
LDLR-Mediated Targeting and Productive Uptake of siRNA-Peptide Ligand Conjugates In Vitro and In Vivo

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Reagents and Material
2.2. Purification and Analytical Methods
2.2.1. Peptide-Based Products
2.2.2. Oligo-Based Products
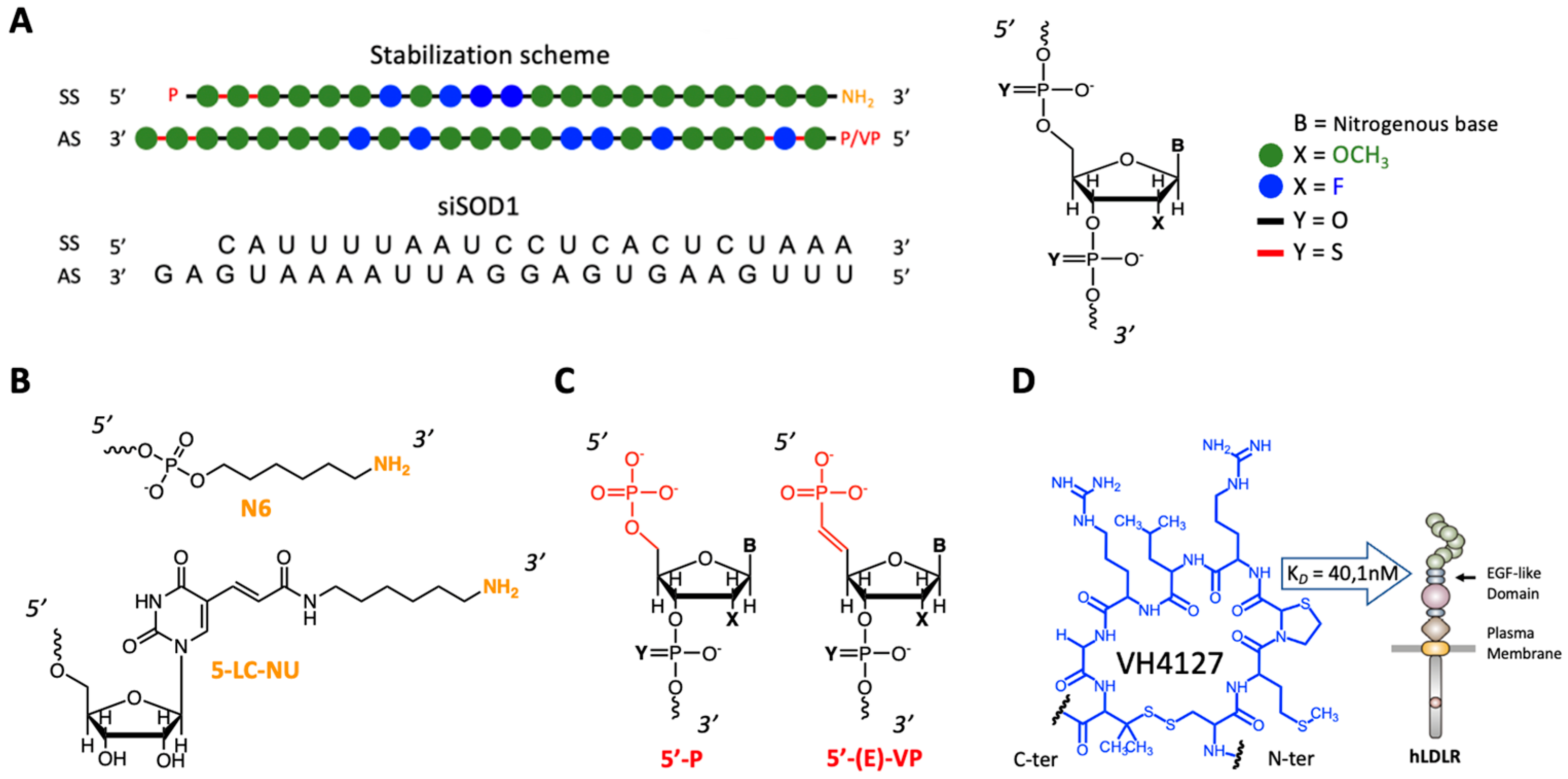
2.3. Sequence and Modifications of siRNAs
2.4. Preparation of Peptide Ligand Precursors
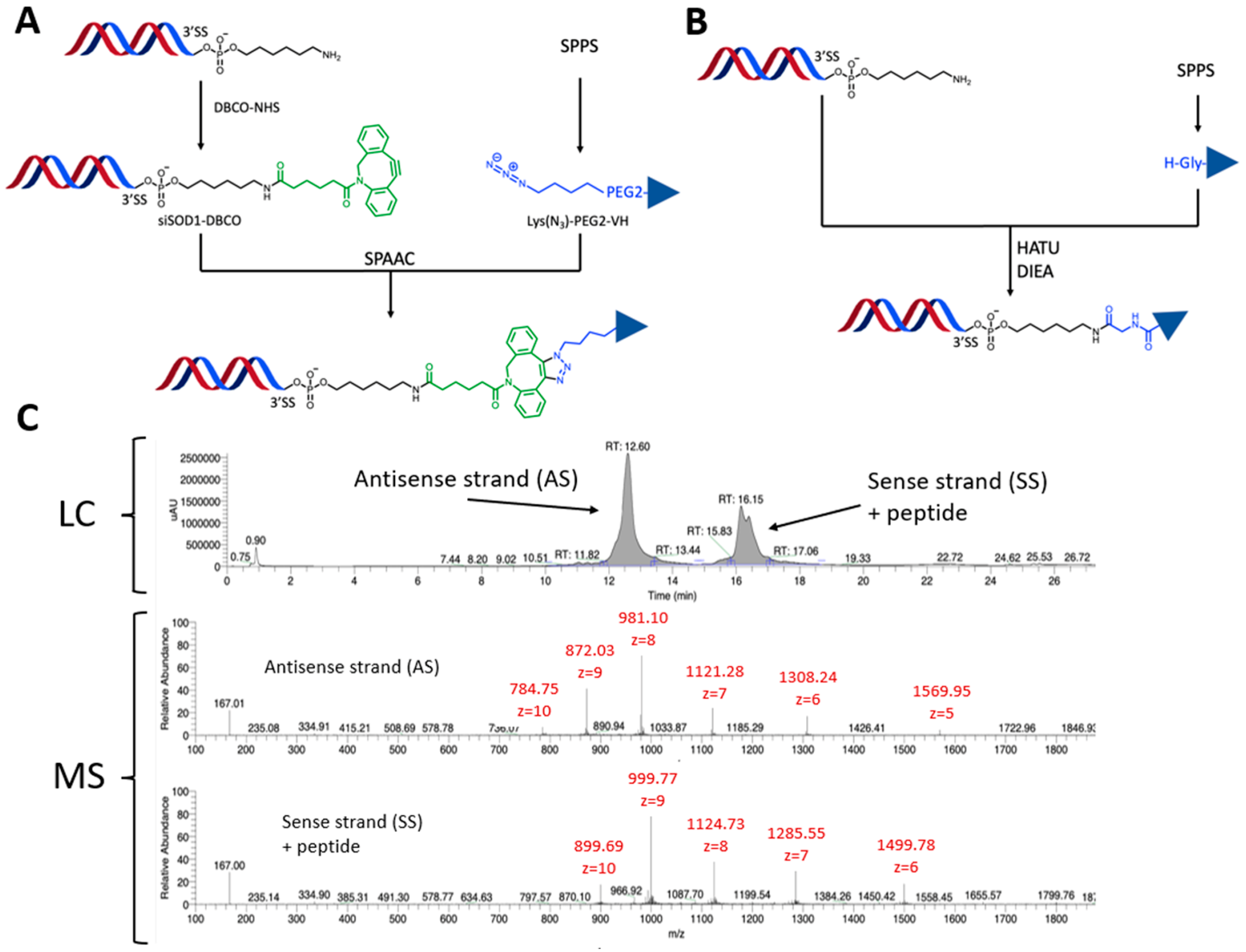
2.5. Preparation of siSOD1-Peptide Conjugates
2.5.1. siSOD1-Peptide Conjugate Synthesis Using SPAAC
2.5.2. siSOD1-Peptide Conjugate Synthesis Using Direct Amidation
2.6. Surface Plasmon Resonance
2.7. Cell Culture and Reagents
2.8. In Vitro Validation of LDLR-Expressing Neuro-2a Cells
2.9. Cell Transfection
2.10. Uptake and Free Uptake Gene-Silencing Experiments
2.11. Animal Handling
2.12. RNA Extraction and cDNA Synthesis
2.13. Real-Time Quantitative PCR
2.14. Data Analysis
3. Results
3.1. Molecular Design and Synthesis of siSOD1-Peptide Conjugates
3.2. LDLR-Binding Affinity of siSOD1-Peptide Conjugates Using Surface Plasmon Resonance (SPR)
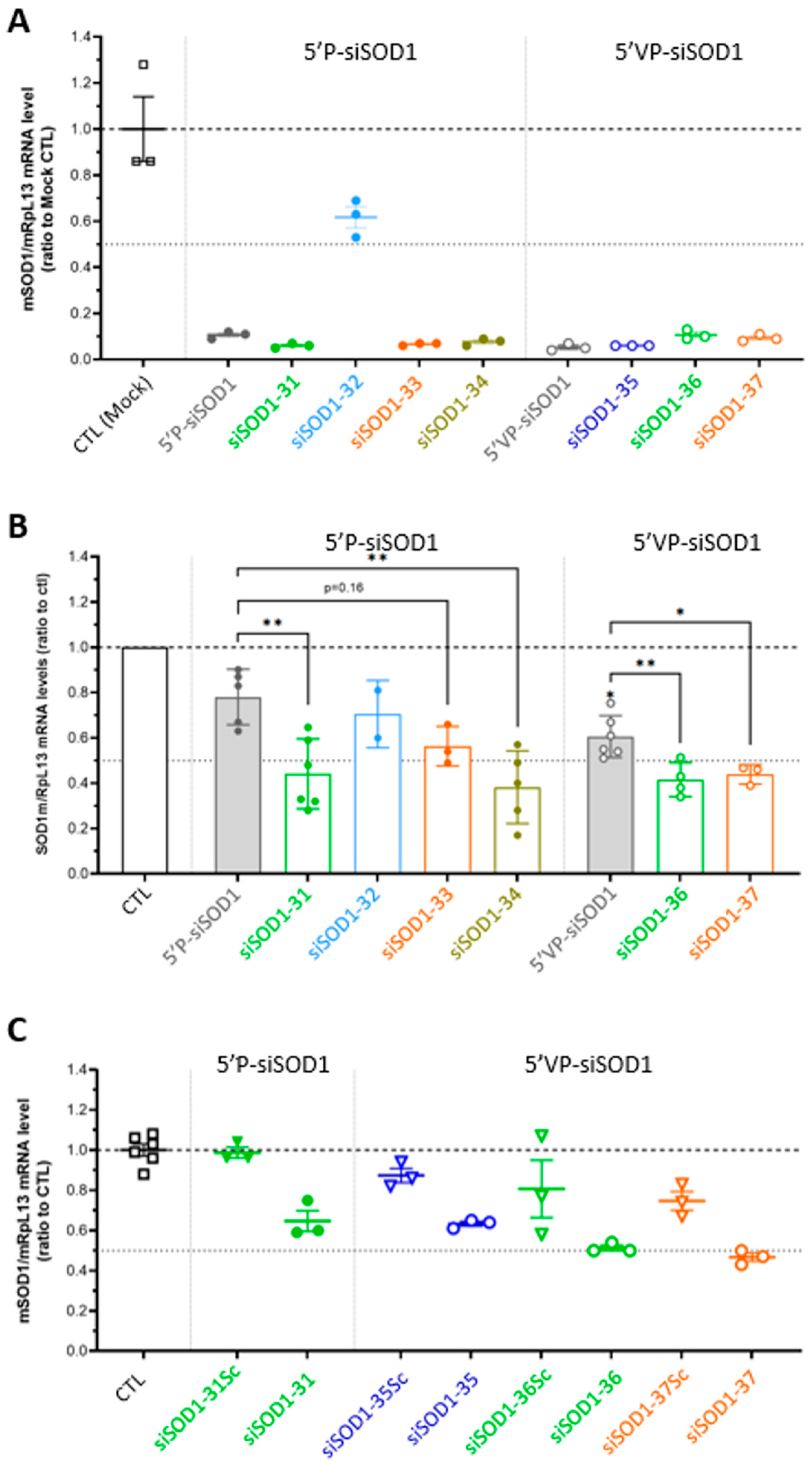
3.3. Gene-Silencing Potency of siSOD1-Peptide Conjugates In Vitro in Murine Neuro-2A Cells
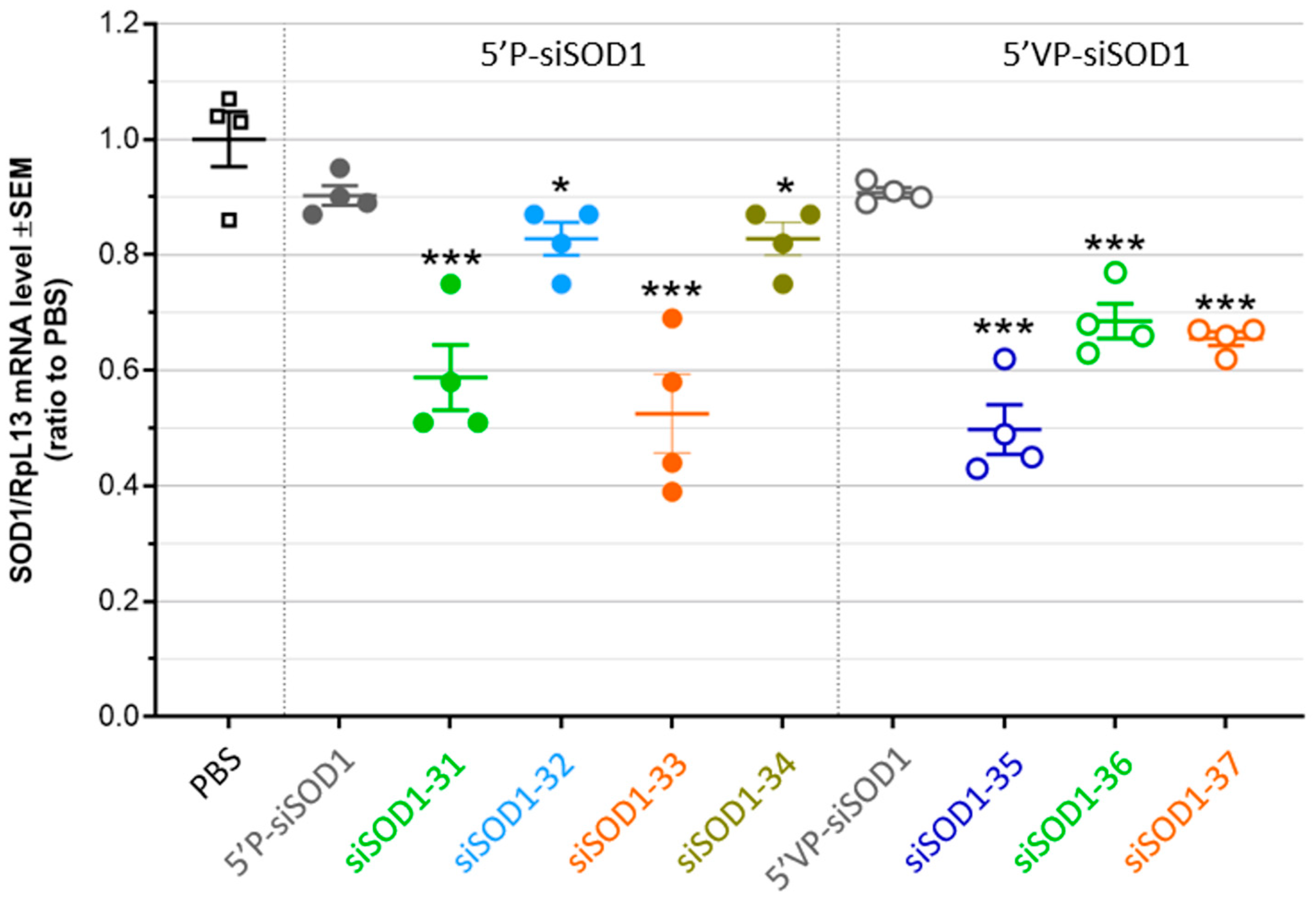
3.4. Gene-Silencing Potency of siSOD1-Peptide Conjugates after Systemic Administration in Mice
4. Discussion and Perspectives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef]
- Neumeier, J.; Meister, G. siRNA Specificity: RNAi Mechanisms and Strategies to Reduce Off-Target Effects. Front. Plant Sci. 2021, 11, 526455. [Google Scholar] [CrossRef]
- Semizarov, D.; Frost, L.; Sarthy, A.; Kroeger, P.; Halbert, D.N.; Fesik, S.W. Specificity of short interfering RNA determined through gene expression signatures. Proc. Natl. Acad. Sci. USA 2003, 100, 6347–6352. [Google Scholar] [CrossRef]
- Fitzgerald, K.; White, S.; Borodovsky, A.; Bettencourt, B.R.; Strahs, A.; Clausen, V.; Wijngaard, P.; Horton, J.D.; Taubel, J.; Brooks, A.; et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N. Engl. J. Med. 2017, 376, 41–51. [Google Scholar] [CrossRef]
- Huang, Y.; Cheng, Q.; Ji, J.-L.; Zheng, S.; Du, L.; Meng, L.; Wu, Y.; Zhao, D.; Wang, X.; Lai, L.; et al. Pharmacokinetic Behaviors of Intravenously Administered siRNA in Glandular Tissues. Theranostics 2016, 6, 1528–1541. [Google Scholar] [CrossRef]
- Godinho, B.M.; Knox, E.G.; Hildebrand, S.; Gilbert, J.W.; Echeverria, D.; Kennedy, Z.; Haraszti, R.A.; Ferguson, C.M.; Coles, A.H.; Biscans, A.; et al. PK-modifying anchors significantly alter clearance kinetics, tissue distribution, and efficacy of therapeutics siRNAs. Mol. Ther. Nucleic Acids 2022, 29, 116–132. [Google Scholar] [CrossRef]
- Dong, Y.; Siegwart, D.J.; Anderson, D.G. Strategies, design, and chemistry in siRNA delivery systems. Adv. Drug Deliv. Rev. 2019, 144, 133–147. [Google Scholar] [CrossRef]
- Kim, B.; Park, J.-H.; Sailor, M.J. Rekindling RNAi Therapy: Materials Design Requirements for In Vivo siRNA Delivery. Adv. Mater. 2019, 31, e1903637. [Google Scholar] [CrossRef]
- Bertucci, A.; Kim, K.-H.; Kang, J.; Zuidema, J.M.; Lee, S.H.; Kwon, E.J.; Kim, D.; Howell, S.B.; Ricci, F.; Ruoslahti, E.; et al. Tumor-Targeting, MicroRNA-Silencing Porous Silicon Nanoparticles for Ovarian Cancer Therapy. ACS Appl. Mater. Interfaces 2019, 11, 23926–23937. [Google Scholar] [CrossRef]
- Soule, B.; Tirucherai, G.; Kavita, U.; Kundu, S.; Christian, R. Safety, tolerability, and pharmacokinetics of BMS-986263/ND-L02-s0201, a novel targeted lipid nanoparticle delivering HSP47 siRNA, in healthy participants: A randomised, placebo-controlled, double-blind, phase 1 study. J. Hepatol. 2018, 68, S112. [Google Scholar] [CrossRef]
- Golan, T.; Hubert, A.; Shemi, A.; Segal, A.; Dancour, A.; Khvalevsky, E.Z.; Ben-David, E.; Raskin, S.; Goldes, Y.; Inbar, Y.; et al. A phase I trial of a local delivery of siRNA against k-ras in combination with chemotherapy for locally advanced pancreatic adenocarcinoma. J. Clin. Oncol. 2013, 31, 4037. [Google Scholar] [CrossRef]
- Kim, B.; Sun, S.; Varner, J.A.; Howell, S.B.; Ruoslahti, E.; Sailor, M.J. Securing the Payload, Finding the Cell, and Avoiding the Endosome: Peptide-Targeted, Fusogenic Porous Silicon Nanoparticles for Delivery of siRNA. Adv. Mater. 2019, 31, e1902952. [Google Scholar] [CrossRef]
- Smidt, J.M.; Lykke, L.; Stidsen, C.E.; Pristovšek, N.; Gothelf, K.V. Synthesis of peptide–siRNA conjugates via internal sulfonylphosphoramidate modifications and evaluation of their in vitro activity. Nucleic Acids Res. 2023, 52, 49–58. [Google Scholar] [CrossRef]
- Lee, J.W.; Choi, J.; Kim, E.H.; Kim, S.H.; Yang, Y. Design of siRNA Bioconjugates for Efficient Control of Cancer-Associated Membrane Receptors. ACS Omega 2023, 8, 36435–36448. [Google Scholar] [CrossRef]
- Cen, B.; Wei, Y.; Huang, W.; Teng, M.; He, S.; Li, J.; Wang, W.; He, G.; Bai, X.; Liu, X.; et al. An Efficient Bivalent Cyclic RGD-PIK3CB siRNA Conjugate for Specific Targeted Therapy against Glioblastoma In Vitro and In Vivo. Mol. Ther. Nucleic Acids 2018, 13, 220–232. [Google Scholar] [CrossRef]
- Janas, M.M.; Zlatev, I.; Liu, J.; Jiang, Y.; Barros, A.S.; Sutherland, E.J.; Davis, W.P.; Liu, J.; Brown, C.R.; Liu, X.; et al. Safety evaluation of 2′-deoxy-2′-fluoro nucleotides in GalNAc-siRNA conjugates. Nucleic Acids Res. 2019, 47, 3306–3320. [Google Scholar] [CrossRef]
- Dindo, M.; Conter, C.; Oppici, E.; Ceccarelli, V.; Marinucci, L.; Cellini, B. Molecular basis of primary hyperoxaluria: Clues to innovative treatments. Urolithiasis 2019, 47, 67–78. [Google Scholar] [CrossRef]
- Gökirmak, T.; Nikan, M.; Wiechmann, S.; Prakash, T.P.; Tanowitz, M.; Seth, P.P. Overcoming the challenges of tissue delivery for oligonucleotide therapeutics. Trends Pharmacol. Sci. 2021, 42, 588–604. [Google Scholar] [CrossRef]
- Debacker, A.J.; Voutila, J.; Catley, M.; Blakey, D.; Habib, N. Delivery of Oligonucleotides to the Liver with GalNAc: From Research to Registered Therapeutic Drug. Mol. Ther. 2020, 28, 1759–1771. [Google Scholar] [CrossRef]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarría, L.; et al. Delivery of oligonucleotide-based therapeutics: Challenges and opportunities. EMBO Mol. Med. 2021, 13, e13243. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Receptor-mediated endocytosis: Insights from the lipoprotein receptor system. Proc. Natl. Acad. Sci. USA 1979, 76, 3330–3337. [Google Scholar] [CrossRef]
- Ghosh, R.N.; Gelman, D.L.; Maxfield, F.R. Quantification of low density lipoprotein and transferrin endocytic sorting in HEp2 cells using confocal microscopy. J. Cell Sci. 1994, 107 Pt 8, 2177–2189. [Google Scholar] [CrossRef]
- Dunn, K.W.; E McGraw, T.; Maxfield, F.R. Iterative fractionation of recycling receptors from lysosomally destined ligands in an early sorting endosome. J. Cell Biol. 1989, 109 Pt 2, 3303–3314. [Google Scholar] [CrossRef]
- David, M.; Lécorché, P.; Masse, M.; Faucon, A.; Abouzid, K.; Gaudin, N.; Varini, K.; Gassiot, F.; Ferracci, G.; Jacquot, G.; et al. Identification and characterization of highly versatile peptide-vectors that bind non-competitively to the low-density lipoprotein receptor for in vivo targeting and delivery of small molecules and protein cargos. PLoS ONE 2018, 13, e0191052. [Google Scholar] [CrossRef]
- Jacquot, G.; Lécorché, P.; Malcor, J.-D.; Laurencin, M.; Smirnova, M.; Varini, K.; Malicet, C.; Gassiot, F.; Abouzid, K.; Faucon, A.; et al. Optimization and in Vivo Validation of Peptide Vectors Targeting the LDL Receptor. Mol. Pharm. 2016, 13, 4094–4105. [Google Scholar] [CrossRef]
- Malcor, J.-D.; Payrot, N.; David, M.; Faucon, A.; Abouzid, K.; Jacquot, G.; Floquet, N.; Debarbieux, F.; Rougon, G.; Martinez, J.; et al. Chemical Optimization of New Ligands of the Low-Density Lipoprotein Receptor as Potential Vectors for Central Nervous System Targeting. J. Med. Chem. 2012, 55, 2227–2241. [Google Scholar] [CrossRef]
- Foster, D.J.; Brown, C.R.; Shaikh, S.; Trapp, C.; Schlegel, M.K.; Qian, K.; Sehgal, A.; Rajeev, K.G.; Jadhav, V.; Manoharan, M.; et al. Advanced siRNA Designs Further Improve In Vivo Performance of GalNAc-siRNA Conjugates. Mol. Ther. 2018, 26, 708–717. [Google Scholar] [CrossRef]
- Fantoni, N.Z.; El-Sagheer, A.H.; Brown, T. A Hitchhiker’s Guide to Click-Chemistry with Nucleic Acids. Chem. Rev. 2021, 121, 7122–7154. [Google Scholar] [CrossRef]
- Zayas, J.; Annoual, M.; Das, J.K.; Felty, Q.; Gonzalez, W.G.; Miksovska, J.; Sharifai, N.; Chiba, A.; Wnuk, S.F. Strain Promoted Click Chemistry of 2- or 8-Azidopurine and 5-Azidopyrimidine Nucleosides and 8-Azidoadenosine Triphosphate with Cyclooctynes. Application to Living Cell Fluorescent Imaging. Bioconjug. Chem. 2015, 26, 1519–1532. [Google Scholar] [CrossRef]
- Alam, M.R.; Ming, X.; Fisher, M.; Lackey, J.G.; Rajeev, K.G.; Manoharan, M.; Juliano, R.L. Multivalent Cyclic RGD Conjugates for Targeted Delivery of Small Interfering RNA. Bioconjug. Chem. 2011, 22, 1673–1681. [Google Scholar] [CrossRef]
- Varini, K.; Lécorché, P.; Sonnette, R.; Gassiot, F.; Broc, B.; Godard, M.; David, M.; Faucon, A.; Abouzid, K.; Ferracci, G.; et al. Target engagement and intracellular delivery of mono- and bivalent LDL receptor-binding peptide-cargo conjugates: Implications for the rational design of new targeted drug therapies. J. Control. Release 2019, 314, 141–161. [Google Scholar] [CrossRef]
- Nakagawa, O.; Ming, X.; Carver, K.; Juliano, R. Conjugation with Receptor-Targeted Histidine-Rich Peptides Enhances the Pharmacological Effectiveness of Antisense Oligonucleotides. Bioconjug. Chem. 2014, 25, 165–170. [Google Scholar] [CrossRef]
- Parmar, R.; Willoughby, J.L.S.; Liu, J.; Foster, D.J.; Brigham, B.; Theile, C.S.; Charisse, K.; Akinc, A.; Guidry, E.; Pei, Y.; et al. 5′-(E)-Vinylphosphonate: A Stable Phosphate Mimic Can Improve the RNAi Activity of siRNA–GalNAc Conjugates. ChemBioChem 2016, 17, 985–989. [Google Scholar] [CrossRef]
- Haraszti, R.A.; Roux, L.; Coles, A.H.; Turanov, A.A.; Alterman, J.F.; Echeverria, D.; Godinho, B.M.; Aronin, N.; Khvorova, A. 5′-Vinylphosphonate improves tissue accumulation and efficacy of conjugated siRNAs in vivo. Nucleic Acids Res. 2017, 45, 7581–7592. [Google Scholar] [CrossRef]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef]
- Gilleron, J.; Paramasivam, P.; Zeigerer, A.; Querbes, W.; Marsico, G.; Andree, C.; Seifert, S.; Amaya, P.; Stöter, M.; Koteliansky, V.; et al. Identification of siRNA delivery enhancers by a chemical library screen. Nucleic Acids Res. 2015, 43, 7984–8001. [Google Scholar] [CrossRef]
- Acier, A.; Godard, M.; Gassiot, F.; Finetti, P.; Rubis, M.; Nowak, J.; Bertucci, F.; Iovanna, J.L.; Tomasini, R.; Lécorché, P.; et al. LDL receptor-peptide conjugate as in vivo tool for specific targeting of pancreatic ductal adenocarcinoma. Commun. Biol. 2021, 4, 987. [Google Scholar] [CrossRef]
- Firer, M.A.; Gellerman, G. Targeted drug delivery for cancer therapy: The other side of antibodies. J. Hematol. Oncol. 2012, 5, 70. [Google Scholar] [CrossRef]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef]
- Ämmälä, C.; Drury, W.J.; Knerr, L.; Ahlstedt, I.; Stillemark-Billton, P.; Wennberg-Huldt, C.; Andersson, E.-M.; Valeur, E.; Jansson-Löfmark, R.; Janzén, D.; et al. Targeted delivery of antisense oligonucleotides to pancreatic β-cells. Sci. Adv. 2018, 4, eaat3386. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle–mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef]
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014, 42, 8796–8807. [Google Scholar] [CrossRef]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F.; Setten, R.L.; Cui, X.-S.; Jadhav, S.G. Delivery of RNA Therapeutics: The Great Endosomal Escape! Nucleic Acid Ther. 2022, 32, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Knerr, L.; Prakash, T.P.; Lee, R.; Iii, W.J.D.; Nikan, M.; Fu, W.; Pirie, E.; De Maria, L.; Valeur, E.; Hayen, A.; et al. Glucagon Like Peptide 1 Receptor Agonists for Targeted Delivery of Antisense Oligonucleotides to Pancreatic Beta Cell. J. Am. Chem. Soc. 2021, 143, 3416–3429. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef]
- Kuo, C.; Nikan, M.; Yeh, S.T.; Chappell, A.E.; Tanowitz, M.; Seth, P.P.; Prakash, T.P.; Mullick, A.E. Targeted Delivery of Antisense Oligonucleotides Through Angiotensin Type 1 Receptor. Nucleic Acid Ther. 2022, 32, 300–311. [Google Scholar] [CrossRef]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release 2016, 237, 1–13. [Google Scholar] [CrossRef]
- Nikan, M.; Tanowitz, M.; Dwyer, C.A.; Jackson, M.; Gaus, H.J.; Swayze, E.E.; Rigo, F.; Seth, P.P.; Prakash, T.P. Targeted Delivery of Antisense Oligonucleotides Using Neurotensin Peptides. J. Med. Chem. 2020, 63, 8471–8484. [Google Scholar] [CrossRef]
- Brown, C.R.; Gupta, S.; Qin, J.; Racie, T.; He, G.; Lentini, S.; Malone, R.; Yu, M.; Matsuda, S.; Shulga-Morskaya, S.; et al. Investigating the pharmacodynamic durability of GalNAc–siRNA conjugates. Nucleic Acids Res. 2020, 48, 11827–11844. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Foillard, S.; Lammers, T.; Schiffelers, R.M.; Doris, E.; Hennink, W.E.; Storm, G. SiRNA delivery with functionalized carbon nanotubes. Int. J. Pharm. 2011, 416, 419–425. [Google Scholar] [CrossRef]
- Dowdy, S.F. Endosomal escape of RNA therapeutics: How do we solve this rate-limiting problem? RNA 2023, 29, 396–401. [Google Scholar] [CrossRef]
- Du Rietz, H.; Hedlund, H.; Wilhelmson, S.; Nordenfelt, P.; Wittrup, A. Imaging small molecule-induced endosomal escape of siRNA. Nat. Commun. 2020, 11, 1809. [Google Scholar] [CrossRef]
- Lee, H.-J.; Huang, Y.-W.; Chiou, S.-H.; Aronstam, R.S. Polyhistidine facilitates direct membrane translocation of cell-penetrating peptides into cells. Sci. Rep. 2019, 9, 9398. [Google Scholar] [CrossRef]
- Alagia, A.; Eritja, R. siRNA and RNAi optimization. Wiley Interdiscip. Rev. RNA 2016, 7, 316–329. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequences | RP-HPLC, Retention Time (min) | HPLC-UV Purity (%) | Theoretical Mass (Da) | ESI-MS (m/z) [M,H]+ (Calculated) |
|---|---|---|---|---|
| Pr-K(N3)-PEG2-[cMThzRLRGPen]c-NH2 | rt = 3.36 min | 91.7% | 1332.60 Da | 1333.51 Da |
| Pr-[cMThzRLRGPen]c-PEG2-K(N3)-NH2 | rt = 3.61 min | 96.7% | 1332.60 Da | 1333.00 Da |
| Pr-[cMThzRLRGPenG]c-OH | rt = 2.90 min | 97.0% | 1091.45 Da | 1092.69 Da |
| Pr-K(N3)-PEG2-[cMThzRLRGPen]c-H2GH2GH2GH2-NH2 | rt = 2.68 min | 90.5% | 2244.95 Da | 2245.44 Da |
| Sense Strand + Peptide | Antisense Strand | |||||
|---|---|---|---|---|---|---|
| RP-HPLC, Retention Time Gradient | Theoretical Molecular Weight | ESI-MS (m/z) | RP-HPLC, Retention Time Gradient | Theoretical Molecular Weight | ESI-MS (m/z) | |
| siSOD1-31 | 16.32 min 16.54 min | 8709.66 Da | 8710.80 Da 8710.65 Da | 12.64 min | 7855.17 Da | 7855.73 Da |
| siSOD1-31Sc | 14.18 min 14.33 min | 8662.56 Da | 8663.22 Da 8663.03 Da | 11.44 min | 7855.17 Da | 7854.96 Da |
| siSOD1-32 | 15.45 min 15.59 min | 8709.66 | 8709.80 Da 8709.60 Da | 12.24 min | 7855.17 Da | 7855.70 Da |
| siSOD1-33 | 16.15 min 16.40 min | 9004.91 Da | 9005.42 Da 9005.29 Da | 12.60 min | 7855.17 Da | 7856.04 Da |
| siSOD1-34 | 16.30 min 16.50 min | 9977.95 Da | 9978.06 Da 9978.08 Da | 12.64 min | 7855.17 Da | 7856.19 Da |
| siSOD1-35 | 9.30 min | 8166.97 Da | 8166.66 Da | 7.99 min | 7851.18 Da | 7850.70 Da |
| siSOD1-35Sc | 8.12 min | 8088.89 Da | 8088.74 da | 8.03 min | 7851.18 Da | 7850.79 Da |
| siSOD1-36 | 15.47 min 15.64 min | 8709.66 Da | 8710.05 Da 8710.25 Da | 12.17 min | 7851.18 Da | 7851.35 Da |
| siSOD1-36Sc | 13.56 min 13.98 min | 8694.86 Da | 8694.86 Da 8694.20 Da | 11.47 min | 7851.18 Da | 7850.92 Da |
| siSOD1-37 | 15.94 min 16.11 min | 9004.91 Da | 9004.75 Da 9004.89 Da | 12.19 min | 7851.18 Da | 7850.92 Da |
| siSOD1-37Sc | 14.38 min 14.49 min | 8941.94 Da | 8942.50 Da 8942.35 Da | 11.51 min | 7851.18 Da | 7850.86 Da |
| Code | siSOD1 | Peptide Ligand | Characterization | Affinities | |||
|---|---|---|---|---|---|---|---|
| Retention Time (min) 1 | Purity (%) 2 | Identity (%) 3 | SPR (nM) 4 | ||||
| VH4127 | / | Pr-VH4127-NH2 | / | / | / | 40.1 ± 9.1 | |
| 5′P | siSOD1-31 | P-(5′AS)-siSOD1-(3′SS)-N6 | Pr-K(N3)-PEG2-VH4127-NH2 | TrAS = 12.64 min TrSS = 16.32–16.54 min | AS: 83.7% SS: 84.8% | AS: 99.99% SS: 99.98% | 104.0 ± 11.7 |
| siSOD1-32 | P-(5′AS)-siSOD1-(3′SS)-N6 | Pr-VH4127-PEG2-K(N3)-NH2 | TrAS = 12.24 min TrSS = 15.45–15.59 min | AS: 62.9% SS: 77.6% | AS: 99.98% SS: 99.99% | 86.8 ± 17.1 | |
| siSOD1-33 | P-(5′AS)-siSOD1-(3′SS)-5-LC-NU | Pr-K(N3)-PEG2-VH4127-NH2 | TrAS = 12.60 min TrSS = 16.15–16.40 min | AS: 89.5% SS: 85.1% | AS: 99.98% SS: 99.99% | 12.8 ± 1.9 | |
| siSOD1-34 | P-(5′AS)-siSOD1-(3′SS)-N6 | Pr-K(N3)-PEG2-VH4127-H8G3-NH2 | TrAS = 12.64 min TrSS = 16.30–16.50 min | AS: 93.1% SS: 81.7% | AS: 99.98% SS: 99.99% | ND | |
| 5′VP | siSOD1-35 | VP-(5′AS)-siSOD1-(3′SS)-N6 | Pr-VH4127-G-OH | TrAS = 7.99 min TrSS = 9.30 min | AS: 88.2% SS: 96.3% | AS: 99.98% SS: 99.98% | 11.3 ± 2.9 |
| siSOD1-36 | VP-(5′AS)-siSOD1-(3′SS)-N6 | Pr-K(N3)-PEG2-VH4127-NH2 | TrAS = 12.17 min TrSS = 15.47–15.64 min | AS: 72.6% SS: 95.8% | AS: 99.98% SS: 99.99% | 25 ± 1.2 | |
| siSOD1-37 | VP-(5′AS)-siSOD1-(3′SS)-5-LC-NU | Pr-K(N3)-PEG2-VH4127-NH2 | TrAS = 13.19 min TrSS = 15.94–16.11 min | AS: 93.8% SS: 67.8% | AS: 99.98% SS: 99.98% | 31.9 ± 1.6 | |
| Code | In Vitro KD on N2A | In Vivo KD | ||
|---|---|---|---|---|
| Transfection | Free Uptake | Liver | ||
| 5′P | 5′P-siSOD1 | >90% | 20% | 10% |
| siSOD1-31 | >90% | 55% | 40% | |
| siSOD1-32 | 40% | 30% | 15% | |
| siSOD1-33 | >90% | 45% | 45% | |
| siSOD1-34 | >90% | 60% | 15% | |
| 5′VP | 5′VP-siSOD1 | >90% | 40% | 10% |
| siSOD1-35 | >90% | 40% | 50% | |
| siSOD1-36 | >90% | 60% | 30% | |
| siSOD1-37 | >90% | 60% | 35% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broc, B.; Varini, K.; Sonnette, R.; Pecqueux, B.; Benoist, F.; Masse, M.; Mechioukhi, Y.; Ferracci, G.; Temsamani, J.; Khrestchatisky, M.; et al. LDLR-Mediated Targeting and Productive Uptake of siRNA-Peptide Ligand Conjugates In Vitro and In Vivo. Pharmaceutics 2024, 16, 548. https://doi.org/10.3390/pharmaceutics16040548
Broc B, Varini K, Sonnette R, Pecqueux B, Benoist F, Masse M, Mechioukhi Y, Ferracci G, Temsamani J, Khrestchatisky M, et al. LDLR-Mediated Targeting and Productive Uptake of siRNA-Peptide Ligand Conjugates In Vitro and In Vivo. Pharmaceutics. 2024; 16(4):548. https://doi.org/10.3390/pharmaceutics16040548
Chicago/Turabian StyleBroc, Baptiste, Karine Varini, Rose Sonnette, Belinda Pecqueux, Florian Benoist, Maxime Masse, Yasmine Mechioukhi, Géraldine Ferracci, Jamal Temsamani, Michel Khrestchatisky, and et al. 2024. "LDLR-Mediated Targeting and Productive Uptake of siRNA-Peptide Ligand Conjugates In Vitro and In Vivo" Pharmaceutics 16, no. 4: 548. https://doi.org/10.3390/pharmaceutics16040548
APA StyleBroc, B., Varini, K., Sonnette, R., Pecqueux, B., Benoist, F., Masse, M., Mechioukhi, Y., Ferracci, G., Temsamani, J., Khrestchatisky, M., Jacquot, G., & Lécorché, P. (2024). LDLR-Mediated Targeting and Productive Uptake of siRNA-Peptide Ligand Conjugates In Vitro and In Vivo. Pharmaceutics, 16(4), 548. https://doi.org/10.3390/pharmaceutics16040548