
Heparin Dosing Regimen Optimization in Veno-Arterial Extracorporeal Membrane Oxygenation: A Pharmacokinetic Analysis
 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Data Management
2.3. Clinical Management
2.4. Anticoagulation Management
2.5. Biological Sampling and Analysis
2.6. Pharmacokinetic Model Development
2.7. Pharmacokinetic Model Selection and Evaluation
2.8. Individualized Regimen Estimation
2.9. PK Simulations
3. Results
3.1. Patients
3.2. Pharmacokinetic Model
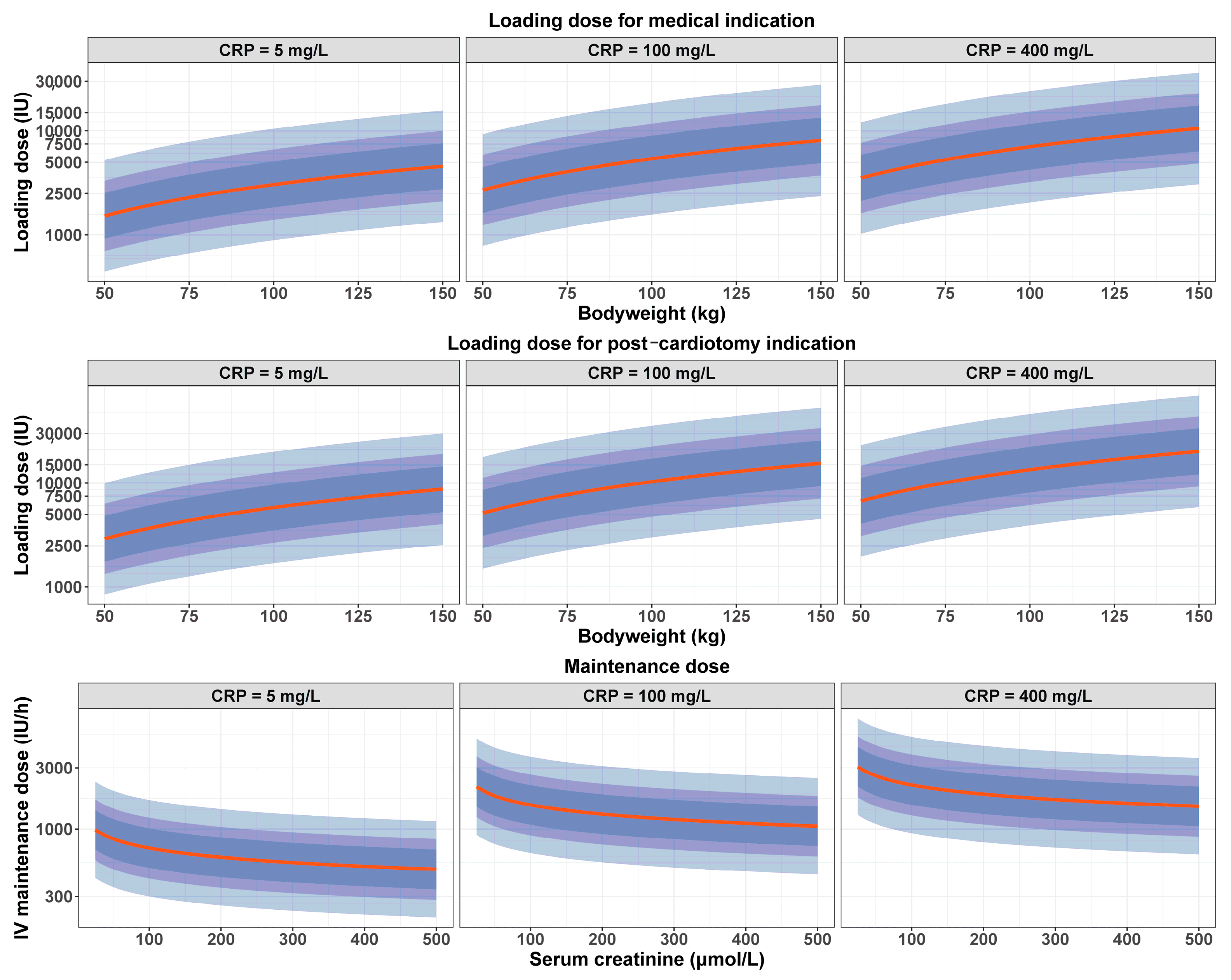
3.3. Optimized Dosing Regimen
3.4. PK Simulations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rao, P.; Khalpey, Z.; Smith, R.; Burkhoff, D.; Kociol, R.D. Venoarterial Extracorporeal Membrane Oxygenation for Cardiogenic Shock and Cardiac Arrest. Circ. Heart Fail. 2018, 11, e004905. [Google Scholar] [CrossRef] [PubMed]
- ESLO Registry. 2023. Available online: https://www.elso.org/registry/elsoliveregistrydashboard.aspx (accessed on 19 October 2023).
- Plötz, F.B.; van Oeveren, W.; Bartlett, R.H.; Wildevuur, C.R. Blood activation during neonatal extracorporeal life support. J. Thorac. Cardiovasc. Surg. 1993, 105, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Millar, J.E.; Fanning, J.P.; McDonald, C.I.; McAuley, D.F.; Fraser, J.F. The inflammatory response to extracorporeal membrane oxygenation (ECMO): A review of the pathophysiology. Crit. Care 2016, 20, 387. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mazzeffi, M.A.; Tanaka, K.; Roberts, A.; Rector, R.; Menaker, J.; Kon, Z.; Deatrick, K.B.; Kaczorowski, D.; Griffith, B.; Herr, D. Bleeding, Thrombosis, and Transfusion with Two Heparin Anticoagulation Protocols in Venoarterial ECMO Patients. J. Cardiothorac. Vasc. Anesth. 2019, 33, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.B.V.; Ryerson, L.M.; Ratano, D.; Fan, E.; Faraoni, D.; Annich, G.M. 2021 ELSO Adult and Pediatric Anticoagulation Guidelines. ASAIO J. 2022, 68, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Aubron, C.; DePuydt, J.; Belon, F.; Bailey, M.; Schmidt, M.; Sheldrake, J.; Murphy, D.; Scheinkestel, C.; Cooper, D.J.; Capellier, G.; et al. Predictive factors of bleeding events in adults undergoing extracorporeal membrane oxygenation. Ann. Intensive Care 2016, 6, 97. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moussa, M.D.; Soquet, J.; Lamer, A.; Labreuche, J.; Gantois, G.; Dupont, A.; Abou-Arab, O.; Rousse, N.; Liu, V.; Brandt, C.; et al. Evaluation of Anti-Activated Factor X Activity and Activated Partial Thromboplastin Time Relations and Their Association with Bleeding and Thrombosis during Veno-Arterial ECMO Support: A Retrospective Study. J. Clin. Med. 2021, 10, 2158. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Toulon, P.; Smahi, M.; De Pooter, N. APTT therapeutic range for monitoring unfractionated heparin therapy. Significant impact of the anti-Xa reagent used for correlation. J. Thromb. Haemost. 2021, 19, 2002–2006. [Google Scholar] [CrossRef] [PubMed]
- Sy, E.; Sklar, M.C.; Lequier, L.; Fan, E.; Kanji, H.D. Anticoagulation practices and the prevalence of major bleeding, thromboembolic events, and mortality in venoarterial extracorporeal membrane oxygenation: A systematic review and meta-analysis. J. Crit. Care 2017, 39, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; Bauer, K.A.; Donati, M.B.; Gould, M.; Samama, M.M.; Weitz, J.I. Parenteral anticoagulants: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008, 133 (Suppl. S6), 141S–159S, Erratum in Chest 2008, 134, 473. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, B.; Fredenburgh, J.C.; Rischke, J.; Hirsh, J.; Young, E.; Weitz, J.I. Effect of nonspecific binding to plasma proteins on the antithrombin activities of unfractionated heparin, low-molecular-weight heparin, and dermatan sulfate. Circulation 1997, 95, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; van der Poll, T. Inflammation and coagulation. Crit. Care Med. 2010, 38 (Suppl. S2), S26–S34. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, J.; Chaux, R.; Hodin, S.; Bourayou, S.; Gibert, A.; Philippot, R.; Molliex, S.; Zufferey, P.J.; Delavenne, X.; Ollier, E. Population pharmacokinetic model of cefazolin in total hip arthroplasty. Sci. Rep. 2021, 11, 19763. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Delattre, M.; Poursat, M.A. An iterative algorithm for joint covariate and random effect selection in mixed effects models. Int. J. Biostat. 2020, 16, 20190082. [Google Scholar] [CrossRef] [PubMed]
- Brendel, K.; Comets, E.; Laffont, C.; Mentré, F. Evaluation of different tests based on observations for external model evaluation of population analyses. J. Pharmacokinet. Pharmacodyn. 2010, 37, 49–65. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lanoiselée, J.; Zufferey, P.J.; Hodin, S.; Tamisier, N.; Gergelé, L.; Palao, J.C.; Campisi, S.; Molliex, S.; Morel, J.; Delavenne, X.; et al. Pharmacokinetic Model for Cefuroxime Dosing during Cardiac Surgery under Cardiopulmonary Bypass. Antimicrob. Agents Chemother. 2020, 64, e01687-20. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Derbalah, A.; Duffull, S.; Moynihan, K.; Al-Sallami, H. The Influence of Haemostatic System Maturation on the Dose-Response Relationship of Unfractionated Heparin. Clin. Pharmacokinet. 2021, 60, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Al-Sallami, H.; Newall, F.; Monagle, P.; Ignjatovic, V.; Cranswick, N.; Duffull, S. Development of a population pharmacokinetic-pharmacodynamic model of a single bolus dose of unfractionated heparin in paediatric patients. Br. J. Clin. Pharmacol. 2016, 82, 178–184. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Salem, A.M.; Niu, T.; Li, C.; Moffett, B.S.; Ivaturi, V.; Gopalakrishnan, M. Reassessing the Pediatric Dosing Recommendations for Unfractionated Heparin Using Real-World Data: A Pharmacokinetic-Pharmacodynamic Modeling Approach. J. Clin. Pharmacol. 2022, 62, 733–746. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Tian, G.; Ren, Y.; Sun, Z.; Lu, W.; Hou, X. Pharmacokinetic model of unfractionated heparin during and after cardiopulmonary bypass in cardiac surgery. J. Transl. Med. 2015, 13, 45. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Delavenne, X.; Ollier, E.; Chollet, S.; Sandri, F.; Lanoiselée, J.; Hodin, S.; Montmartin, A.; Fuzellier, J.F.; Mismetti, P.; Gergelé, L. Pharmacokinetic/pharmacodynamic model for unfractionated heparin dosing during cardiopulmonary bypass. Br. J. Anaesth. 2017, 118, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.M.; Smith, T.; Wilkes, J.; Bailly, D.K.; Heyrend, C.; Profsky, M.; Yellepeddi, V.K.; Gopalakrishnan, M. Pharmacokinetic Modeling Using Real-World Data to Optimize Unfractionated Heparin Dosing in Pediatric Patients on Extracorporeal Membrane Oxygenation and Evaluate Target Achievement-Clinical Outcomes Relationship. J. Clin. Pharmacol. 2023, 64, 30–44. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.M.; Jacobson, L.J.; Hay, W.W., Jr.; Hathaway, W.E. Heparin clearance in the newborn. Pediatr. Res. 1981, 15, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Andrew, M.; Marzinotto, V.; Massicotte, P.; Blanchette, V.; Ginsberg, J.; Brill-Edwards, P.; Burrows, P.; Benson, L.; Williams, W.; David, M.; et al. Heparin therapy in pediatric patients: A prospective cohort study. Pediatr. Res. 1994, 35, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Newall, F.; Johnston, L.; Ignjatovic, V.; Monagle, P. Unfractionated heparin therapy in infants and children. Pediatrics 2009, 123, e510–e518. [Google Scholar] [CrossRef] [PubMed]
- Young, E.; Prins, M.; Levine, M.N.; Hirsh, J. Heparin binding to plasma proteins, an important mechanism for heparin resistance. Thromb. Haemost. 1992, 67, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Peng, R.; Pei, S.; Gao, S.; Sun, Y.; Cheng, G.; Yu, D.; Wang, X.; Gao, Z.; Ji, B.; et al. Neutrophil extracellular traps are increased after extracorporeal membrane oxygenation support initiation and present in thrombus: A preclinical study using sheep as an animal model. Thromb. Res. 2023, 221, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Willers, A.; Arens, J.; Mariani, S.; Pels, H.; Maessen, J.G.; Hackeng, T.M.; Lorusso, R.; Swol, J. New Trends, Advantages and Disadvantages in Anticoagulation and Coating Methods Used in Extracorporeal Life Support Devices. Membranes 2021, 11, 617. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Heizer, J.W.; Schardt, T.Q.; Murphy, M.E.; Branchford, B.R. Unfractionated heparin dosing requirements in the presence of inflammation during the first six months of life. Thromb. Res. 2019, 177, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Poletti, L.F.; Bird, K.E.; Marques, D.; Harris, R.B.; Suda, Y.; Sobel, M. Structural aspects of heparin responsible for interactions with von Willebrand factor. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Kostousov, V.; Nguyen, K.; Hundalani, S.G.; Teruya, J. The influence of free hemoglobin and bilirubin on heparin monitoring by activated partial thromboplastin time and anti-Xa assay. Arch. Pathol. Lab. Med. 2014, 138, 1503–1506. [Google Scholar] [CrossRef] [PubMed]
- Machin, D.; Devine, P. The effect of temperature and aprotinin during cardiopulmonary bypass on three different methods of activated clotting time measurement. J. Extra Corpor. Technol. 2005, 37, 265–271. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Inoue, M.; Hirose, Y.; Gamou, M.; Goto, M. Effect of hemodilution with dextran and albumin on activated clotting time (ACT). Masui 1997, 46, 809–812. [Google Scholar] [PubMed]
- Bragadottir, G.; Redfors, B.; Ricksten, S.E. Assessing glomerular filtration rate (GFR) in critically ill patients with acute kidney injury—True GFR versus urinary creatinine clearance and estimating equations. Crit. Care 2013, 17, R108. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]



{kind=link}
{kind=link}
{kind=link}
| Total Population | Development Cohort | Validation Cohort | |
|---|---|---|---|
| n = 74 | n = 64 | n = 10 | |
| Age (years) | 52 ± 13 | 51 ± 12 | 53 ± 15 |
| Male gender | 53 (71.6) | 44 (68.8) | 9 (90) |
| Total body weight (kg) | 75 (54–122) | 75 (54–122) | 67 (56–105) |
| Comorbidities | |||
| Diabetes mellitus | 13 (17.6) | 12 (18.8) | 1 (10) |
| Chronic kidney disease | 17 (23) | 16 (25) | 1 (10) |
| Strokes | 2 (2.7) | 2 (3.1) | 0 (0) |
| Ischemic cardiopathy | 16 (21.6) | 15 (23.4) | 1 (10) |
| Hypertension | 16 (21.6) | 15 (23.4) | 1 (10) |
| Hypercholesterolemia | 16 (21.6) | 12 (18.7) | 4 (40) |
| Atrial fibrillation | 10 (13.5) | 9 (14.1) | 1 (10) |
| P2Y12 inhibitors during ECMO | 16 (21.6) | 12 (18.7) | 4 (40) |
| Lactate on admission (mmol/L) | 5 (1–23) a | 5.1 (1–23) a | 5.1 (1.7–12.4) |
| CRRT during ECMO | 34 (45.9) | 29 (45.3) | 5 (50) |
| VA-ECMO indication | |||
| PC-LCOS | 29 (39.2) | 26 (40.6) | 3 (30) |
| Myocardial infarction | 24 (32.4) | 19 (29.7) | 5 (50) |
| Myocarditis | 4 (5.4) | 3 (4.7) | 1 (10) |
| Acute on chronic heart disease | 6 (8.1) | 5 (7.8) | 1 (10) |
| Others b | 11 (14.9) | 11 (17.2) | 0 (0) |
| Description of VA-ECMO support | |||
| ECMO duration (days) | 7 (1–30) | 7 (1–30) | 7.5 (2–18) |
| Cumulative ECMO duration (days) | 573 | 495 | 78 |
| Peripheral ECMO | 70 (94.6) | 60 (93.7) | 10 (100) |
| Mean UFH dose (IU/kg/h) | 11.39 | 11.35 | 11.64 |
| Weaning categories | |||
| Successful weaning | 45 (60.8) | 39 (61.0) | 6 (60) |
| Heart transplantation | 5 (6.8) | 5 (7.8) | 0 (0) |
| Left or bi-ventricular assist devices | 3 (4.0) | 2 (3.1) | 1 (10) |
| Death under ECMO | 21 (28.4) | 18 (28.1) | 3 (30) |
| Covariates during VA-ECMO | |||
| C-reactive protein (mg/L) | 107 (3–547) | 113 (3–547) | 86 (7–345) |
| Fibrinogen (g/L) | 4 (1–11.1) | 5 (1–10) | 3.7 (1.4–11.1) |
| Serum creatinine (µmol/L) | 114.9 (26.5–530.4) | 114.9 (26.5–530.4) | 88.4 (53–415.5) |
| Parameter | Estimate (% RSE) |
|---|---|
| CL (L/h) = θ1 × (Scr/115)θ2 × (CRP/100)θ3 | |
| θ1 | 3.41 (7.04) |
| θ2 | −0.237 (0.18) |
| θ3 | 0.258 (0.66) |
| V (L) = θ4Indic×θ5 × (BW/80)θ6 × (CRP/110)θ7 | |
| θ4 | 8.65 (15.7) |
| θ5 | 0.647 |
| θ6 | 1 (fixed) |
| θ7 | 0.191 (0.45) |
| ΩCL (SD) | 0.52 (10.3) |
| ΩV (SD) | 0.75 (13.2) |
| BICc | −432.52 |
| Additive residual variability (SD) | 0.07 (5.42) |
| Proportional residual variability (SD) | 0.41 (3.62) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanoiselée, J.; Mourer, J.; Jungling, M.; Molliex, S.; Thellier, L.; Tabareau, J.; Jeanpierre, E.; Robin, E.; Susen, S.; Tavernier, B.; et al. Heparin Dosing Regimen Optimization in Veno-Arterial Extracorporeal Membrane Oxygenation: A Pharmacokinetic Analysis. Pharmaceutics 2024, 16, 770. https://doi.org/10.3390/pharmaceutics16060770
Lanoiselée J, Mourer J, Jungling M, Molliex S, Thellier L, Tabareau J, Jeanpierre E, Robin E, Susen S, Tavernier B, et al. Heparin Dosing Regimen Optimization in Veno-Arterial Extracorporeal Membrane Oxygenation: A Pharmacokinetic Analysis. Pharmaceutics. 2024; 16(6):770. https://doi.org/10.3390/pharmaceutics16060770
Chicago/Turabian StyleLanoiselée, Julien, Jérémy Mourer, Marie Jungling, Serge Molliex, Lise Thellier, Julien Tabareau, Emmanuelle Jeanpierre, Emmanuel Robin, Sophie Susen, Benoit Tavernier, and et al. 2024. "Heparin Dosing Regimen Optimization in Veno-Arterial Extracorporeal Membrane Oxygenation: A Pharmacokinetic Analysis" Pharmaceutics 16, no. 6: 770. https://doi.org/10.3390/pharmaceutics16060770
APA StyleLanoiselée, J., Mourer, J., Jungling, M., Molliex, S., Thellier, L., Tabareau, J., Jeanpierre, E., Robin, E., Susen, S., Tavernier, B., Vincentelli, A., Ollier, E., & Moussa, M. D. (2024). Heparin Dosing Regimen Optimization in Veno-Arterial Extracorporeal Membrane Oxygenation: A Pharmacokinetic Analysis. Pharmaceutics, 16(6), 770. https://doi.org/10.3390/pharmaceutics16060770