
Cryo-Milled β-Glucan Nanoparticles for Oral Drug Delivery

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Characterization
3.1. Optimization of Polymeric Films and Freezer Mill Process Settings
3.2. Particle Size, Zeta Potential, Surface Morphology, and Entrapment Efficiency
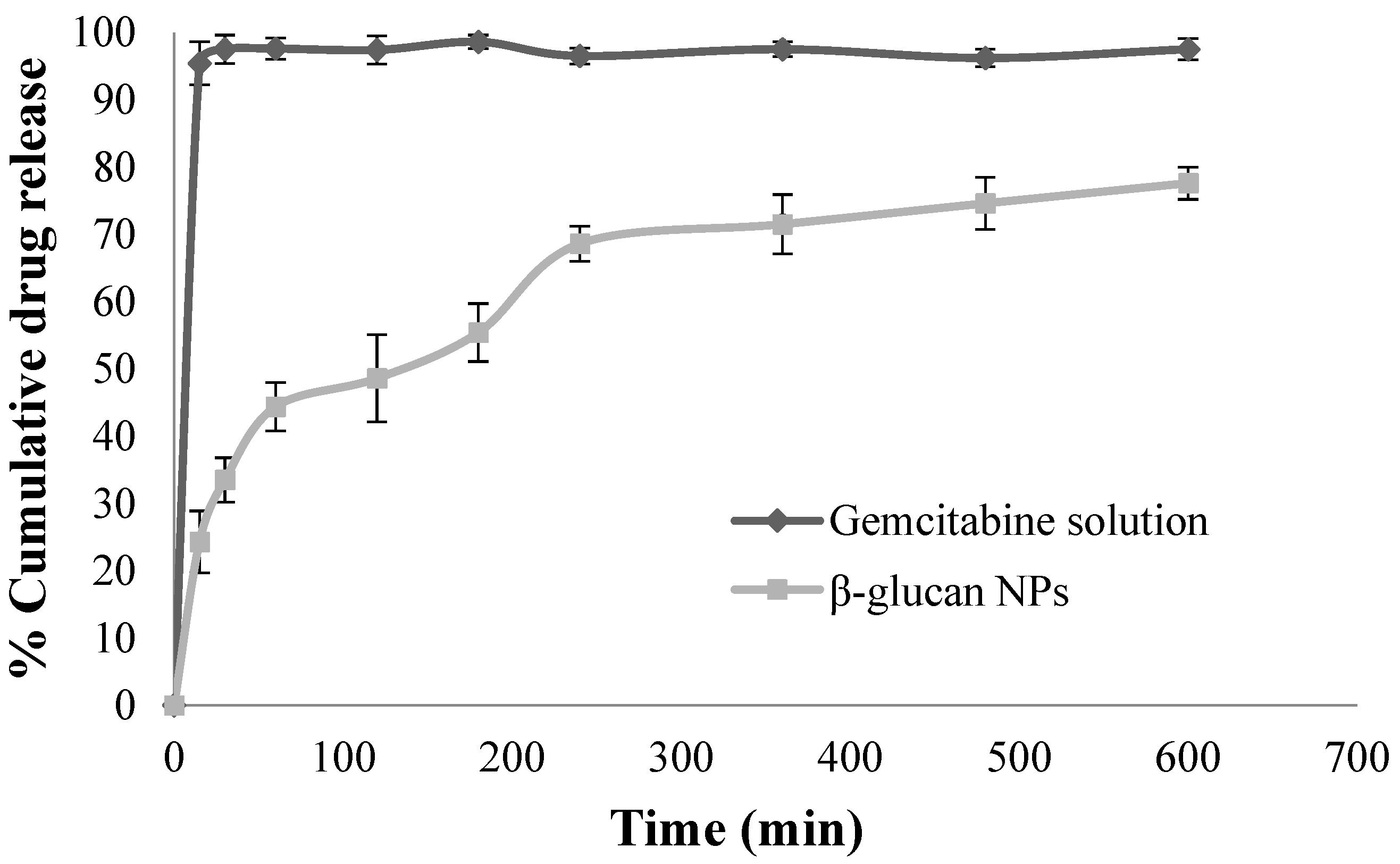
3.3. In Vitro Drug Release Studies
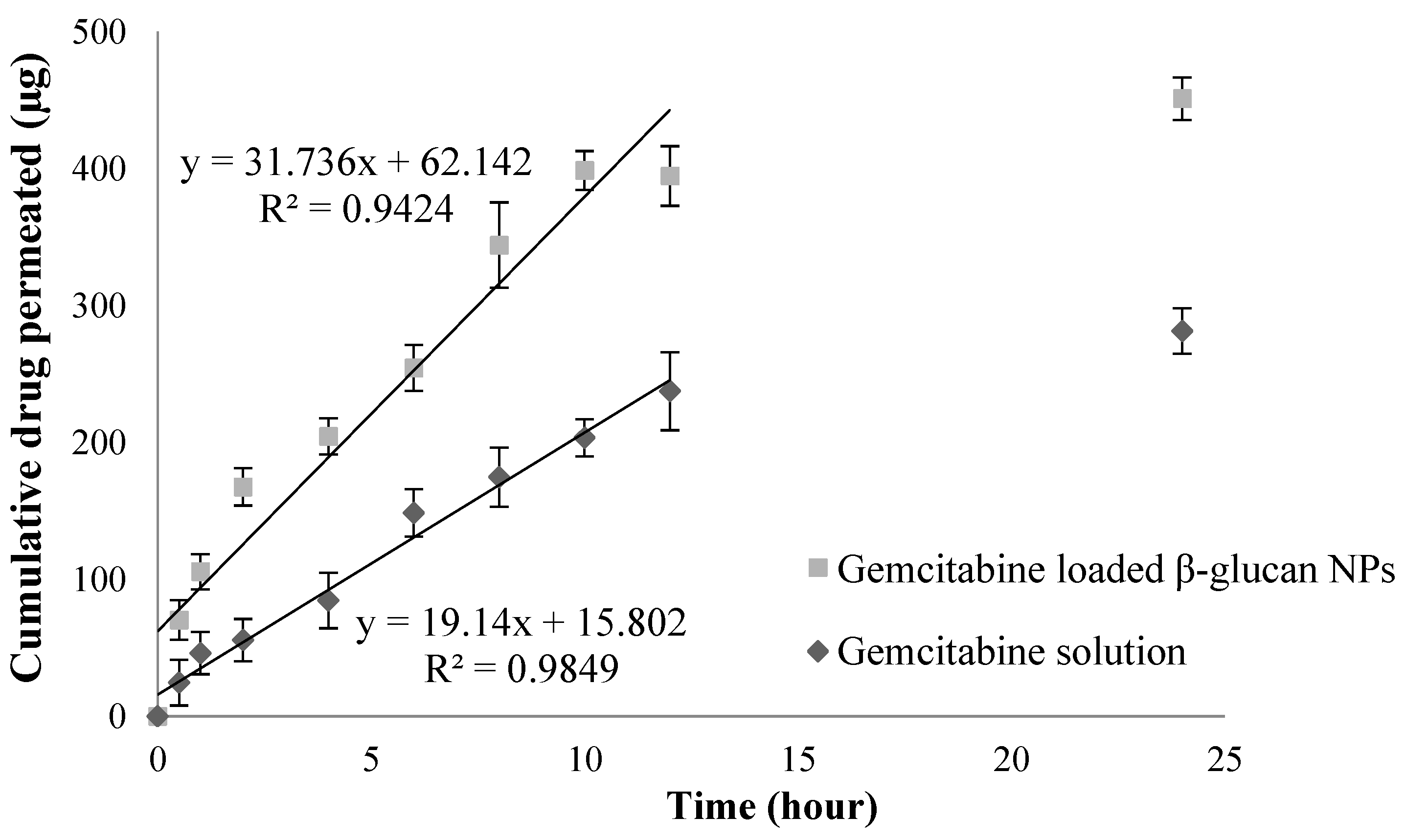
3.4. Ex Vivo Permeation Studies
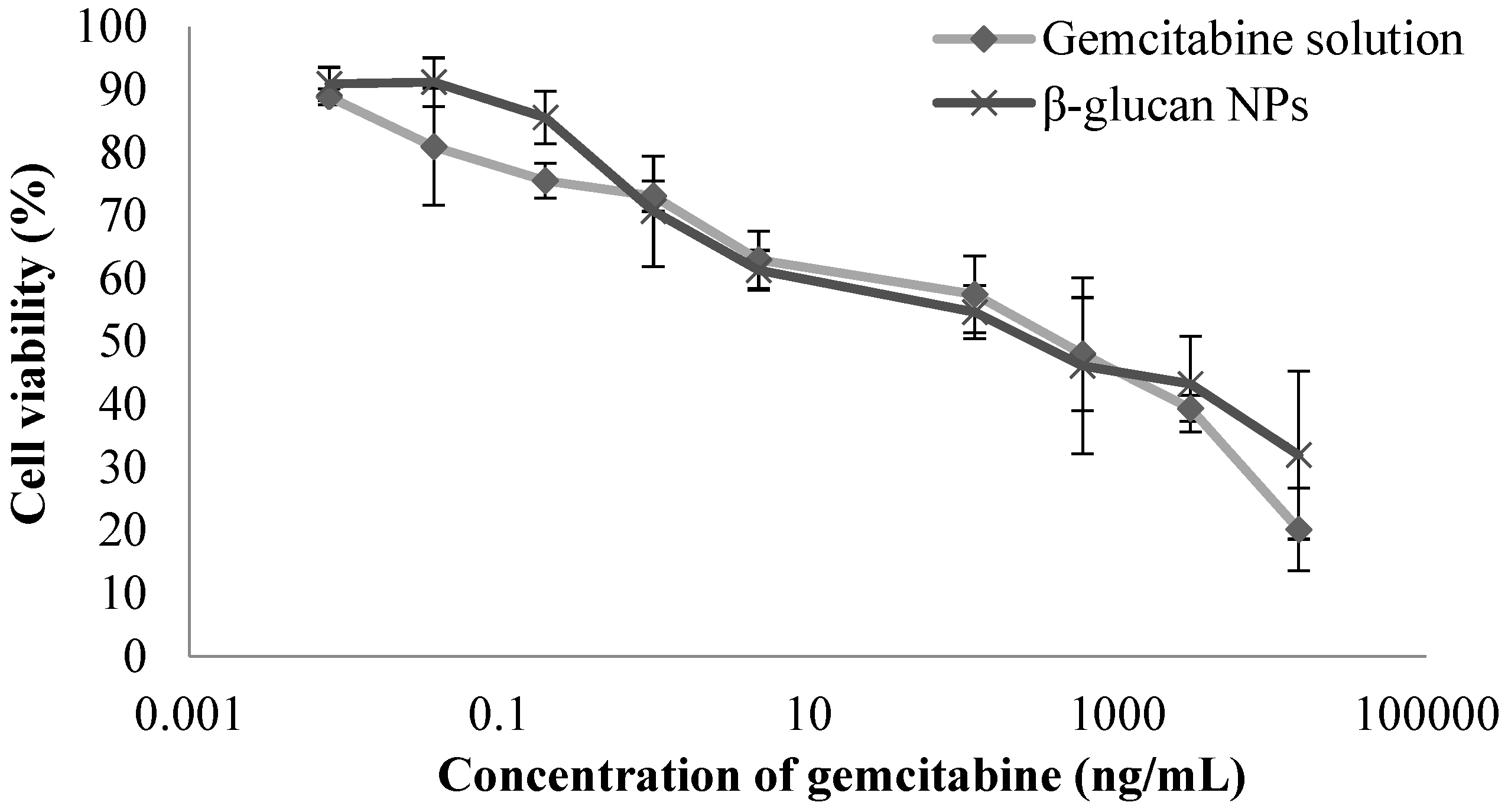
3.5. Cell Culture and Cytotoxicity Studies
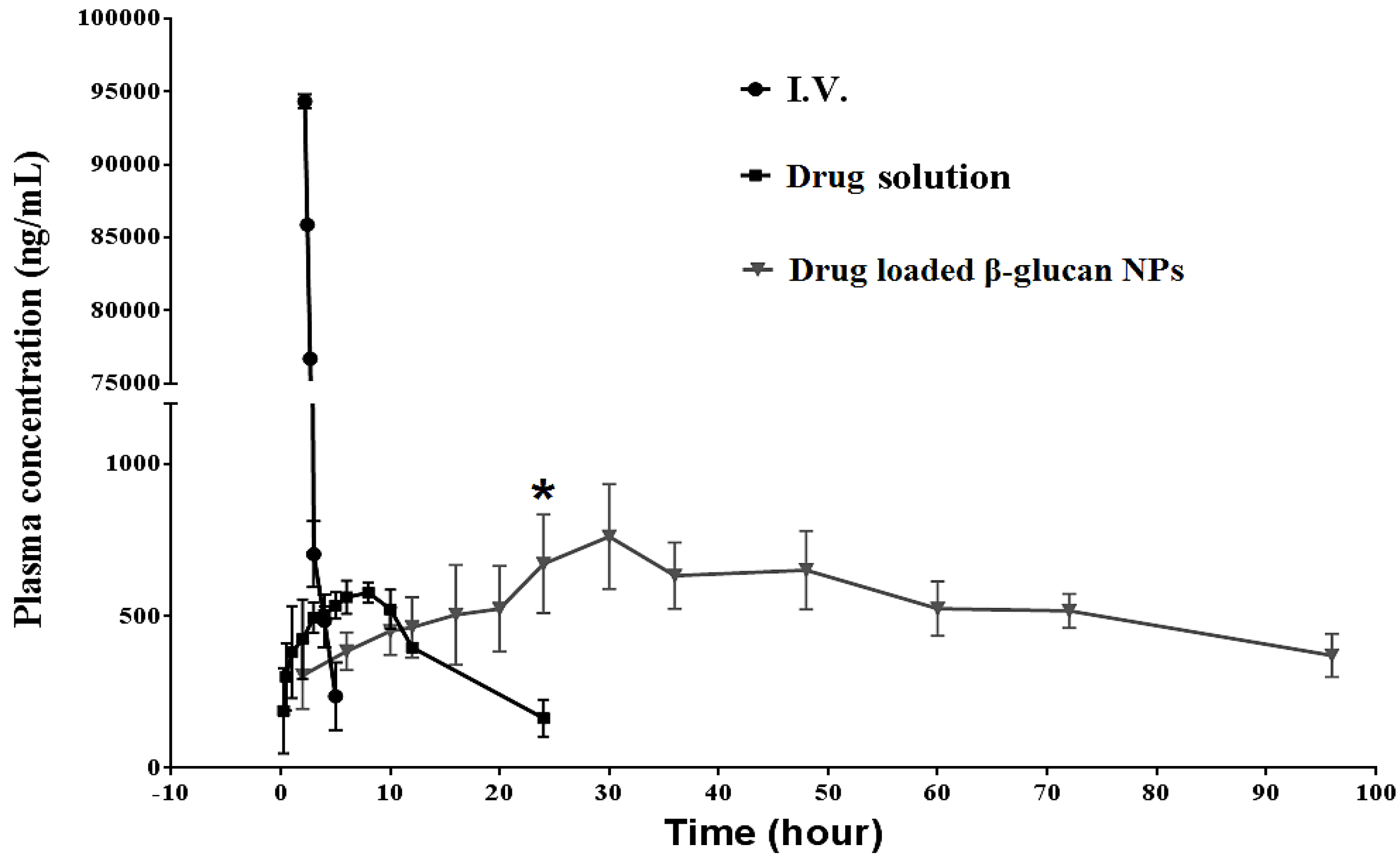
3.6. In Vivo Pharmacokinetic Studies Using Sprague Dawley Rats
3.7. In Vivo Acute Toxicity Studies
3.8. In Vivo Pharmacodynamic Studies Using BALB/c Nude Mice
3.9. Statistical Analysis
4. Results and Discussion
4.1. Optimization of Polymeric Films, and Freezer Mill Settings
4.2. Particle Size, Zeta Potential, Surface Morphology, and Entrapment Efficiency
4.3. In Vitro Drug Release Studies
4.4. Ex Vivo Permeation Studies
4.5. Cell Culture and Cytotoxicity Studies
4.6. In Vivo Pharmacokinetic Studies Using Sprague Dawley Rats
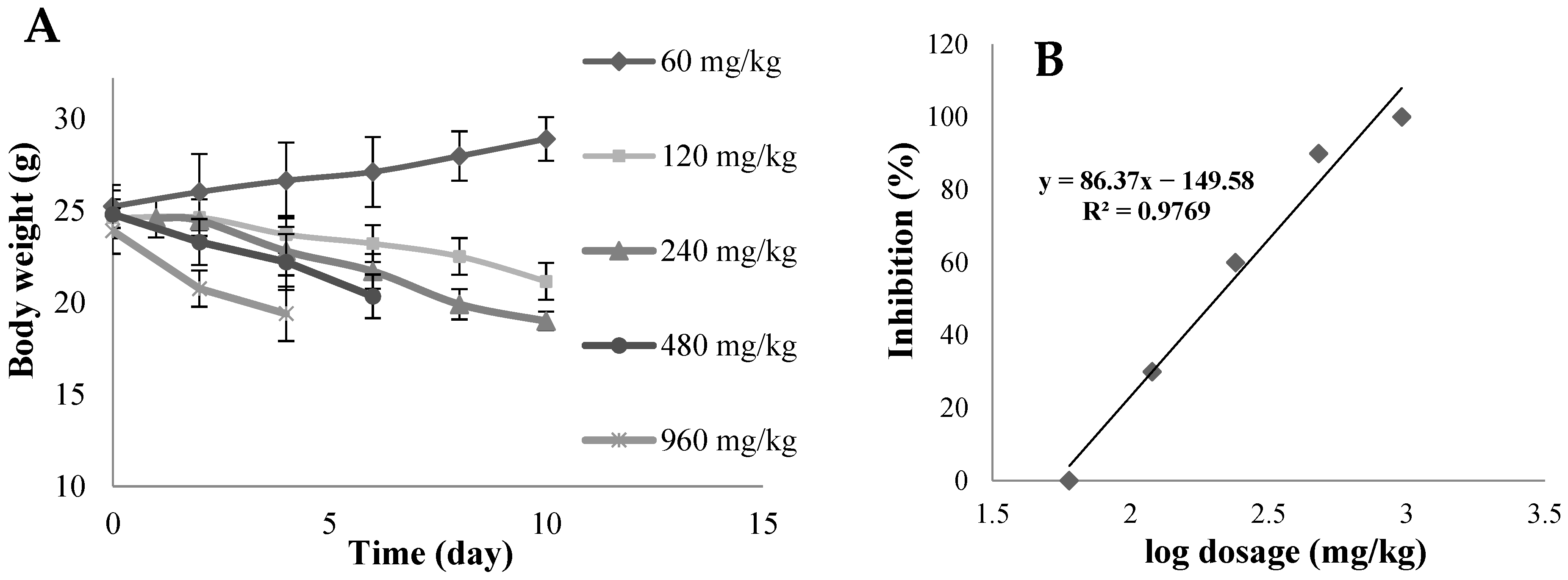
4.7. In Vivo Acute Toxicity Studies
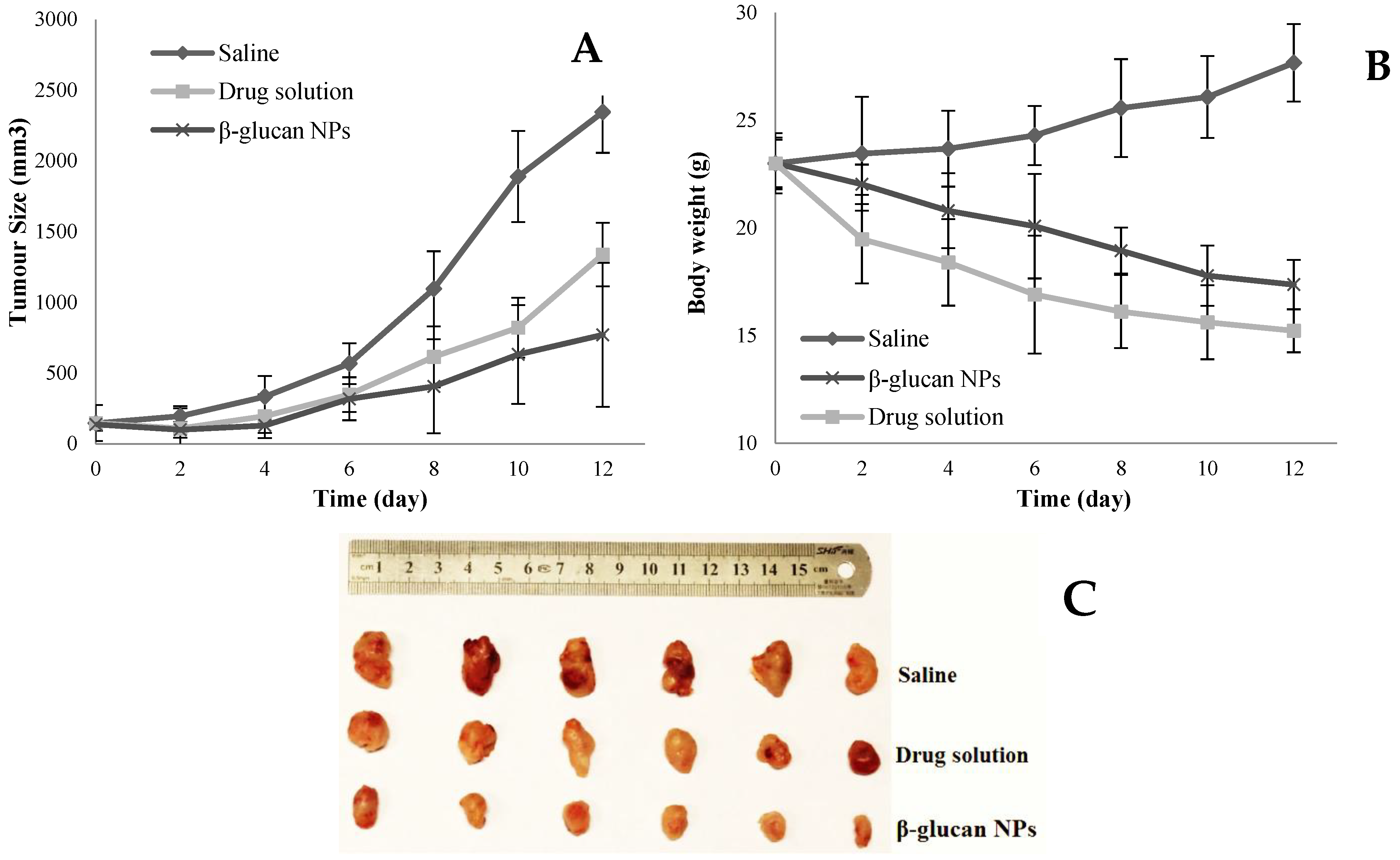
4.8. In Vivo Pharmacodynamic Studies Using BALB/c Nude Mice
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mei, L.; Zhang, Z.; Zhao, L.; Huang, L.; Yang, X.-L.; Tang, J.; Feng, S.-S. Pharmaceutical nanotechnology for oral delivery of anticancer drugs. Adv. Drug Deliv. Rev. 2013, 65, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Kang, W.; Li, W.; Chen, S.; Gao, Y. Oral delivery of protein and peptide drugs: From non-specific formulation approaches to intestinal cell targeting strategies. Theranostics 2022, 12, 1419. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.; Cho, Y.W.; Park, K. Nanoparticles for oral delivery: Targeted nanoparticles with peptidic ligands for oral protein delivery. Adv. Drug Deliv. Rev. 2013, 65, 822–832. [Google Scholar] [CrossRef]
- Al, G.M.; Liu, M.; Wen, J. Ligands for oral delivery of peptides across the blood-brain-barrier. Acta Mater. Medica 2022, 1, 106–123. [Google Scholar]
- Win, K.Y.; Feng, S.-S. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials 2005, 26, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Rodriguez, S.A.; Allemann, E.; Fessi, H.; Doelker, E. Polymeric nanoparticles for oral delivery of drugs and vaccines: A critical evaluation of in vivo studies. Crit. Rev. Ther. Drug Carr. Syst. 2005, 22, 419–464. [Google Scholar] [CrossRef]
- Spleis, H.; Sandmeier, M.; Claus, V.; Schnürch, A.B. Surface design of nanocarriers: Key to more efficient oral drug delivery systems. Adv. Colloid Interface Sci. 2023, 313, 102848. [Google Scholar] [CrossRef]
- Song, N.; Liu, W.; Tu, Q.; Liu, R.; Zhang, Y.; Wang, J. Preparation and in vitro properties of redox-responsive polymeric nanoparticles for paclitaxel delivery. Colloids Surf. B Biointerfaces 2011, 87, 454–463. [Google Scholar] [CrossRef]
- Pradhan, R.; Poudel, B.K.; Ramasamy, T.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Docetaxel-loaded polylactic acid-co-glycolic acid nanoparticles: Formulation, physicochemical characterization and cytotoxicity studies. J. Nanosci. Nanotechnol. 2013, 13, 5948–5956. [Google Scholar] [CrossRef]
- Bensaid, F.; Boullay, O.T.D.; Amgoune, A.; Pradel, C.; Reddy, L.H.; Didier, E.; Sablé, S.; Louit, G.; Bazile, D.; Bourissou, D. Y-Shaped mPEG-PLA Cabazitaxel Conjugates: Well-Controlled Synthesis by Organocatalytic Approach and Self-Assembly into Interface Drug-Loaded Core–Corona Nanoparticles. Biomacromolecules 2013, 14, 1189–1198. [Google Scholar] [CrossRef]
- Ajithkumar, A.; Andersson, R.; Siika-aho, M.; Tenkanen, M.; Åman, P. Isolation of cellotriosyl blocks from barley β-glucan with endo-1, 4-β-glucanase from Trichoderma reesei. Carbohydr. Polym. 2006, 64, 233–238. [Google Scholar] [CrossRef]
- Wu, Y.; Li, P.Y.; Jiang, Z.Z.; Sun, X.L.; He, H.Q.; Yan, P.J.; Xu, Y.; Liu, Y. Bioinspired yeast-based β-glucan system for oral drug delivery. Carbohydr. Polym. 2023, 319, 121163. [Google Scholar] [CrossRef] [PubMed]
- Min, C.J.; Jeong, D.; Piao, J.; Kim, K.; Nguyen, A.B.L.; Kwon, N.J.; Lee, M.K.; Yu, J.H.; Jung, S. Hydroxypropyl cyclic β-(1→2)-d-glucans and epichlorohydrin β-cyclodextrin dimers as effective carbohydrate-solubilizers for polycyclic aromatic hydrocarbons. Carbohydr. Res. 2015, 401, 82–88. [Google Scholar]
- Li, S.; Li, W.; Yang, X.; Gao, Y.; Chen, G. Dietary-polysaccharide-modified fish-oil-based double emulsion as a functional colloidal formulation for oral drug delivery. Pharmaceutics 2022, 14, 2844. [Google Scholar] [CrossRef] [PubMed]
- Huong, L.M.; Thu, H.P.; Thuy, N.T.B.; Ha, T.T.H.; Thi, H.T.M.; Trang, M.T.; Hang, T.T.N.; Nghi, D.H.; Phuc, N.X.; Quang, D.T. Preparation and Antitumor-promoting Activity of Curcumin Encapsulated by 1,3-β-Glucan Isolated from Vietnam Medicinal Mushroom Hericium erinaceum. Chem. Lett. 2011, 40, 846–848. [Google Scholar] [CrossRef]
- Qi, S.; Weuts, I.; De Cort, S.; Stokbroekx, S.; Leemans, R.; Reading, M.; Belton, P.; Craig, D.Q.M. An investigation into the crystallisation behaviour of an amorphous cryomilled pharmaceutical material above and below the glass transition temperature. J. Pharm. Sci. 2010, 99, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Chieng, N.; Zujovic, Z.; Bowmaker, G.; Rades, T.; Saville, D. Effect of milling conditions on the solid-state conversion of ranitidine hydrochloride form 1. Int. J. Pharm. 2006, 327, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Pérez, C.R.; González, M.D.A.; Sarria, F.R.; López, M.C.H.; Gallego, J.M.C. Investigating the room-and cryo-milling impact in lignocellulosic biomass and its consequence over pyrolysis and oxidative treatments. J. Clean. Prod. 2024, 437, 140761. [Google Scholar] [CrossRef]
- Derakhshandeh, K.; Fathi, S. Role of chitosan nanoparticles in the oral absorption of Gemcitabine. Int. J. Pharm. 2012, 437, 172–177. [Google Scholar] [CrossRef]
- Vandana, M.; Sahoo, S.K. Long circulation and cytotoxicity of PEGylated gemcitabine and its potential for the treatment of pancreatic cancer. Biomaterials 2010, 31, 9340–9356. [Google Scholar] [CrossRef]
- Trickler, W.J.; Khurana, J.; Nagvekar, A.A.; Dash, A.K. Chitosan and glyceryl monooleate nanostructures containing gemcitabine: Potential delivery system for pancreatic cancer treatment. AAPS Pharm. Sci. Technol. 2010, 11, 392–401. [Google Scholar] [CrossRef]
- Zhou, L.J.; Du, K.X.; Dai, Y.H.; Zeng, Y.M.; Luo, Y.B.; Ren, M.D.; Pan, W.B.; Liu, Y.; Zhang, L.; Zhu, R.; et al. Metabolic reprogramming based on RNA sequencing of gemcitabine-resistant cells reveals the FASN gene as a therapeutic for bladder cancer. J. Transl. Med. 2024, 22, 55. [Google Scholar] [CrossRef]
- Semalty, M.; Semalty, A.; Kumar, G. Formulation and characterization of mucoadhesive buccal films of glipizide. Indian J. Pharm. Sci. 2008, 70, 43. [Google Scholar] [CrossRef]
- Chen, G.; Svirskis, D.; Lu, W.; Ying, M.; Huang, Y.; Wen, J. N-trimethyl chitosan nanoparticles and CSKSSDYQC peptide: N-trimethyl chitosan conjugates enhance the oral bioavailability of gemcitabine to treat breast cancer. J. Control. Release 2018, 10, 142–153. [Google Scholar] [CrossRef]
- Nabil, W.N.N.; Xi, Z.; Liu, M.; Li, Y.; Yao, M.; Liu, T.; Dong, Q.; Xu, H. Advances in therapeutic agents targeting quiescent cancer cells. Acta Mater. Medica 2022, 8, 56–71. [Google Scholar] [CrossRef]
- Youdim, K.A.; Avdeef, A.; Abbott, N.J. In vitro trans-monolayer permeability calculations: Often forgotten assumptions. Drug Discov. Today 2003, 8, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Svirskis, D.; Lu, W.; Ying, M.; Li, H.; Liu, M.; Wen, J. N-trimethyl chitosan coated nano-complexes enhance the oral bioavailability and chemotherapeutic effects of gemcitabine. Carbohydr. Polym. 2021, 273, 118592. [Google Scholar] [CrossRef]
- Fotakis, G.; Timbrell, J.A. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef]
- Hussain, A.; Sasidharan, S.; Ahmed, T.; Ahmed, M.; Sharma, C. Clove (Syzygium aromaticum) extract potentiates gemcitabine cytotoxic effect on human cervical cancer cell line. Int. J. Cancer Res. 2009, 5, 95–104. [Google Scholar] [CrossRef][Green Version]
- Wolz, E.; Pfannkuch, F.; Straub, J.; van Huygevoort, A. Assessment of Acute Oral Toxicity with Ro 26-7624/000 (EGCG) in the Rat (Acute Toxic Class Method); Report; Roche Vitamins Ltd.: Basel, Switzerland, 2001. [Google Scholar]
- Rehman, U.; Shoaib, M.; Raza, Z.A.; Rehan, Z.A.; Bakhtiyar, M.J.; Sharif, F.; Yousaf, M. Citric acid crosslinked biocompatible silk fibroin-mediated porous chitosan films for sustained drug release application. Mater. Today Commun. 2023, 34, 105373. [Google Scholar] [CrossRef]
- Kowalczuk, J.; Tritt-Goc, J.; Piślewski, N. The swelling properties of hydroxypropyl methyl cellulose loaded with tetracycline hydrochloride: Magnetic resonance imaging study. Solid State Nucl. Magn. Reson. 2004, 25, 35–41. [Google Scholar] [CrossRef]
- Bourtoom, T. Plasticizer effect on the properties of biodegradable blend film from rice starch-chitosan. Sonklanakarin J. Sci. Technol. 2008, 30, 149. [Google Scholar]
- Dhanapal, A.; Sasikala, P.; Rajamani, L.; Kavitha, V.; Yazhini, G.; Banu, M.S. Edible films from polysaccharides. Food Sci. Qual. Manag. 2012, 3, 9–18. [Google Scholar]
- Gaisford, S.; Dennison, M.; Tawfik, M.; Jones, M.D. Following mechanical activation of salbutamol sulphate during ball-milling with isothermal calorimetry. Int. J. Pharm. 2010, 393, 75–79. [Google Scholar] [CrossRef]
- Yang, X.; Li, W.Q.; Li, S.Z.; Chen, S.M.; Hu, Z.; He, Z.Y.; Zhu, X.Q.; Niu, X.S.; Zhou, X.M.; Li, H.H.; et al. Fish oil-based microemulsion can efficiently deliver oral peptide blocking PD-1/PD-L1 and simultaneously induce ferroptosis for cancer immunotherapy. J. Control. Release 2024, 365, 654–667. [Google Scholar] [CrossRef]
- Oliveira, M.B.; Mano, J.F. Polymer-based microparticles in tissue engineering and regenerative medicine. Biotechnol. Prog. 2011, 27, 897–912. [Google Scholar] [CrossRef] [PubMed]
- Lotina, A.S.; Portela, R.; Baeza, P.; Rodriguez, V.A.; Villarroel, M.; Ávila, P. Zeta potential as a tool for functional materials development. Catal. Today 2023, 423, 113862. [Google Scholar] [CrossRef]
- Zhao, W.; Li, M.; Zhang, Z.; Peng, H.-X. EMI shielding effectiveness of silver nanoparticle-decorated multi-walled carbon nanotube sheets. Int. J. Smart Nano Mater. 2010, 1, 249–260. [Google Scholar] [CrossRef]
- Lash, L.H. Mitochondrial glutathione in toxicology and disease of the kidneys. Toxicol. Res. 2012, 1, 39–46. [Google Scholar] [CrossRef]
- Arias, J.L.; López-Viota, M.; López-Viota, J.; Delgado, Á.V. Development of iron/ethylcellulose (core/shell) nanoparticles loaded with diclofenac sodium for arthritis treatment. Int. J. Pharm. 2009, 382, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Pragnesh, D.; Pradip, N.; Macwan, M.; Kamaliya, B. Biodegradable Gg-cl-poly (NIPAm-co-AA)/-o-MWCNT based hydrogel for combined drug delivery system of metformin and sodium diclofenac: In vitro studies. RSC Adv. 2023, 13, 22875–22885. [Google Scholar]
- Esquivel, S.V.; Himanshu, N.; Bhatt, N.; Diwan, R.; Habib, A.; Lee, W.Y.; Khatun, Z.; Nurunnabi, M. β-Glucan and Fatty Acid Based Mucoadhesive Carrier for Gastrointestinal Tract Specific Local and Sustained Drug Delivery. Biomolecules 2023, 13, 768. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, J.V.; Nugraha, C.; Ng, X.W.; Venkatraman, S. Sustained-release from nanocarriers: A review. J. Control. Release 2014, 193, 122–138. [Google Scholar] [CrossRef]
- Rezvanjou, S.N.; Niavand, M.R.; Shayesteh, O.H.; Yeganeh, E.M.; Moghadam, D.A.; Derakhshandeh, K.; Mahjub, R. Preparation and characterisation of self-emulsifying drug delivery system (SEDDS) for enhancing oral bioavailability of metformin hydrochloride using hydrophobic ion pairing complexation. J. Microencapsul. 2023, 40, 53–66. [Google Scholar] [CrossRef]
- Veltkamp, S.A.; Pluim, D.; van Tellingen, O.; Beijnen, J.H.; Schellens, J.H.M. Extensive metabolism and hepatic accumulation of gemcitabine after multiple oral and intravenous administration in mice. Drug Metab. Dispos. 2008, 36, 1606–1615. [Google Scholar] [CrossRef]
- Joshi, G.; Kumar, A.; Sawant, K. Enhanced bioavailability and intestinal uptake of Gemcitabine HCl loaded PLGA nanoparticles after oral delivery. Eur. J. Pharm. Sci. 2014, 60, 80–89. [Google Scholar] [CrossRef]
- Hao, W.-H.; Wang, J.-J.; Hsueh, S.-P.; Hsu, P.-J.; Chang, L.-C.; Hsu, C.-S.; Hsu, K.-Y. In vitro and in vivo studies of pharmacokinetics and antitumor efficacy of D07001-F4, an oral gemcitabine formulation. Cancer Chemother. Pharmacol. 2013, 71, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Ejeh, S.A.; Abu, H.A.; Onyeyili, P.A.; Abenga, J.N.; Ogbe, R.J.; Abalaka, S.E. Acute and sub-acute toxicological evaluation of ethanol extract of Alchornea cordifolia leaves in Wistar rats. Sci. Afr. 2023, 19, e01575. [Google Scholar] [CrossRef]
- Sonaje, K.; Lin, Y.-H.; Juang, J.-H.; Wey, S.-P.; Chen, C.-T.; Sung, H.-W. In vivo evaluation of safety and efficacy of self-assembled nanoparticles for oral insulin delivery. Biomaterials 2009, 30, 2329–2339. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle Size (d.nm) | PdI | Zeta Potential (mV) | Entrapment Efficiency (%) | |
|---|---|---|---|---|
| MC NPs | 555.0 ± 32.2 | 0.24 ± 0.13 | −0.6 ± 0.7 | 63.9 ± 1.8 |
| β-Glucan NPs | 447.6 ± 14.2 | 0.18 ± 0.07 | −1.2 ± 0.5 | 64.3 ± 2.1 |
| EC NPs | 472.6 ± 21.7 | 0.32 ± 0.11 | −20.1 ± 1.2 | 56.3 ± 1.8 |
| Chitosan NPs | 643 ± 52.4 | 0.13 ± 0.11 | +6.9 ± 1.4 | 60.2 ± 1.3 |
| Zero Order | First Order | Higuchi Model | Korsmeyer–Peppas Model | ||||||
|---|---|---|---|---|---|---|---|---|---|
| β-Glucan NPs | r2 | k0 | r2 | k1 | r2 | kh | r2 | n | kk |
| 0.73 | 28.45 | 0.88 | 1.86 | 0.94 | 20.75 | 0.96 | 0.52 | 1.04 | |
| PK Parameters | Gemcitabine (i.v.) | Gemcitabine Solution (Oral) | β-Glucan NPs (Oral) |
|---|---|---|---|
| Cmax (ng/mL) | 94,152 ± 3435 | 577.11 ± 98.23 | 761.04 ± 214.32 |
| Tmax (h) | 0.25 | 8 | 30 |
| AUC0-inf (ng·h/mL) | 93,050 ± 1459 | 9176 ± 785 | 46,457 ± 2124 |
| T1/2 (h) | 1.18 ± 0.85 | 9.40 ± 2.13 | 69.98 ± 20.50 |
| Oral bioavailability | 100% | 9.86% | 49.92% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, G.; Liu, Y.; Svirskis, D.; Li, H.; Ying, M.; Lu, W.; Wen, J. Cryo-Milled β-Glucan Nanoparticles for Oral Drug Delivery. Pharmaceutics 2024, 16, 546. https://doi.org/10.3390/pharmaceutics16040546
Chen G, Liu Y, Svirskis D, Li H, Ying M, Lu W, Wen J. Cryo-Milled β-Glucan Nanoparticles for Oral Drug Delivery. Pharmaceutics. 2024; 16(4):546. https://doi.org/10.3390/pharmaceutics16040546
Chicago/Turabian StyleChen, Guanyu, Yi Liu, Darren Svirskis, Hongyu Li, Man Ying, Weiyue Lu, and Jingyuan Wen. 2024. "Cryo-Milled β-Glucan Nanoparticles for Oral Drug Delivery" Pharmaceutics 16, no. 4: 546. https://doi.org/10.3390/pharmaceutics16040546
APA StyleChen, G., Liu, Y., Svirskis, D., Li, H., Ying, M., Lu, W., & Wen, J. (2024). Cryo-Milled β-Glucan Nanoparticles for Oral Drug Delivery. Pharmaceutics, 16(4), 546. https://doi.org/10.3390/pharmaceutics16040546