novel Class of Human ADAM8 Inhibitory Antibodies for Treatment of Triple-Negative Breast Cancer. Pharmaceutics 2024, 16, 536. https://doi.org/10.3390/pharmaceutics16040536
Mineva ND, Pianetti S, Das SG, Srinivasan S, Billiald NM, Sonenshein GE. A Novel Class of Human ADAM8 Inhibitory Antibodies for Treatment of Triple-Negative Breast Cancer. Pharmaceutics. 2024; 16(4):536. https://doi.org/10.3390/pharmaceutics16040536
Chicago/Turabian StyleMineva, Nora D., Stefania Pianetti, Sonia G. Das, Srimathi Srinivasan, Nicolas M. Billiald, and Gail E. Sonenshein. 2024. "A Novel Class of Human ADAM8 Inhibitory Antibodies for Treatment of Triple-Negative Breast Cancer" Pharmaceutics 16, no. 4: 536. https://doi.org/10.3390/pharmaceutics16040536
APA StyleMineva, N. D., Pianetti, S., Das, S. G., Srinivasan, S., Billiald, N. M., & Sonenshein, G. E. (2024). A Novel Class of Human ADAM8 Inhibitory Antibodies for Treatment of Triple-Negative Breast Cancer. Pharmaceutics, 16(4), 536. https://doi.org/10.3390/pharmaceutics16040536