
Whole-Body Physiologically Based Pharmacokinetic Modeling Framework for Tissue Target Engagement of CD3 Bispecific Antibodies

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
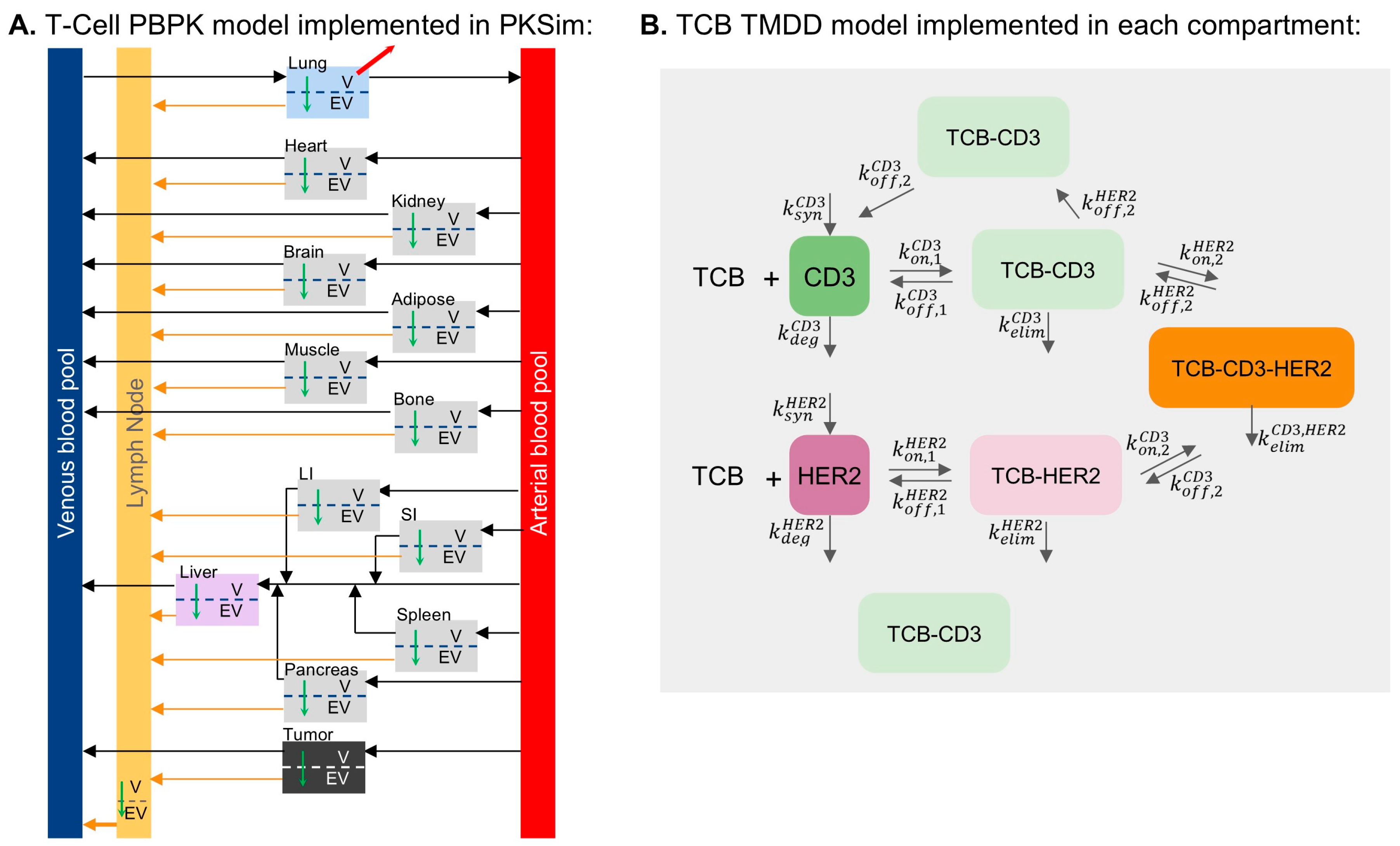
2.1. T-Cell and TCB PBPK Model Development
- Calibration of T-cell distribution parameters. This stage is described in Section 2.2.
- Calibration of parameters for the distribution of gD-only and gD-CD3 TCB. This stage is described in Section 2.3.
- Calibration of parameters for the distribution HER2-CD3 TCB. This stage is described in Section 2.3.
- The complete set of parameters are summarized in Supplementary Table S1.
2.2. Calibration of T-Cell Distribution
2.3. Calibration of gD/CD3 TCB and HER2/CD3 TCB
- Synapse internalization through trogocytosis was set to be slower (1/3) than internalization of TCB receptor complexes due to the assumed additional energy required to “strip” the receptor from the non-internalizing cell. Bigger differences in this factor led to little differentiation between the HER2-CD3L and HER2/CD3H scenarios.
- An avidity factor (×0.01) was introduced to account for increased binding affinity due to proximity between T-cells and tumor cells during synapse formation, enhancing the likelihood of receptor rebinding. This had a low impact on TCB PK but a high impact on synapse formation.
2.4. Model-Based Evaluations and Analyses
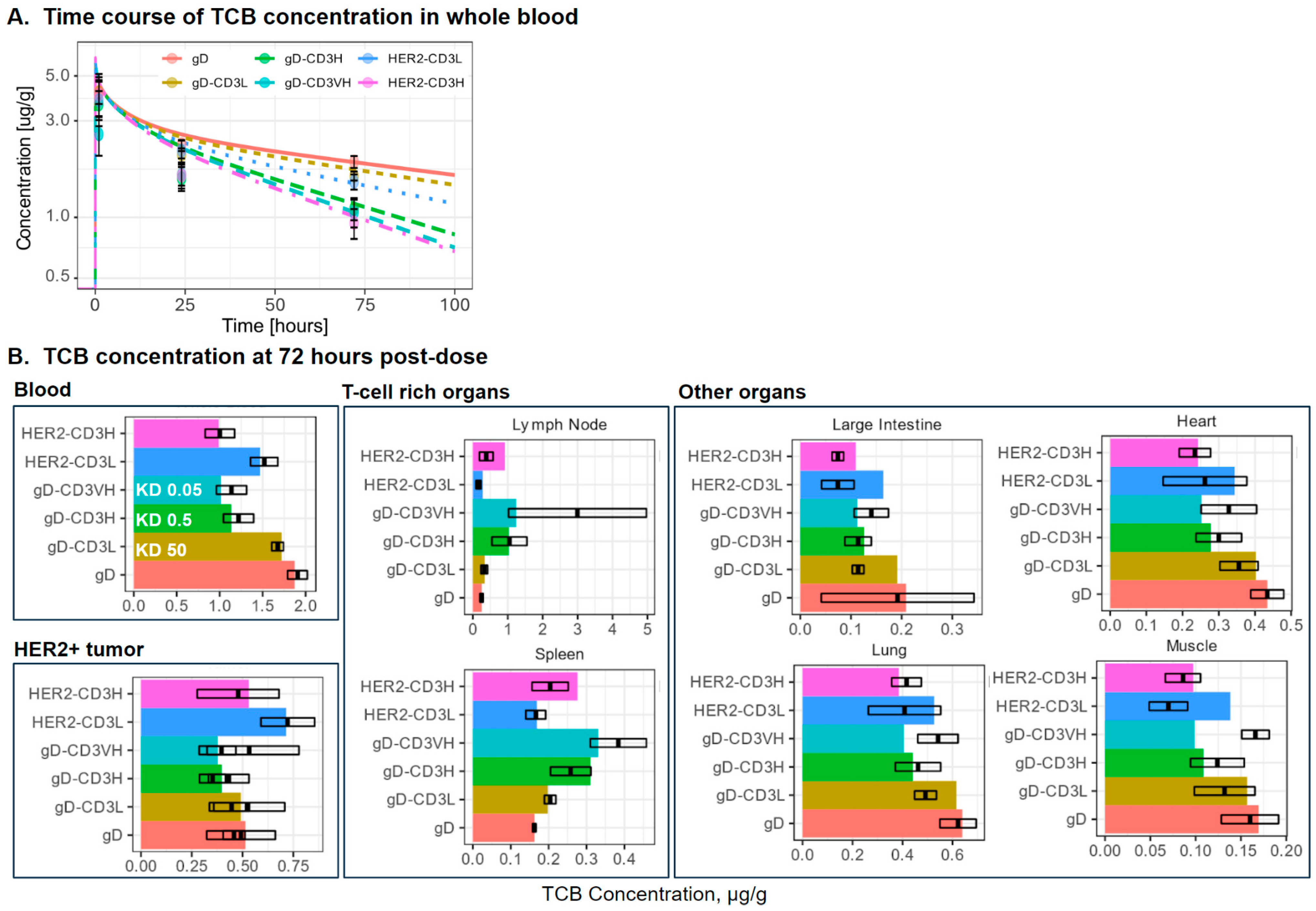
2.4.1. Evaluation of TCB Distribution
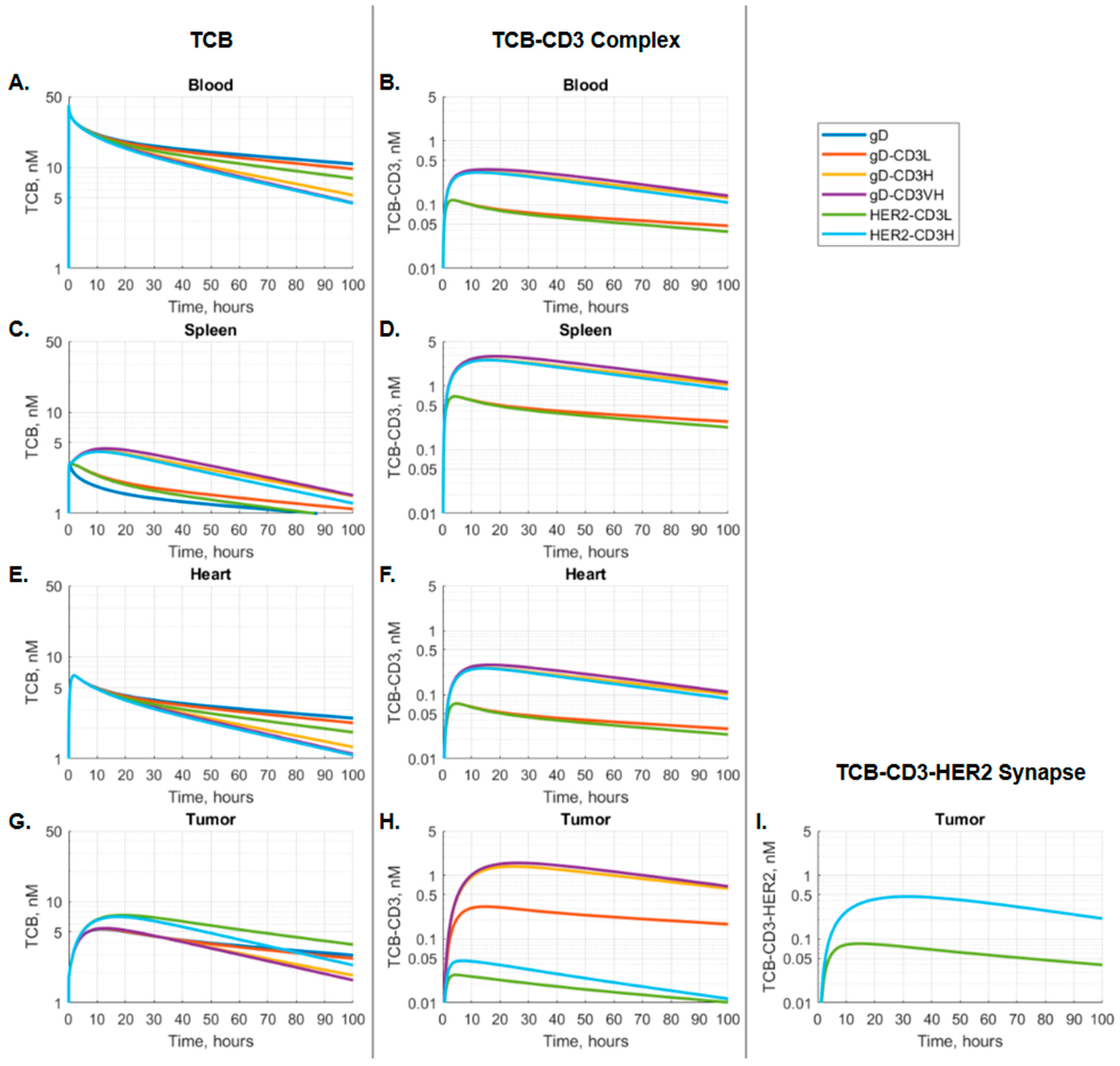
2.4.2. Model-Based Evaluation of Binding Kinetics and Synapse Formation
2.5. Software Used
3. Results
3.1. PBPK Model Simulations of Distribution of T-Cell, gD-CD3 TCB, and HER2-CD3 TCB
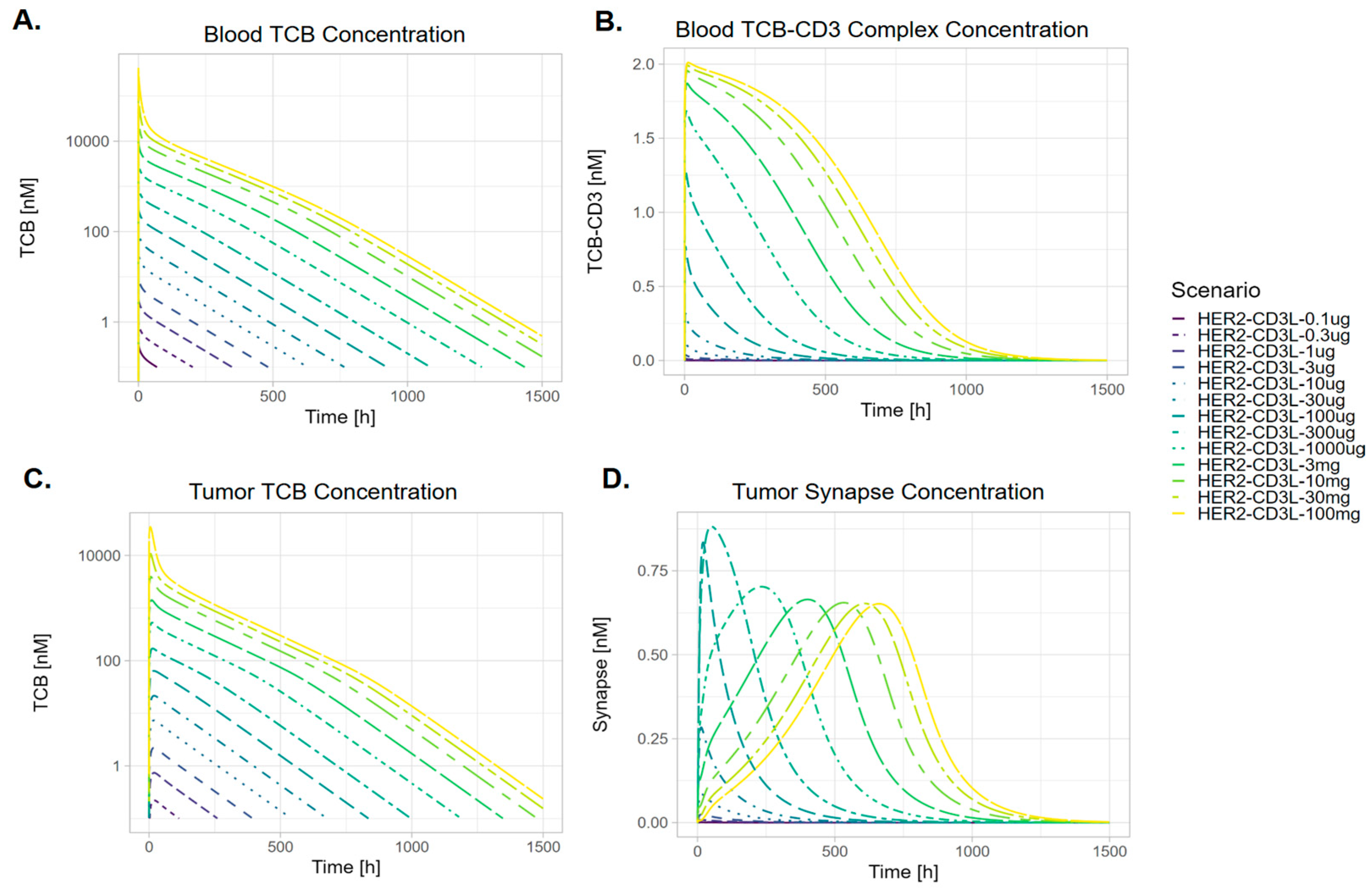
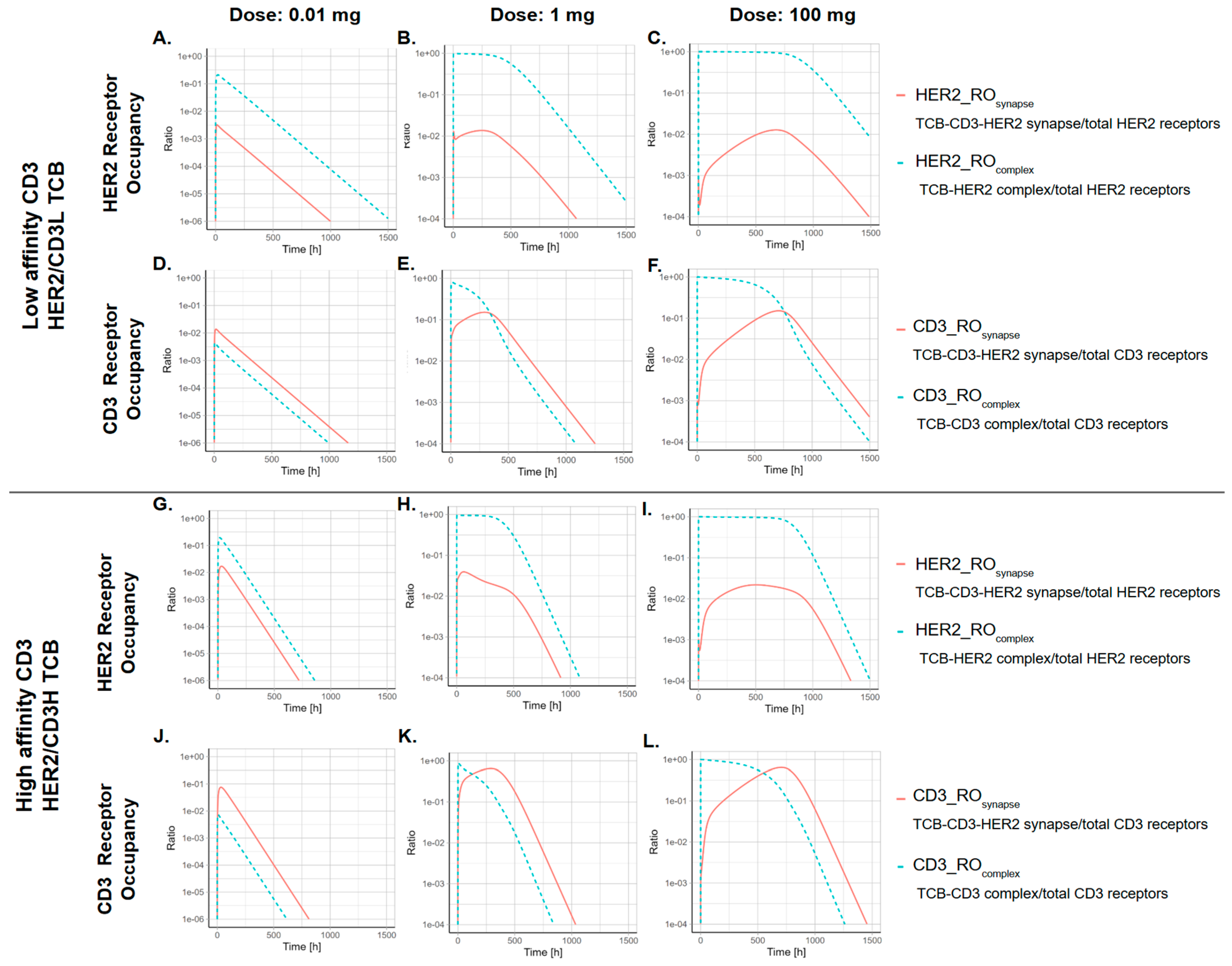
3.2. The Kinetics of TCB-CD3-HER2 Synapse Is Dose-Dependent
3.3. The CD3 Arm Affinity Influences the Kinetics of TCB-CD3-HER2 Synapse
3.4. The Kinetics of TCB-HER2-CD3 Synapse Is Dependent on the Ratio Between the Effector-to-Target Cell Ratio
4. Discussion
4.1. CD3 Affinity on Pharmacological Drivers of Efficacy vs. On-Target Safety
4.2. Blood PK Metric as Surrogate for Dose–Response Characterization
4.3. Tumor E–T Ratio
4.4. Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kassner, J.; Abdellatif, B.; Yamshon, S.; Monge, J.; Kaner, J. Current Landscape of CD3 Bispecific Antibodies in Hematologic Malignancies. Trends Cancer 2024, 10, 708–732. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Dees, S.; Grewal, I.S. Overcoming the Challenges Associated with CD3+ T-Cell Redirection in Cancer. Br. J. Cancer 2021, 124, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Arvedson, T.; Bailis, J.M.; Britten, C.D.; Klinger, M.; Nagorsen, D.; Coxon, A.; Egen, J.G.; Martin, F. Targeting Solid Tumors with Bispecific T Cell Engager Immune Therapy. Annu. Rev. Cancer Biol. 2022, 6, 17–34. [Google Scholar] [CrossRef]
- Zasedateleva, T.; Schaller, S.; de Lange, E.C.M.; de Witte, W.E.A. Local Depletion of Large Molecule Drugs due to Target Binding in Tissue Interstitial Space. CPT Pharmacomet. Syst. Pharmacol. 2024, 13, 2068–2086. [Google Scholar] [CrossRef]
- Warnders, F.J.; Waaijer, S.J.H.; Pool, M.; Lub-de Hooge, M.N.; Friedrich, M.; Terwisscha van Scheltinga, A.G.T.; Deegen, P.; Stienen, S.K.; Pieslor, P.C.; Cheung, H.K.; et al. Biodistribution and PET Imaging of Labeled Bispecific T Cell-Engaging Antibody Targeting EpCAM. J. Nucl. Med. 2016, 57, 812–817. [Google Scholar] [CrossRef]
- Crișan, G.; Moldovean-Cioroianu, N.S.; Timaru, D.-G.; Andrieș, G.; Căinap, C.; Chiș, V. Radiopharmaceuticals for PET and SPECT Imaging: A Literature Review over the Last Decade. Int. J. Mol. Sci. 2022, 23, 5023. [Google Scholar] [CrossRef]
- Manafi-Farid, R.; Ataeinia, B.; Ranjbar, S.; Jamshidi Araghi, Z.; Moradi, M.M.; Pirich, C.; Beheshti, M. ImmunoPET: Antibody-Based PET Imaging in Solid Tumors. Front. Med. 2022, 9, 916693. [Google Scholar] [CrossRef]
- Moek, K.L.; Waaijer, S.J.H.; Kok, I.C.; Suurs, F.V.; Brouwers, A.H.; Menke-van der Houven van Oordt, C.W.; Wind, T.T.; Gietema, J.A.; Schröder, C.P.; Mahesh, S.V.K.; et al. 89Zr-Labeled Bispecific T-Cell Engager AMG 211 PET Shows AMG 211 Accumulation in CD3-Rich Tissues and Clear, Heterogeneous Tumor Uptake. Clin. Cancer Res. 2019, 25, 3517–3527. [Google Scholar] [CrossRef] [PubMed]
- Mandikian, D.; Takahashi, N.; Lo, A.A.; Li, J.; Eastham-Anderson, J.; Slaga, D.; Ho, J.; Hristopoulos, M.; Clark, R.; Totpal, K.; et al. Relative Target Affinities of T-Cell-Dependent Bispecific Antibodies Determine Biodistribution in a Solid Tumor Mouse Model. Mol. Cancer Ther. 2018, 17, 776–785. [Google Scholar] [CrossRef]
- Suurs, F.V.; Lorenczewski, G.; Stienen, S.; Friedrich, M.; de Vries, E.G.E.; de Groot, D.J.A.; Lub-de Hooge, M.N. The Biodistribution of a CD3 and EpCAM Bispecific T-Cell Engager Is Driven by the CD3 Arm. J. Nucl. Med. 2020, 61, 1594–1601. [Google Scholar] [CrossRef]
- Müller, T.; Tasser, C.; Tesar, M.; Fucek, I.; Schniegler-Mattox, U.; Koch, J.; Ellwanger, K. Selection of Bispecific Antibodies with Optimal Developability Using FcRn-Ph-HPLC as an Optimized FcRn Affinity Chromatography Method. mAbs 2023, 15, 2245519. [Google Scholar] [CrossRef]
- Khot, A.; Matsueda, S.; Thomas, V.A.; Koya, R.C.; Shah, D.K. Measurement and Quantitative Characterization of Whole-Body Pharmacokinetics of Exogenously Administered T Cells in Mice. J. Pharmacol. Exp. Ther. 2019, 368, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Nikitich, A.; Helmlinger, G.; Peskov, K.; Bocharov, G. Mathematical Modeling of Endogenous and Exogenously Administered T Cell Recirculation in Mouse and Its Application to Pharmacokinetic Studies of Cell Therapies. Front. Immunol. 2024, 15, 1357706. [Google Scholar] [CrossRef]
- Zhang, X.; Lumen, A.; Wong, H.; Connarn, J.; Dutta, S.; Upreti, V.V. A Mechanistic Physiologically-Based Pharmacokinetic Platform Model to Guide Adult and Pediatric Intravenous and Subcutaneous Dosing for Bispecific T Cell Engagers. Clin. Pharmacol. Ther. 2024, 115, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, T.; Kim, M.-S.; Piatkov, K.; Wang, H.; Zhu, A.Z.X. Leveraging a Physiologically-Based Quantitative Translational Modeling Platform for Designing B Cell Maturation Antigen-Targeting Bispecific T Cell Engagers for Treatment of Multiple Myeloma. PLoS Comput. Biol. 2022, 18, e1009715. [Google Scholar] [CrossRef]
- Jiang, X.; Chen, X.; Jaiprasart, P.; Carpenter, T.J.; Zhou, R.; Wang, W. Development of a Minimal Physiologically-Based Pharmacokinetic/Pharmacodynamic Model to Characterize Target Cell Depletion and Cytokine Release for T Cell-Redirecting Bispecific Agents in Humans. Eur. J. Pharm. Sci. 2020, 146, 105260. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, I.; Gadkar, K.; Stefanich, E.; Li, C.-C.; Sun, L.L.; Chu, Y.-W.; Ramanujan, S. Mitigating the Risk of Cytokine Release Syndrome in a Phase I Trial of CD20/CD3 Bispecific Antibody Mosunetuzumab in NHL: Impact of Translational System Modeling. NPJ Syst. Biol. Appl. 2020, 6, 28. [Google Scholar] [CrossRef]
- Susilo, M.E.; Li, C.-C.; Gadkar, K.; Hernandez, G.; Huw, L.-Y.; Jin, J.Y.; Yin, S.; Wei, M.C.; Ramanujan, S.; Hosseini, I. Systems-Based Digital Twins to Help Characterize Clinical Dose-Response and Propose Predictive Biomarkers in a Phase I Study of Bispecific Antibody, Mosunetuzumab, in NHL. Clin. Transl. Sci. 2023, 16, 1134–1148. [Google Scholar] [CrossRef]
- Lippert, J.; Burghaus, R.; Edginton, A.; Frechen, S.; Karlsson, M.; Kovar, A.; Lehr, T.; Milligan, P.; Nock, V.; Ramusovic, S.; et al. Open Systems Pharmacology Community—An Open Access, Open Source, Open Science Approach to Modeling and Simulation in Pharmaceutical Sciences. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 878–882. [Google Scholar] [CrossRef]
- Open Systems Pharmacology. GitHub. Available online: https://github.com/Open-Systems-Pharmacology (accessed on 26 January 2025).
- Open Systems Pharmacology Community. Open Systems Pharmacology. Available online: http://www.open-systems-pharmacology.org (accessed on 26 January 2025).
- Niederalt, C.; Kuepfer, L.; Solodenko, J.; Eissing, T.; Siegmund, H.-U.; Block, M.; Willmann, S.; Lippert, J. A Generic Whole Body Physiologically Based Pharmacokinetic Model for Therapeutic Proteins in PK-Sim. J. Pharmacokinet. Pharmacodyn. 2018, 45, 235–257. [Google Scholar] [CrossRef]
- PK-Sim: PK-Sim® Is a Comprehensive Software Tool for Whole-Body Physiologically Based Pharmacokinetic Modeling. GitHub. Available online: https://github.com/Open-Systems-Pharmacology/PK-Sim (accessed on 26 January 2025).
- MoBi: MoBi® Is a Software Tool for Multiscale Physiological Modeling and Simulation. GitHub. Available online: https://github.com/Open-Systems-Pharmacology/MoBi (accessed on 26 January 2025).
- Shah, D.K.; Betts, A.M. Antibody Biodistribution Coefficients: Inferring Tissue Concentrations of Monoclonal Antibodies Based on the Plasma Concentrations in Several Preclinical Species and Human. mAbs 2013, 5, 297–305. [Google Scholar] [CrossRef]
- Mandikian, D.; Figueroa, I.; Oldendorp, A.; Rafidi, H.; Ulufatu, S.; Schweiger, M.G.; Couch, J.A.; Dybdal, N.; Joseph, S.B.; Prabhu, S.; et al. Tissue Physiology of Cynomolgus Monkeys: Cross-Species Comparison and Implications for Translational Pharmacology. AAPS J. 2018, 20, 107. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Melder, R.J.; Baxter, L.T.; Jain, R.K. Physiologically Based Kinetic Model of Effector Cell Biodistribution in Mammals: Implications for Adoptive Immunotherapy. Cancer Res. 1996, 56, 3771–3778. [Google Scholar] [PubMed]
- Schropp, J.; Khot, A.; Shah, D.K.; Koch, G. Target-Mediated Drug Disposition Model for Bispecific Antibodies: Properties, Approximation, and Optimal Dosing Strategy. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Wang, H.; Sove, R.J.; Jafarnejad, M.; Tsai, C.-H.; Wang, J.; Giragossian, C.; Popel, A.S. A Quantitative Systems Pharmacology Model of T Cell Engager Applied to Solid Tumor. AAPS J. 2020, 22, 85. [Google Scholar] [CrossRef]
- Martínez-Martín, N.; Fernández-Arenas, E.; Cemerski, S.; Delgado, P.; Turner, M.; Heuser, J.; Irvine, D.J.; Huang, B.; Bustelo, X.R.; Shaw, A.; et al. T Cell Receptor Internalization from the Immunological Synapse Is Mediated by TC21 and RhoG GTPase-Dependent Phagocytosis. Immunity 2011, 35, 208–222. [Google Scholar] [CrossRef]
- Rubin, R.H.; Baltimore, D.; Chen, B.K.; Wilkinson, R.A.; Fischman, A.J. In Vivo Tissue Distribution of CD4 Lymphocytes in Mice Determined by Radioimmunoscintigraphy with an 111In-Labeled Anti-CD4 Monoclonal Antibody. Proc. Natl. Acad. Sci. USA 1996, 93, 7460–7463. [Google Scholar] [CrossRef]
- Melder, R.J.; Munn, L.L.; Stoll, B.R.; Marecos, E.M.; Baxter, L.T.; Weissleder, R.; Jain, R.K. Systemic Distribution and Tumor Localization of Adoptively Transferred Lymphocytes in Mice: Comparison with Physiologically Based Pharmacokinetic Model. Neoplasia 2002, 4, 3–8. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Staflin, K.; Zuch de Zafra, C.L.; Schutt, L.K.; Clark, V.; Zhong, F.; Hristopoulos, M.; Clark, R.; Li, J.; Mathieu, M.; Chen, X.; et al. Target Arm Affinities Determine Preclinical Efficacy and Safety of Anti-HER2/CD3 Bispecific Antibody. JCI Insight 2020, 5, e133757. [Google Scholar] [CrossRef]
- Li, C.-C.; Bender, B.; Wilkins, J.; Li, F.; Turner, D.C.; Wang, B.; Deng, R.; Vadhavkar, S.; Li, Z.; Kwan, A.; et al. A Novel Step-up Dosage Regimen for Enhancing the Benefit-to-Risk Ratio of Mosunetuzumab in Relapsed or Refractory Follicular Lymphoma. Clin. Pharmacol. Ther. 2025, 117, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Djebli, N.; Morcos, P.N.; Jaminion, F.; Guerini, E.; Kratochwil, N.A.; Justies, N.; Schick, E.; Kwan, A.; Humphrey, K.; Lundberg, L.; et al. Population Pharmacokinetics and Exposure-Response Analyses for Glofitamab in Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma (R/R NHL): Confirmation of Efficacy and CRS Mitigation in Patients with Step-up Dosing. Blood 2020, 136 (Suppl. S1), 1–2. [Google Scholar] [CrossRef]
- Radtke, K.K.; Bender, B.C.; Li, Z.; Turner, D.C.; Roy, S.; Belousov, A.; Li, C.-C. Clinical Pharmacology of Cytokine Release Syndrome with T-Cell-Engaging Bispecific Antibodies: Current Insights and Drug Development Strategies. Clin. Cancer Res. 2025, 31, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Wu, L.S.; Lin, S.X.W.; Xu, Y.; Chen, Y.; Iwaki, Y.; Kobos, R.; Stephenson, T.; Kemmerer, K.; Uhlar, C.M.; et al. Population Pharmacokinetics and Exposure-Response with Teclistamab in Patients with Relapsed/Refractory Multiple Myeloma: Results from MajesTEC-1. Target Oncol. 2023, 18, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research, FDA. Multi-Disciplinary Review and Evaluation BLA 761291: Teclistamab 2021; Center for Drug Evaluation and Research, FDA: Silver Spring, MD, USA, 2021.
- Center for Drug Evaluation and Research, FDA. Multi-Disciplinary Review and Evaluation BLA 761345: Elranatamab 2022; Center for Drug Evaluation and Research, FDA: Silver Spring, MD, USA, 2022.
- Center for Drug Evaluation and Research, FDA. Multi-Disciplinary Review and Evaluation BLA 761263. Lunsumio (Mosunetuzumab); Center for Drug Evaluation and Research, FDA: Silver Spring, MD, USA, 2022.
- Center for Drug Evaluation and Research, FDA. Multi-Disciplinary Review and Evaluation BLA 761309. Glofitamab-gxbm (Columvi); Center for Drug Evaluation and Research, FDA: Silver Spring, MD, USA, 2022.
- Betts, A.; Haddish-Berhane, N.; Shah, D.K.; van der Graaf, P.H.; Barletta, F.; King, L.; Clark, T.; Kamperschroer, C.; Root, A.; Hooper, A.; et al. A Translational Quantitative Systems Pharmacology Model for CD3 Bispecific Molecules: Application to Quantify T Cell-Mediated Tumor Cell Killing by P-Cadherin LP DART®. AAPS J. 2019, 21, 66. [Google Scholar] [CrossRef]
- Liao, X.; Qi, T.; Zhou, J.; Liu, C.; Cao, Y. Optimizing Clinical Translation of Bispecific T-Cell Engagers through Context Unification with a Quantitative Systems Pharmacology Model. Clin. Pharmacol. Ther. 2024, 116, 415–425. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Wesche, H. T-Cell-Engaging Antibodies for the Treatment of Solid Tumors: Challenges and Opportunities. Curr. Opin. Oncol. 2022, 34, 552–558. [Google Scholar] [CrossRef]
- Ouyang, P.; Wang, L.; Wu, J.; Tian, Y.; Chen, C.; Li, D.; Yao, Z.; Chen, R.; Xiang, G.; Gong, J.; et al. Overcoming Cold Tumors: A Combination Strategy of Immune Checkpoint Inhibitors. Front. Immunol. 2024, 15, 1344272. [Google Scholar] [CrossRef]
- Lewis Phillips, G.; Guo, J.; Kiefer, J.R.; Proctor, W.; Yadav, D.B.; Dybdal, N.; Shen, B.-Q. Trastuzumab does not bind rat or mouse ErbB2/neu: Implications for selection of non-clinical safety models for trastuzumab-based therapeutics. Breast Cancer Res. Treat. 2022, 191, 303–317. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Susilo, M.E.; Schaller, S.; Jiménez-Franco, L.D.; Kulesza, A.; de Witte, W.E.A.; Chen, S.-C.; Boswell, C.A.; Mandikian, D.; Li, C.-C. Whole-Body Physiologically Based Pharmacokinetic Modeling Framework for Tissue Target Engagement of CD3 Bispecific Antibodies. Pharmaceutics 2025, 17, 500. https://doi.org/10.3390/pharmaceutics17040500
Susilo ME, Schaller S, Jiménez-Franco LD, Kulesza A, de Witte WEA, Chen S-C, Boswell CA, Mandikian D, Li C-C. Whole-Body Physiologically Based Pharmacokinetic Modeling Framework for Tissue Target Engagement of CD3 Bispecific Antibodies. Pharmaceutics. 2025; 17(4):500. https://doi.org/10.3390/pharmaceutics17040500
Chicago/Turabian StyleSusilo, Monica E., Stephan Schaller, Luis David Jiménez-Franco, Alexander Kulesza, Wilhelmus E. A. de Witte, Shang-Chiung Chen, C. Andrew Boswell, Danielle Mandikian, and Chi-Chung Li. 2025. "Whole-Body Physiologically Based Pharmacokinetic Modeling Framework for Tissue Target Engagement of CD3 Bispecific Antibodies" Pharmaceutics 17, no. 4: 500. https://doi.org/10.3390/pharmaceutics17040500
APA StyleSusilo, M. E., Schaller, S., Jiménez-Franco, L. D., Kulesza, A., de Witte, W. E. A., Chen, S.-C., Boswell, C. A., Mandikian, D., & Li, C.-C. (2025). Whole-Body Physiologically Based Pharmacokinetic Modeling Framework for Tissue Target Engagement of CD3 Bispecific Antibodies. Pharmaceutics, 17(4), 500. https://doi.org/10.3390/pharmaceutics17040500