Abstract
The capability of a given substance to change its spin state by the action of a stimulus, such as a change in temperature, is by itself a very challenging property. Its interest is increased by the potential applications and the need to find sustainable functional materials. 3D transition metal complexes, mainly with octahedral geometry, display this property when coordinated to particular sets of ligands. The prediction of this behavior has been attempted by many authors. It is, however, made very difficult because spin crossover (SCO), as it is called, occurs most often in the solid state, where besides complexes, counter ions, and solvents are also present in many cases. Intermolecular interactions definitely play a major role in SCO. In this review, we decided to analyze SCO in mono- and binuclear transition metal complexes containing halogens as ligands or as substituents of the ligands. The aim was to try and find trends in the properties which might be correlated to halogen substitution patterns. Besides a revision of the properties, we analyzed structures and other information. We also tried to build a simple model to run Density Functional Theory (DFT) calculations and calculate several parameters hoping to find correlations between calculated indices and SCO data. Although there are many experimental studies and single-crystal X-ray diffraction structures, there are only few examples with the F, Cl, Br and series. When their intermolecular interactions were not very different, T1/2 (temperature with 50% high spin and 50% low spin states) usually increased with the calculated ligand field parameter (Δoct) within a given family. A way to predict SCO remains elusive.
1. Introduction
When ligands bind to a metal to form an octahedral complex the five d degenerate levels of the free ion are split in two sets, the t2g, with lower energy, and the eg, with higher energy. Their occupation depends on the balance between the energy of these two sets (Δoct) and the electron pairing energy (P). While for 4d and 5d metal centers Δoct is in general much higher than P, the same does not happen with 3d derivatives. As a result, for d electron counts between 4 and 7, two occupation modes arise. In the low spin (LS), the pairing energy is smaller than Δoct and the electrons will pair in the t2g levels, before occupying eg. On the other hand, the inverse situation leads to high spin (HS) complexes, where electrons will half occupy the t2g and then the eg set, before starting to pair. Occupation of M-L σ* eg levels leads to a significant lengthening of the M-L bonds, to which the environment can adjust in variable ways. Not surprisingly, for some complexes, Δoct and P will be similar, so that the two electronic states (HS and LS) can be occupied and therefore octahedral complexes of 3d4 to 3d7 transition metals may switch from one to the other when an external stimulus, such as temperature, light, pressure or a magnetic field, are applied. Cambi and co-workers were the first to interpret this phenomenon, as a result of their observation of unusual magnetic properties of several Fe(III) dithiocarbamate complexes. They recognized that the change of temperature led to interconversion of spin states [1]. Although this discovery was reported in 1931, only in the last 25 years was there a huge increase in spin crossover (SCO) research, prompted by the increasing miniaturization of electronic devices and the approach of the superparamagnetic limit. This effort is concentrated in a very small number of transition metal centers, the most common being Fe(II) [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24] Fe(III) [11,12,13,14,15,18,23,25,26,27], and Co(II) [12,28,29]. The Jahn–Teller effect occurring in the HS state of Mn(III) contributes to the interest of this ion, even though it is much less common [30]. Fe(III) displays a higher redox stability than the previous ions [27], being thus a good candidate for applications in the production of SCO materials, an area increasingly more appealing in SCO research [31].
The concerted efforts of a large community of researchers working in this field helped to understand how very subtle changes affect this property. Several models inspired on the experimental accomplishments have been used to describe, explain and hopefully predict SCO in molecules. Theoretical models, such as the more popular Ising-like and the thermodynamic model, started to be developed to rationalize experiments and understand the workings of SCO. These models, however, reached their limits when the size of the systems increased significantly (e.g., nanoparticles). This subject has been addressed by several authors and has recently been reviewed by Borshch [32], by Gudyma, Maksymov and Bobák [33], and by Pavlik and Linares [34].
Although the SCO phenomenon is molecule based, when it is observed in the solid state there are no isolated molecules, but instead well packed molecules in the crystal lattice. The components of this lattice, such as solvates, host molecules, and counter ions, when present, are very important to control the nature of the interactions between the metal centers, and the degree of communication between them. The nature and structure of the ligands is probably the most important factor when it comes to promote interactions between metal centers. The ability to establish intermolecular interactions is almost as important as having the right ligand-field strength to tune the relationship between Δoct and P, providing the right coordination environment. The strongest intermolecular interaction will be electrostatic when the solids are ionic. Hydrogen bonds, ranging from the classic ones, involving the more electronegative elements, to the weak ones, where C-H is a ubiquitous donor, may occur between ligands and host molecules or solvates. They will be charge assisted, and stronger in ionic solids. Specific groups may enhance π-π and C-H⋅⋅⋅π interactions [35], and the presence of halogens may even give rise to halogen bonds [36]. Even though some of these interactions may appear to be very weak, cooperativity effects in the solid may give them relevant roles. It has been shown that inclusion of a cationic Fe(III) complex in a halogen-bonded supramolecular network influences the spin crossover [37], but these effects are still starting to be studied. Although not always possible, it should be desirable to obtain non-solvated compounds, since the formation of unstable lattices that can lose their solvates might cause irreversible changes in the structure and therefore in the magnetic behavior of the compound. The large number of relevant factors contributes to a variety of SCO patterns (Figure 1), from left to right and top to bottom: abrupt, gradual, two stepped, with hysteresis, and incomplete. The temperature at which the number of LS and HS states is the same is called T1/2 (shown in top center).
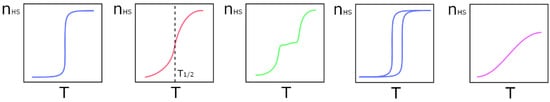
Figure 1.
Different types of spin crossover curves: Left to right—abrupt and complete, gradual and complete, stepped and complete, abrupt and complete with hysteresis, and gradual and incomplete.
When the aim is to analyze the behavior of isolated molecules, SCO has been either studied in solution [38,39,40,41,42,43] or by depositing them on supports [44,45,46,47,48,49]. In solution, the distribution of spin states is assigned using a Boltzmann distribution, and a spin equilibrium is expected at different temperatures. However, depending on the nature of the ligand used, interactions between molecules may occur and SCO systems with different degrees of communication between metal centers have sometimes been reported [50,51,52,53,54,55]. The molecules deposited on supports have been subjected to electric fields to produce systems that change their spin state by an application of a more suitable stimulus towards application into spintronics [47,48,49,56,57,58,59,60].
SCO complexes have for some time been considered as promising precursors for the next generation of data storage materials [61,62,63,64]. This application was seen as the most important and relevant in the usage of SCO materials in spintronic devices. However, this proved to be challenging in the short to medium term, and molecular and materials scientists have been moving their focus towards the development of sensor and actuators with SCO molecules and materials [64,65].
Quantum chemical methods, namely DFT calculations [66], have been used in order to shed more light on the SCO phenomenon. As mentioned above, HS octahedral complexes display longer interatomic distances between the metal center and the ligating atom, owing to the higher electron population in M-L anti-bonding (eg*) orbitals. The SCO process will require a single to multiple electron deexcitation into the essentially non-bonding t2g molecular orbitals (MOs). Such a process can occur in steps, HS→IS→LS, whereby an intermediate spin state (IS) is formed, but may be too short-lived to be detected by experimental techniques, or via concurrent electron deexcitation. The pathway will be determined by the energetics of the hypersurface spin-state crossings and the magnitude of the spin-orbit coupling. Potential energy surfaces between differing spin states are orthogonal with a non-relativistic description, but with spin-orbit coupling the spin states can admix and the complex may acquire a different magnetic moment. Indeed, this phenomenon is also recognizably present in chemical changes known as ‘spin forbidden reactions’ [67].
Computational studies may pursue thermodynamic or kinetic data. The LS-HS energy gap is typically the most sought-after thermodynamic result from these calculations, to try and rank the SCO ability of each species. It can be obtained from a single-point energy evaluation of both spin states performed on the crystal structure coordinates, which will yield values in the Franck–Condon region, or, alternatively, geometry optimizations may be carried out for both spin states yielding (lower) energy differences between the adiabatic surface minima.
A crucial quantity to describe the kinetics of hypersurface crossing is the minimum energy crossing point (MECP), defined as the minimum energy that allows the two potential surfaces to cross. For example, in a d5 complex the two possible options would be a sextet HS and a doublet LS. The SCO pathways can be either 6HS→4IS→2LS or 6HS→2LS, as depicted in Figure 2. In the former case there would be two MECPs, one for the sextet to quartet crossing and another for the quartet to doublet crossing. In the latter possibility only one MECP is required. Both pathways may indeed compete, should all MECPs be similar in energy. If, however, the MECPs for the intermediate spin states have higher energy than the MECP between the HS and LS states, the SCO would be resolved into a synchronous double (de-)excitation and no IS state would be detected.
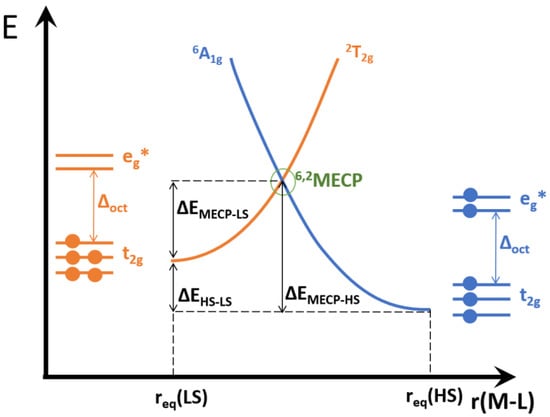
Figure 2.
A scheme illustrating a minimum energy crossing point between two d5 spin-state potential energy surfaces, and highlighting the relevant energy differences. The relative height of the two surface minima (6A1g and 2T2g) may interchange.
In this review, we aimed at searching SCO complexes with halogens and similar substituents in the ligands. One advantage of halogens is their facility to participate in organic reactions and therefore modify existing groups transforming them in other functional groups, which in turn will change the electronic and molecular structure of the initial complexes, as well as their solid-state packing. On the other hand, they can promote halogen-bonding interactions, which add to other relevant interactions for cooperativity and act on the SCO behavior, and to be applied in crystal engineering. The octahedral complexes from d4 to d7 metal centers bearing such substituents will be addressed in the order of increasing d count, starting with systems in the solid state and dealing with the few examples occurring in solution at the end. Only mono- and binuclear species will be considered, since polymeric solids will present very different challenges. We believe that development of efficient functional materials will significantly contribute to sustainability.
We performed DFT calculations (B3LYP*/def2-SV(P) level, see also Section 4: Computational Studies and 6: Computational Methods) to complement this review, whenever experimental structural data are available especially for families of complexes. The energies of the HS and LS states, as well as the MECPs (Figure 2), were calculated for a selection of mononuclear complexes to try to establish a trend in the thermodynamics and kinetics of the SCO phenomenon. The calculations can be based on the reported single-crystal X-ray structures, which include intermolecular effects, and then compared with those resulting from geometry optimization of a “cut” from the structure, such as the neutral complex, the cation-anion pair, or the previous and one explicit solvent molecule. They would show us how reliable the latter approach might be in the absence of experimental structural data. We tried to put together a large number of data in order to derive reliable trends.
2. Results. Solid State.
2.1. d4 Complexes, HS (S = 2) and LS (S = 1)—Mn(III)
There are not many examples of SCO for d4 complexes, the only examples known containing Mn(III). The HS form of these octahedral complexes exhibits a Jahn–Teller distortion.
Wang, Ferbinteanu, Huang and co-workers [68] synthesized [Mn(5-Br-sal-N-1,5,8,12)]ClO4, a Mn(III) complex of the hexadentate ligand L1=5-Br-sal-N-1,5,8,12, N1-2-hydroxybenzylideneaminopropylaminoethyl-N3-2-hydroxybenzylidenepropane-1,3-diamine (Scheme 1) and measured its magnetic properties. Unsolvated crystals were obtained from a mixture of methanol and ethanol. Single-crystal X-ray crystallography was performed at different temperatures and showed that the asymmetric unit contained two independent manganese(III) complex cations (Mn1 and Mn2). An interesting feature of this system was that only one of the [Mn(5-Br-sal-N-1,5,8,12)]+ units displayed a spin transition, in such a way that at 100 K one unit was in the HS state (Mn1) and the other in the LS state (Mn2). Magnetic studies agreed with these structural findings. Computational calculations gave some insight into the spin-conversion mechanism, by analyzing the energy and relating it to ligand-field strength. A key element of the process is the higher strain associated with the coordinated ligand in the LS form. It was proposed that its mechanical relaxation initiates the LS-HS process, as thermal movements increases. The unusual compressed octahedral geometry, resulting from the Jahn–Teller effect in the HS form, affects the hexadentate encapsulating ligand and favors the spin conversion. Hydrogen bonds with the perchlorate are present, but the introduction of the bromo-substituent on the ligand did not give rise to any special property changes, despite short Br⋅⋅⋅O contacts in the crystal structure.
Scheme 1.
Ligands L1-L3 (protonated form).
In Figure 3 we show our optimized geometries of the cationic complexes in the LS and the HS forms in similar views (the optimization was performed in a single [Mn(5-Br-sal-N-1,5,8,12)]ClO4 unit, but the anion is not shown). The LS species is much closer to a perfect octahedron, the Mn-Namine (2.081, 2.049 Å) bonds being only marginally longer than the Mn-Namide ones (2.015, 1.988 Å). Mn-O bonds are shorter. The angles are in the range ~173–178° and ~85–92°. In the HS form there is one eg* electron, so that bond lengthening and Jahn–Teller effect are expected. Indeed, the four Mn-N bonds increase significantly to 2.228, 2.323 Å (Mn-Namine) and 2.123, 2.135 Å (Mn-Nimine), while the Mn-O bonds barely change. The angles vary between ~155–175° and 81–123°. These values confirm the proposal of the authors that there is an unusual compression distortion.
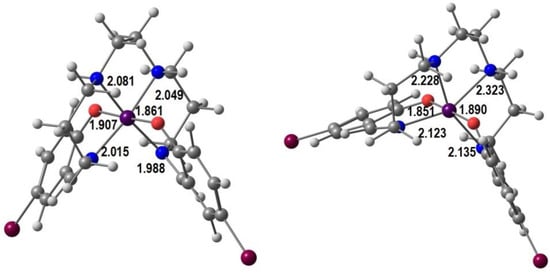
Figure 3.
B3LYP*/def2-SV(P) DFT optimized LS (left) and HS (right) structures of [Mn(5-Br-sal-N-1,5,8,12)]+ with bond distances (Å).
Morgan and co-workers examined in detail the effect of the anion in salts of the cationic complex [Mn(3,5-Br2-sal-N-1,5,8,12)]+ (L2 = 3,5-Br2-sal-N-1,5,8,12) [69], having isolated, as crystals, complexes with several anions in different conditions. None of these salts showed a complete LS to HS transition by 300 K, reflecting the stabilization of the rare S = 1 state in this ligand environment. The hexafluorophosphate salt [MnL2]PF6.0.5CH3OH (2a) remains in the LS electronic configuration between 10–300 K, while the other five salts, [MnL2]NO3·C2H5OH (2b), [MnL2]BF4·C2H5OH (2c), [MnL2]CF3SO3·C2H5OH (2d), [MnL2]ClO4·C2H5OH (2e) and [MnL2]ClO4·0.5CH3CN (2f), show gradual and incomplete SCO behavior. The authors could not find a clear relationship between the SCO behavior and the intermolecular interactions in the different salts and solvates. Indeed, the LS salt [MnL2]PF6.0.5CH3OH does not exhibit strong hydrogen bonding in the crystal structure (only weak NH⋅⋅⋅F). In 2b and 2d at 100 K (LS form) strong NH⋅⋅⋅O bonds, between the cations and the nitrate or triflate, are responsible for the formation of 1D hydrogen bonded chains, which become weaker or break on warming (293 K). The BF4− in 2c forms at 100 K weak hydrogen bonds with the co-crystallized solvent C2H5OH, which weaken upon warming. The two perchlorate salts, obtained from ethanol and acetonitrile, exhibit different hydrogen-bond patterns. At 100 K [MnL2]ClO4·C2H5OH (2e) displays solvent-anion bonds not involving the cation. This is the salt with the highest conversion to HS, already visible at 100 K. [MnL2]ClO4·0.5CH3CN (2f), however, shows NH⋅⋅⋅O cation-anion bonds, similar to those of 2a. These detailed studies emphasize the difficulties associated with an understanding of SCO. The authors did not conclude about any contribution of the halogens to the SCO. We performed calculations on some of these compounds (Table 1).

Table 1.
Calculated energy differences (kcal mol−1) between HS and LS states of several salts and solvates of complexes [Mn(3,5-Br2-sal-N-1,5,8,12)]+ (2) in the X-ray structure and in the optimized (opt) geometries, between MECP and LS and HS, Δoct (cm−1) and T1/2 (K).
In these complexes the halogen is always Br, but the N-H⋅⋅⋅A interactions between the cation and the anions A and/or solvent vary. The calculations show clearly that all the parameters depend on the anion and, for the same anion, on the solvent. The experimental T1/2 does not follow any of the energy differences (ΔEHS-LS or ΔEMECP-L/HS) but there is a very good correlation with Δoct. The species with the highest Δoct does not undergo SCO, showing that the solvent may be determining.
Morgan et al. extended their work [70] and that of Ferbinteanu and Huang [68] to other halogenated ligands (L1, L2, Scheme 1), synthesizing the NO3−, ClO4− or PF6− salts of the cationic complexes [Mn(3,5-Cl2-sal-N-1,5,8,12)]+, [Mn(5-Cl-sal-N-1,5,8,12)]+ and [Mn(5-Br-sal-N-1,5,8,12)]+. MeOH and EtOH solvated crystals were obtained for the first complex and unsolvated solids for the other two. They found three distinct types of behavior for the three cations: (1) the salts of 3,5-diBr complexes were LS, with a gradual SCO starting at room temperature; (2) the salts with 5-Cl displayed stepped and incomplete SCO; and (3) the 5-Br salts were HS. The interpretation of the magnetic behavior was sought in a careful examination of the intermolecular interactions in the packing diagrams of all compounds. In the 5-Br/Cl complexes one NH⋅⋅⋅O bond formed in the LS site, but it disappeared in the HS site contradicting the previous observations (5-Br complex) [68]. Both showed π-π stacking. The crystal structure of 3,5-Cl2 complexes contained solvates which participated in packing interactions (hydrogen bonds). It was found that in general, in the absence of hydrogen bonding, the compounds were more likely to remain in the HS state. However, solvated complexes tend to be more stable in the LS state. The role of the different halogens was not considered.
Collet, Morgan et al. also isolated [71] the NTf2− salt of the [Mn(3,5-Cl2-sal-N-1,5,8,12)]+ cation (L2 ligand), which undergoes two structural phase transitions along an incommensurate low-temperature structure. According to the magnetic studies, the spin-state changes gradually down to 210 K and plateaus between 210 and 160 K, which suggest in this intermediate phase a HS:LS ratio close to 1:1. Further cooling below 160 K leads to an abrupt (over a 7 K range) collapse in the magnetic moment, which continues to 10 K. A 14 K hysteresis window is also seen when warming at 5 K/min, with an abrupt transition between the low-temperature and intermediate phases. The authors emphasized the influence of structural order/disorder transitions involving counter ion or ligand on the interchanging spin rates of these complex cations. The electronic ordering in the intermediate phase depends on the coupling between electronic and structural states, the molecular size and the nature of the anion layer playing a large role. Single spin states (HS or LS) are favored when cations are coupled within a cationic layer and the anion layers prevent interactions between the cations belonging to different layers. This anisotropic coupling favors layered structures and highlights the role of the anion in the formation of the striped HS-LS structure. The NTf2− anions are disordered in the crystal structure.
Harding and co-workers reported [72] the OTf− salts of two Mn(III) [Mn(L3)2]+ complexes containing the 5-X–N-(8-quinolyl)salicylaldimine ligands (Hqsal-X, L3, X = Cl and Br). Crystals of these complexes could be isolated from the oxidation of Mn(II) but were very sensitive to degradation. Their magnetic profiles show, for both Mn(III) complexes, SCO with spin state changing gradually over the temperature measured. Crystals of the reduced neutral Mn(II) compound were obtained. Analysis of the crystal packing revealed strong supramolecular interactions. It was clear that, despite the halogen involvement in supramolecular interactions (CH⋅⋅⋅X), the crystal packing did not significantly alter by changing it. Computational studies showed a HS–LS gap in the Mn(III) complexes of −0.36 and −0.37 kcal mol−1, reflecting the spin crossover observed in these systems. The HOMO and LUMO orbitals of the two Mn(III) complexes were found to be very similar. Computational studies suggested that the geometry of the ligand does not prevent the possibility of SCO, contrary to what was found for the Morgan systems [73,74].
2.2. d5 Complexes, HS (S = 5/2) and LS (S = 1/2)—Fe(III)
Brewer and co-workers [75] described the formation of Fe(III) complexes with tetradentate and pentadentate Schiff-base ligands derived from salen and their following reaction with a nickel(II) imidazolate complex containing the ligand 5-{o-[(5-chloro-2-hydroxyphenyl)phenylmethyleneamino]-phenyliminomethyl}imidazole) (L4, Scheme 2), to yield three imidazolate-bridged heterodinuclear complexes. In one of them, a chloride anion was coordinated to Fe(III). Techniques such as variable-temperature magnetic susceptibility, Electron Paramagnetic Resonance (EPR) and Mössbauer spectroscopy were used to characterize the octahedral Fe(III) compounds. Magnetic susceptibility data showed gradual spin crossover with magnetic moments varying from 3.5 μB at 80 K to 4.5 μB at 298 K for the chloride compound. The authors noted that related complexes with a N3O3 donor set were in general HS, so that it was interesting to observe that the present compound exhibited SCO. This behavior was assigned to the greater donor capability of the anionic imidazolate ligand in the Ni(II) coordination sphere, compared with that of neutral imidazole ligands. Mössbauer and EPR spectroscopy results were in agreement with the gradual nature of the spin crossover.
Scheme 2.
Ligands L4–L12 (protonated form).
Spiccia et al. prepared [76] a series of chromium(III), manganese(II) and iron(III) complexes of dmptacn [1,4-bis(2-pyridylmethyl)-1,4,7-triazacyclononane] (L5, Scheme 2), by reaction of the macrocycle with the corresponding metal chloride salts. Their magnetic properties were examined by magnetic susceptibility, EPR and Mössbauer measurements. The Fe(III) compound displayed its spin lability with SCO between 300 and 180 K. The magnetic profile showed that at 295 K the magnetic susceptibility was 3.58 μB, indicating that the transition is incomplete at low temperatures. The analysis of the spectroscopic data confirmed the previous results.
Real and co-workers [77] synthesized three Fe(III) compounds of the ClO4− salt of the N,N′-4-chloro-o-phenylenebis(3-methoxysalicylideneimine) ligand (L6, Scheme 2) (one hydrated, 6a, one anhydrous, 6b, and another a cationic sodium salt, 6c) and a ClO4− Fe(III) compound (7) of the N,N′-4-chloro-o-phenylenebis(3-ethoxysalicylideneimine) (L7, Scheme 2). The authors found that the hydrated sample 6a shows SCO, the anhydrous 6b is HS and the cationic sample 6c is LS, while the latter (7) is high-spin over the temperature studied. The SCO compound 6a has a T1/2 = 202 K. The model of Slichter and Drickamer was applied to estimate thermodynamic parameters (∆H = 12 kJ mol−1) and the intermolecular interaction parameter Γ = 1.6 kJ mol−1, and ∆S = 60 J mol−1 K−1. The authors noticed that the hydrated compound easily lost the crystallization water molecule affording the HS anhydrous species 6b, either at T > 345 K or in vacuum at room temperature. They studied the influence of the degree of dehydration on the magnetic behavior, and found that the spin conversion was gradually more incomplete and finally vanished as the water loss process ended. The compound could also be completely dehydrated if it was kept for at least two hours under vacuum. The hydration-dehydration process was fully reversible and no aging of the process was detected. This HS species can be reversibly converted in the LS sodium salt 6c. The preference for the LS state in this cation is explained by the role of the imidazole ligands bound to Fe(III) and acting as π acceptors, but constrained to a specific position by binding also to Na(I). The change between 6a and 6b results from the loss of hydrogen bonds, following the release of water in the crystal and preventing cooperativity effects (Scheme 3).
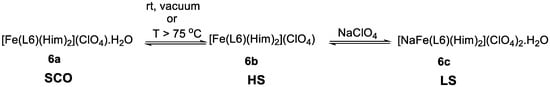
Scheme 3.
Interplay between the three Fe(III) complexes, 6a, 6b, and 6c, with ligand L6.
Boillot and co-workers [78] investigated by SQUID and Mössbauer measurements the magnetic properties of a monohydrated Li salt of an anionic ferric complex [Fe(5Brthsa)2]− with the ligand (H2-5Brthsa = 5-bromosalicylaldehyde thiosemicarbazone) (L8, X = Br, Scheme 2). The authors reported a spin transition with an asymmetric hysteresis. An abrupt transition was observed on heating, but gradual crossover was seen on cooling, without reaching a complete HS state at 380 K. Dyatlova et al. [79] previously reported the anhydrous compound which exhibited a gradual SCO process without any hysteresis, largely depending on the thermal treatment and the extent of air exposure of the sample. It was also noticed that the water of crystallization helps to stabilize the LS of Fe(III) ion in this series of compounds [80]. The authors associated the cooperative character of the spin transition to a crystallographic first-order phase transition. The effect of the water of crystallization on the hydrogen-bond network involving also Fe(III) complexes might explain this phase transition, but no crystal structure could be obtained for the monohydrated species.
In a following work, Floquet, Boillot and co-workers [81] determined the structure of the same monohydrated Li salt of the anionic ferric complex with the ligand (H2-5Brthsa = 5-bromosalicylaldehyde thiosemicarbazone, L8, X = Br) by powder X-ray diffraction at several temperatures. This work aimed at understanding the results of the previous study, namely by obtaining the molecular structure and analyzing the packing in the solid state to interpret the phase transition and the origin of the large asymmetric hysteretic loop in the SCO. The authors concluded that the SCO of the compound is simultaneous with a first-order crystallographic phase transition and that no space group modification occurs during the process. The HS form of the anionic complex exhibits no π-π stacking, but hydrogen bonds, possibly between water OH donors and N acceptors are observed, as well as short contacts involving Br. Besides the bond shortening and unit cell contraction, the LS form packs with different motifs and strong hydrogen bonds (NH⋅⋅⋅O and OH⋅⋅⋅N), keeping the short N⋅⋅⋅Br contacts. The drastic change in packing and the phase transition were considered responsible for the SCO behavior.
Sato, Dunbar and co-workers [82] reported the synthesis of a neutral mononuclear Fe(III) complex [Fe(H-5-Cl-thsa-Me)(5-Cl-thsa-Me)]·H2O, where H2-5-Cl-thsa-Me = 5-chlorosalicylaldehyde methylthiosemicarbazone (L8, X = Cl), and the characterization by powder/single-crystal X-ray diffraction, Mössbauer spectroscopy, differential scanning calorimetry (DSC), and magnetic susceptibility measurements. The single-crystal X-ray analysis showed Namide–H⋯O hydrogen bonds, but did not reveal any participation of the chloro-substituent in the intermolecular interaction. Magnetic measurements reflected a compound in the HS state at room temperature. Upon decreasing the temperature, χMT remains nearly constant down to 275 K after which undergoes a two-step transition. The transition temperatures of each step were calculated as δ(χMT)/δT; T1/2(↓) to be 270 K and 245 K in the cooling mode and T1/2(↑) to be 249 K and 278 K in the heating mode. The hysteresis widths are 4 and 8 K for the two-step spin transition.
Krupska et al. reported [83] the EPR studies of the related spin-crossover compound [2-methyl-5-ethyl-pyridinium][5-chloro-salicylalthiosemicarbazonatoferrate(III)] (L8, X = Cl) under hydrostatic pressure up to 500 MPa in a temperature range of 80-310 K. The authors found that LS complexes were organized in restricted domains of the crystal lattice, instead of being randomly distributed, based on their exchange interactions. This fact is in agreement with previous models rationalizing the spin-crossover phenomenon. They were also able to measure the growth of these domains by direct observation. The HS→LS transition was driven by the application of hydrostatic pressure through a certain threshold resulting in a large pressure-conditioned hysteresis of 110 MPa resulting in claiming a new type of bistable system.
The interest of Murray et al. [84] for halogen effects on SCO resulted in a series of heteroleptic neutral complexes with the formula [Fe(qsal-X)(thsa)]∙nMeCN, where qsal-X− = X-substituted quinolylsalicylaldimine (L3), thsa2− = thiosemicarbazone-salicylaldiminate, X = F, Cl, Br and I (L8). We show in Figure 4 our DFT optimized structures of the HS and LS forms of the Cl derivative of qsal and thsa, in order to emphasize the trends in bond distances in Fe(III) neutral complexes with two tridentate ligands. All distances increase in the HS form: Fe-S by 0.13 Å, Fe-O by 0.05 and 0.06 Å, and Fe-N by 0.19, 0.24, and 0.20 Å, as expected. The HS structure is more distorted, as is apparent from comparison between the two species. For instance, the S-Fe-O angle varies from 174° in the LS to 160° in the HS.
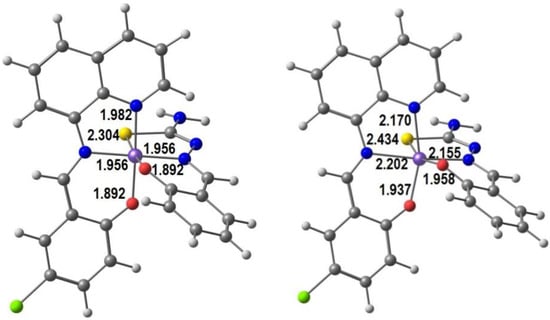
Figure 4.
DFT calculated LS and HS structures of [Fe(qsal-Cl)(thsa)] with bond distances (Å).
The same authors reported that magnetic studies on solid samples of the F, Cl and Br compounds show incomplete SCO, which can be related to MeCN solvent loss. The solvated I complex, on the other hand, remains fully LS up to 360 K. Single crystals of samples exhibiting different degrees of solvation were examined at several temperatures. The authors found only three different structures for the four halogen-substituted derivatives, since the Cl and Br compounds are isostructural. In this family, C-X∙∙∙H interactions involving F, Cl and Br substituents are observed, though the details are different between F and Cl/Br. C-X∙∙∙π interactions, however, are only observed for the I substituent. It should be noted that, although these halogen interactions are weak, they are very important, because they contribute significantly to the overall packing of the structures and consequently to their physical properties. Additionally, the magnetic studies show the spin-crossover temperatures and stabilization of LS states grow from X = F to I, so that it was suggested that the ligand-field strength also increases from F to I. This trend in LS stabilization applies also to the desolvated forms, despite their different crystal packing. It thus appears that the increase in ligand-field strength in this class of compounds [Fe(qsal-X)(thsa)] from X = F ˂ Cl ˂ Br ˂ I results mainly from the electronic structure around the metal center. The SCO behavior seems to be more influenced by the intrinsic properties of the metal center than by crystal packing. B3LYP*/def2-SV(P)DFT calculations were carried out to analyze this aspect (Table 2).
Table 2.
Calculated energy differences (kcal mol−1) between HS and LS states in the optimized (opt) geometries of complexes [Fe(qsalCl)(thsa)], between MECP and LS and HS, Δoct (cm−1) and T1/2 (K).
The calculations indicate an increase in ligand field (Δoct) from F to I, a result in agreement with experiment. Indeed, a parallel increase in T1/2 is observed upon going from F to Cl and Br, until the point that no SCO is observed for the iodine-substituted compound. It can be also noticed that although there is a clear trend on the Δoct values, no correlation is observed with the energy difference between spin states (ΔEHS-LS) or with the kinetic barrier for the spin transition (ΔEMECP-LS/HS).
Sato et al. [85] reported an Fe(III) complex, [FeIII(H-5-Br-thsa)(5-Br-thsa)].H2O (thsa2− = thiosemicarbazone-salicylaldiminate, L8, X = Br), with a novel neutral six-/five-step spin crossover, accompanied by a two-step reversible crystallographic symmetry breaking. DFT calculations aiming at a better understanding of the potential mechanism of the multi-step SCO complemented the experimental work. At 300 K, the χMT value is 4.20 cm3Kmol−1, which is similar to the expected value of 4.375 cm3Kmol−1 of the HS state of FeIII. On further cooling from 300 K to 5 K, the χMT values decreased stepwise from 4.20 cm3Kmol−1 to 0.44 cm3Kmol−1, indicating a complete HS to LS transition. In the cooling mode the first transition step was centered at T1 = 242 K and showed an abrupt fall to a short plateau with χMT value of around 3.50 cm3Kmol−1, characteristic of a 19% spin-state conversion of the Fe(III) centers. The following second to fifth transition steps are relatively gradual transitions centered at 238 K (T2), 211 K (T3), 194 K (T4), and 170 K (T5), respectively, and reflect successive spin transitions occurring in the intermediate states. The sixth conversion step, centered at 144 K (T6), shows an abrupt drop to a plateau with χMT value ~ 0.44 cm3Kmol−1, indicating that complete spin-state conversion was achieved. Upon heating, a hysteresis loop of around 7 K is observed, with five-step transitions centered at 148 K (T1), 173 K (T2), 194 K (T3), 212 K (T4), and 245 K (T5). No special role of the halogen was assigned to this peculiar SCO profile.
Floquet and co-workers [86] reported three neutral thiosemicarbazone Fe(III) complexes [Fe(H5X-thsa)(5X-thsa)].H2O (L8, X = H, Cl and Br) as well as their magnetic susceptibility, DSC and Mössbauer measurements. SCO and small hysteresis loops were observed for the three compounds. The thermal spin transition of the chloro-compound was discontinuous and the LS state to HS state change took place at T1/2 (↑) = 228 K, while the reverse transformation appeared at T1/2 (↓) = 225 K, thus affording a 3 K thermal hysteresis. In the bromo compound, the magnetic behavior seems more complicated. On heating, this compound reveals a discontinuous spin transition with a small hysteresis loop centered at 194 K (T(↑) = 195 K and T(↓) = 193 K). Besides this main transition centered at 194 K, the first derivative curves helped to detect two very partial spin-conversion processes found at T(↑) = 171 K and T(↑) = 206 K, which involve approximately to 8–9% of the solid for each transition. DSC and Mössbauer spectroscopy complemented the study of the magnetic properties. No structural information was added, but the authors stressed how the experimental synthetic details may determine the SCO properties.
Sato et al. reported [87] the compound K[Fe(5-Br-thsa)2] (5-Br-thsa-H2=5-bromosalicylaldehyde thiosemicarbazone, L8). This anionic complex shows different SCO processes in the heating and cooling cycles resulting in a 69 K hysteresis loop (T1/2(↑) = 358 K and T1/2(↓) = 289 K). While the compound shows a one-step transition in heating mode, the cooling mode shows a two-step transition. Single-crystal X-ray diffraction studies were performed at various temperatures and showed no phase transitions accompanying the transitions. The cooperative character of this transformation is attributed to a crystallographic 2D coordination polymeric chain consisting of alternating alkali metal and anionic Fe(III) complexes, where K binds the O, N, and S atoms of the Fe(III) coordination sphere. The additional presence of hydrogen bonding and cation–π interactions allow the 2D network structure to accommodate the geometric changes around the metal during SCO.
Sato et al. described the synthesis [88] of four Fe(III) SCO compounds [Fe(L9)2]+, with salicylaldehyde 2-pyridyl hydrazone-type ligands (L9, X = Cl, Br) and dicarboxylic acid monoanions (tetrachloroterephtalic and tetrabromoterephtalic acids) to investigate the influence of the halogen in the spin-transition temperature of SCO complexes. The crystal structures showed a 1D chain formed by the OH⋅⋅⋅O charge assisted hydrogen bonds between anions, which also form NH⋅⋅⋅O hydrogen bonds with the cations. All the compounds are isostructural. The spin-transition temperature shifts by changing the halogen substituent in the salicylaldehyde 2-pyridyl hydrazone-type ligands and dicarboxylic acids without affecting the molecular arrangement in the crystal packing. The T1/2 values of the compounds with L9 (X = Br) ligands were clearly shifted to higher temperatures than those of L9 (X = Cl). This shift was considered to have been originated from the different electron-withdrawing effect of the halogen substituents. This effect is shown in Table 3, where the results of DFT calculations are shown. When Entries 1 and 2 are compared, only the cation is considered, and Δoct is higher for Br than for Cl. However, the anions also include Cl and Br. The compound with Cl+Cl (Entry 3) has lower T1/2 and Δoct than Br+Cl (5). The agreement is not so good when Cl+Br (4) and Br+Br (6) are compared, but the simplicity of the models may be responsible.
Table 3.
Calculated energy differences (kcal mol−1) between HS and LS states in the optimized (opt) geometries of Cl and Br salts of complexes [Fe(L9)2]+, between MECP and LS and HS, Δoct (cm−1) and T1/2 (K).
Chaudhuri et al. [89] synthesized two complexes with three O,N-coordinated o-iminobenzosemiquinonate radical anions (L10), differing in one substituent (F or tert-butyl), with Fe(III) centers in HS and LS arrangements, and reported their electronic and molecular structures. Electrochemical measurements showed that the radical with the F group became more difficult to oxidize to the quinone form and easier to reduce to the amido-phenolate due to the strong inductive (-I) effect of the fluoro group. This group led to a stabilization of the HS state, in opposition to the tert-butyl substituent, as revealed by temperature dependent magnetic measurements. Further studies concluded that the fluoro complex exhibits strong antiferromagnetic coupling operating between the three ligand radicals (S = 1/2) and three of the unpaired electrons of the HS Fe(III) center (S = 5/2) yielding an S = 1 (two unpaired electrons) as the ground state. Therefore, the authors concluded that the nature of the meta substituents at the aniline moieties modifies the strength of the ligand field acting on the Fe(III) ferric centers of both complexes as expected: electron-withdrawing groups decrease the electron density on the N atom of the aniline fragment whereas electron releasing groups behave in the opposite way. The authors also noticed that the π delocalization in the ring facilitates the transfer of the polar properties of the ortho and para substituents (I effect) through the phenyl ring.
Hauser, Boillot and co-workers [90] studied the solid-state photophysical properties of the ferric catecholate spin-crossover compounds with the TPA = tris(2-pyridylmethyl)-amine ligand and R-Cat = catecholate dianion substituted by R = NO2, Cl, or H (L11, PF6− salt, structure proposed previously [91]). They investigated the photoexcitation and relaxation properties of the three catecholate Fe(III) complexes by laser flash photolysis. Their data, as others previously, revealed a decrease of the charge-transfer energy with the electron-donating character of the catecholate group. The authors have shown the occurrence of a photoexcitation process leading to a LS→HS conversion at low temperatures. Other structures were solved later [92].
More recently, Boillot and co-workers reported [93] the synthesis and crystal structure at 120 and 350 K of the SbF6− salt of the same Fe(III) heteroleptic cation, based on the ligand tris(2-pyridylmethyl)amine (TPA) and 3,4,5,6-tetrachlorocatecholate dianion (TCC2−, L11). It exhibits an incomplete S = 1/2 ⇄ S = 5/2 thermal spin-crossover process centered at 250 K. According to single-crystal X-ray diffraction measurements, the cations are densely packed through π stacking (between the catecholate and the phenyl rings) and van der Waals interactions (Cl4···C17, Cl2···H11, C9···H16). Pairs of Fe(III)–TCC moieties are similarly oriented, and the resulting charge-transfer dipole, from catecholate to iron(III), projects along the crystallographic c axis. Pressure induced SCO was investigated by single-crystal Raman spectroscopy. Raman spectra were measured upon varying the pressure (P = 0–8.1 kbar) at ambient temperature. The piezoconversion, as that induced thermally, is relatively gradual, and the 2/3HS-LS spin transition occurs across 4.5 kbar, which probably reflects a similar SCO mechanism in response to both stimuli.
Belo, Vasco da Gama et al. described [94] a solvated bimetallic compound consisting of the cationic Fe(III) complex [Fe(L3)2]+ with the H-qsal-Cl = N-(8-quinolyl)-5-chlorosalicylaldimine ligand (L3, Scheme 1) and the anionic Ni(II) complex containing the α-tpdt = 2,3-thiophenedithiolate ligand (Scheme 2). The crystal structure at 150 K showed that packing is based on an arrangement of alternate layers of [Fe(L3)2]+ cations and [Ni(α-tpdt)2]− anions. The magnetic measurements and Mössbauer spectroscopy revealed hybrid behavior in the compound, combining ferromagnetic cluster-glass behavior, ascribed to the anions network, and SCO of the cations. Cl⋅⋅⋅H-C charge assisted hydrogen bonds promote the interaction between cations, and many intermolecular interactions are present, including hydrogen bonds with the acetonitrile solvent. No halogen bonds were identified.
Harding et al. [95] studied the effect of the halogen in substituted quinolylsalicylaldimine Fe(III) complexes. The ligand, Hqsal-X (Hqsal-X = 5-X-Nquinolylsalicylaldimine, X = F, Cl, Br, I (L3) reacted with Fe(NCS)3 to form [Fe(qsal-X)2]NCS⋅solvent. The compounds were characterized by single-crystal X-ray crystallography and the magnetic behavior by SQUID magnetometry and Mössbauer spectroscopy. Solution magnetic susceptibility studies were also performed and will be reviewed in the corresponding section (3.). The fluoro, chloro and bromo derivatives are found to undergo SCO with T1/2 increasing along the group, while the iodo analogue is always LS. In the case of the fluoro and chloro derivatives the SCO occurs in two steps with hysteresis in the first step. T1/2(↓) = 157 K and T1/2(↑) = 170 K for the fluoro compound, and T1/2(↓) = 167 K and T1/2(↑) = 177 K for the chloro-compound. The hysteresis loops are 13 K wide for the former and 10 K wide for the latter. Structural studies revealed that all SCO compounds crystallize in the triclinic space group and that the LS example crystallizes in the monoclinic space group. π⋅⋅⋅π interactions between the cationic complexes leading to 1D chains are the most important overall structural motif present in the crystal structure despite the different halogens. The authors found that CH⋅⋅⋅X hydrogen bonds and X⋅⋅⋅π interactions involving the halogens increase cooperativity between the 1D chains, particularly in the fluoro and the chloro cases where the SCO is stepped and abrupt. In the structure of the chloro-compound, an interplay of Cl⋅⋅⋅π interactions defines a structural motif named P4AE (parallel fourfold aryl embrace, Figure 5). A very tight packing, depending on the size of the halogen, was observed for the fluoro compound. The authors concluded that introduction of the halogen results in more loosely packed 1D chains within the 2D planes than in the [Fe(qsal-X)2]NCS⋅sol series, which may be responsible for its less hysteretic SCO when compared to the non-halogenated parent compound.
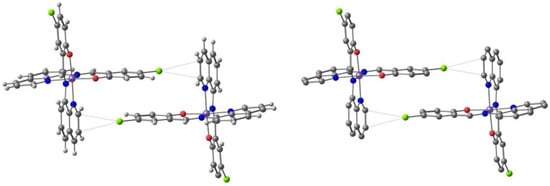
Figure 5.
The Cl⋅⋅⋅π interactions defining the parallel fourfold aryl embrace (P4AE) formed in the crystal structure of [Fe(qsal-X)2]NCS⋅MeOH.
Further work of Harding et al. [96] focused their attention on the iodo substituted quinolylsalicylaldimine (qsal-I, L3, X = I) using a different counter ion, triflate. The methanol solvate of the compound was analyzed by single-crystal X-ray diffraction, SQUID magnetometry, Mössbauer spectroscopy and DSC. The temperature dependence of χMT showed that freshly prepared crystals of the methanol solvate could undergo gradual SCO in the cooling mode with T1/2 = 234 K, but more abrupt SCO upon warming, with T1/2 = 232 K and variable hysteresis. Subsequent cycles led to a slight alteration in the magnetic profile and suggested solvent loss which was confirmed by TGA studies. The unsolvated sample exhibits complete and abrupt SCO in the cooling and warming modes at T1/2 = 224 K and T1/2 = 232 K, respectively, with a hysteresis of 8 K. π⋅⋅⋅π interactions together with anion-cation interactions accounted for the cooperative behavior of the solid. No special reference was made to the role of the halogen. The authors only mentioned possible I⋅⋅⋅π interactions responsible in increasing the dimensionality of the network from 1D to 2D as in Reference [95], although these were clearly very weak.
Harding et al. [97] reported the two-step spin transition with symmetry breaking for the [Fe(qsal-Br)2]NO3·2MeOH (qsal = L3, different anion from [95]). The compound undergoes abrupt two-step symmetry breaking spin crossover, T1/2 (1st step) = 136 K and T1/2 (2nd step) = 232 K with a hysteresis of 16 K and 5 K, respectively, and an unprecedented [HS–LS] plateau of 96 K. This behavior reflects a doubling of the unit cell, originating two independent Fe(III) centers at 175 and 220 K, which undergo successive SCO. The cations form 1D chains through two orthogonal π-π stackings, longer in the HS than in the LS state, complemented by CH⋅⋅⋅O hydrogen bonds involving the anion and the solvent, and their tightness favors the intermediate [HS-LS] state (96 K). The chains also pack in a 2D structure by means of π-π stackings, in a motif seen in 2D sheets of a range of [Fe(qsal-X)2]+ complexes which achieve abrupt SCO (Figure 6). In [Fe(qsal-Br)2]NO3·2MeOH the 2D layers are still linked by strong Br⋯O halogen bonds involving one of the MeOH molecules and weaker C–H⋯Br hydrogen bonds.
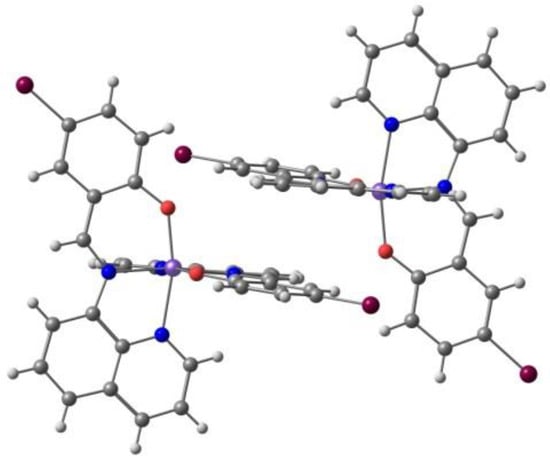
Figure 6.
The π⋅⋅⋅π stacking interactions between two units of the cation showing the close contacts of Br with other atoms (counter ion and solvent are omitted) in the crystal structure of [Fe(qsal-Br)2]NO3·2MeOH.
Takahashi et al. [98] reported the [Fe(qsal-I)2][Ni(dmit)2]·CH3CN·H2O compound, with the iodine analogue of L3, the S containing [Ni(dmit)2]− counter ion (Scheme 2), and acetonitrile as solvent, with the aim of promoting halogen bonds and studying the effect of this less-known intermolecular interaction in spin crossover. They found that the compound exhibited synergy between a SCO transition (Fe(III)) and a spin-Peierls-like singlet (anion). The compound also showed the light-induced excited spin state trapping effect. On cooling, the χMT values gradually decreased down to 170 K, but further lowering the temperature led to an abrupt decrease in the χMT value, observed at T1/2 = 150 K and suggesting the occurrence of a cooperative SCO transition. χMT gradually decreased again below 135 K. On heating the sample, a reverse transition occurred withT1/2 = 156 K and a thermal hysteresis of approximately 6 K.
The weak π-π stacking between cations is responsible for the formation of 1D chains, while the anions are arranged in a 1D zigzag array in another direction. They interact by means of halogen bonds between the iodine atoms in the Fe(III) cation and the sulfur atoms in the [Ni(dmit)2]− anion, Figure 7 (I⋯S 3.690 Å, shorter than the sum of van der Waals radii, 3.78 Å). These halogen bonds and their competition with the inter cations π-π stacking play a crucial role in stabilizing the paramagnetic state of π-spins (anion) and in the synergistic magnetic transition between d- and π-spins.
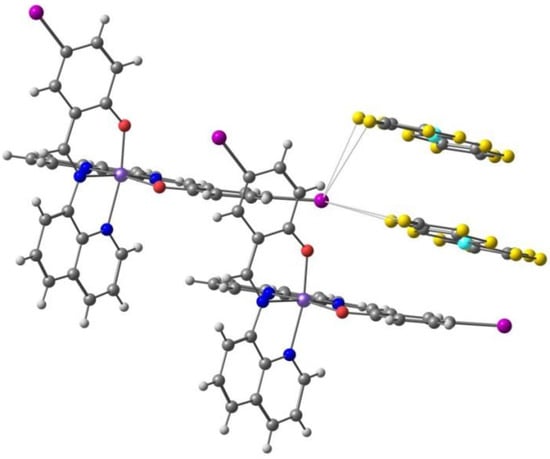
Figure 7.
The π⋅⋅⋅π stacking interactions between two units of the Fe(III) cation and two units of the [Ni(dmit)2]− anion, as well as the I⋅⋅⋅S halogen bonds linking one cation and the anions (solvent omitted) in the crystal structure of [Fe(qsal-I)2][Ni(dmit)2]·CH3CN·H2O.
Waerenborgh et al. [99] continued the work using [Fe(qsal-Br)2][Ni(dmit)2].solv (sol = CH2Cl2 or (CH3)2CO) changing the halogen in L3 to Br and the solvent. The compounds were characterized by single-crystal X-ray crystallography and their magnetic properties determined by SQUID magnetometry and Mössbauer spectroscopy. The magnetic measurements showed that the CH2Cl2 solvate has a gradual SCO with T1/2 around 250 K. The acetone solvate, however, exhibited an abrupt transition with a thermal hysteresis of 13 K close to room temperature (T1/2 (↓) ∼273 K and T1/2 (↑) ∼286 K).
The packing diagrams revealed, as in Reference [98], the π–π assembled layers of cations separated by layers of anions (S⋯S van der Waals interactions). Different weaker interactions may be observed depending on the solvate. The packing is softer in the dichloromethane solvate than in the acetone one, allowing for a smoother, gradual SCO. In the dichloromethane solvate, pairs of solvent occupy cavities in the anion layer and adapt to the geometric changes during the SCO.
The same authors, Waerenborgh and co-workers, also reported [100] the acetonitrile solvate of the [Fe(qsal-Cl)2][Ni(dmit)2].2CH3CN (L3), where Cl was introduced as halogen in the L3 ligand. The crystal structure is based again on arrangements of segregated chains of the [Fe(qsal-Cl)2]+ cations and of the [Ni(dmit)2]− anions. Each kind of unit interacts strongly with the neighboring species, the cations mainly through π-π stacking interactions, and the anions through SS (or SC) contacts. The anions are organized as an arrangement of dimers presenting strong intradimer antiferromagnetic coupling and weaker coupling between the dimers. Solvent molecules establish contacts with cations or anions, giving rise to the formation of sheets of cations and anions. The cationic chains are relatively isolated and only at high temperatures do they present short contacts involving the Cl atoms from the ligands (d = 3.406 Å). Magnetization measurements and Mössbauer spectroscopy were used to characterize the SCO process. With increasing temperature, the magnetization measurements indicate that SCO occurs in two steps, one relatively sharp, at ~233 K, leading from a LS state to a state with 50:50 disordered HS/LS, and a second displaying a more gradual increase of the number of HS states (centered at ~256 K) towards a HS state. This process is different from the one observed for [Fe(qsal-Br)2]NO3·2MeOH in [97] where an ordered [HS-LS] state was detected. Here, at low temperatures, there are no Cl⋯Cl short contacts (dCl…Cl = 4.362 Å), which appear when temperature increases. The magnetic behavior results from the contributions of the cations and anions. The SCO process of this compound, featuring a disordered intermediate phase with a doubled unit cell, may be related to structural constraints that prevent the full LS/HS transformation in a narrow temperature range.
In the previous studies of L3 (qsal) complexes, the role of the halogen was enhanced by the possibility of forming halogen bonds with the S in [Ni(dmit)2]− anions. Both cations and anions could form π⋅⋅⋅π stacks. However, changes in solvent also contributed to modify the subtle interactions defining the weak bonds network in the solids, making it very difficult to trace the factor responsible for the SCO patterns.
Harding and co-workers reported [101] an abrupt SCO close to room temperature with a hysteresis of 30 K (warming: T1/2 = 278 K and cooling: T1/2 = 248 K) observed for the related Fe(III) compound [Fe(qsal-I)2][NTf2] (L3). Structural data reveal, as the most striking difference between the HS and LS structures, a remarkable change in the conformation of the bis(trifluoromethylsulfonyl)amide ([NTf2]−), from a syn conformation with the C–S⋯S–C torsion angle = 8.7(3)1° and 9.3(4)1° at 255 and 275 K, in the HS state, to an intermediate conformation in the LS state with the C–S⋯S–C torsion angle = 94.5(6)1)°. The anti-conformation is not accessible owing to the repulsion between the CF3 group and the quinoline ring. This change is responsible for the high cooperativity and wide hysteresis. The packing consists of two types of 1D chains formed by π–π stacking between the [Fe(qsal-I)2]+ complexes interconnected by C–H⋯π hydrogen bonds.
In another publication, Harding et al. studied [102] the mixed ligand complexes [Fe(qsal-Cl)(qsal-Br)]Y·sol (L3) with several anions and solvents (Y = NCS−, sol = MeOH; PF6−; BPh4−, sol = 2CH2Cl2; OTf−, sol = 0.5MeOH). All the X-ray crystallographic studies of the NCS−, PF6− and OTf− complexes at 123 K show LS Fe(III) centers. In the BPh4 structure, determined at 100, 123 and 269 K, the Fe(III) center appears as LS, mixed HS-LS and essentially HS, respectively. Packing in the complexes is dominated by π⋯π stacking between the cations to yield a 1D chain. C–H···π, C–H···Cl/Br and C–Cl/Br···π interactions link these chains to the anions and solvent, originating highly cooperative supramolecular 3D networks mirroring the homoleptic complexes [Fe(qsal-X)2)]+. Thus, the heteroleptic compounds give SCO profiles between those of the homoleptic ones. SQUID magnetometric studies show almost complete, 50% complete and stepped spin crossover for NCS−, PF6− and BPh4−, respectively, while [Fe(qsal-Cl)(qsal-Br)]OTf·0.5MeOH remains low spin up to 350 K.
In the search for systems displaying broad hysteresis at room temperature, Harding et al. also studied [103] the solvent effects in [Fe(qsal-I)2]OTf.sol (L3) solids with sol = MeOH (3a), EtOH (3b), n-PrOH (3c), i-PrOH (3d), acetone (3e) and MeCN (3f). Structural studies in both spin states for all solids except 3f reveal that the structures rely on two types of π-π stacking, based on two motifs: two cations bridged by two triflate anions (CH···O hydrogen bonds: the triflate embrace) and two cations bridged by solvent molecules. These form 1D chains combining the parallel fourfold aryl embrace (P4AE, Figure 8) and halogen bonds (I-X with X = I, O, π) are also present, to create the 3D tightly packed networks.
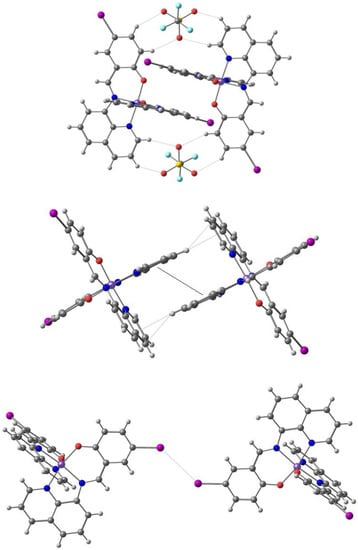
Figure 8.
The triflate embrace formed by C-H⋅⋅⋅O hydrogen bonds (top), the parallel fourfold aryl embrace (P4AE, center) formed by C-H⋅⋅⋅π hydrogen bonds, and the I⋅⋅⋅I halogen bonds (bottom) in the crystal structure of [Fe(qsal-I)2]OTf⋅EtOH (solvent not shown).
In [Fe(qsal-I)2]OTf [96] SCO occurs at T1/2 = 225 K and T1/2 = 234 K (ΔT = 9 K). These values change when there is solvent. Compounds 3a (after MeOH loss) and 3b display abrupt SCO, which becomes gradual for 3c (T1/2 = 199 K) and 3d (T1/2 = 251 K), and incomplete for 3e and 3f (even up to 350 K). The ethanol solvate 3b has the most interesting behavior, with aged samples exhibiting an exceptionally wide hysteresis of 80 K (T1/2 = 139 K and T1/2 = 219 K). However, fresh samples of 3b (first cycle) exhibit stepped SCO with hysteresis varying from 2 to 42 K. Variable-temperature powder X-ray diffraction (VT-PXRD) studies showed that, upon cooling below 180 K, two new phases, 3b-b and 3b-c, form (3b-c is a minor phase). Phase 3b-c (LS) and the HS phase 3b-a undergo a spin transition at T1/2 = 180 K and T1/2 = 215 K, while phase 3b-b exhibits two-step SCO. This study emphasizes that, while solvent may result in only small structural changes, these may have a dramatic effect on SCO characteristics.
Mercuri and co-workers [104] reported a new series of tridentate N-8-quinolyl-salicylaldimine ligands Hqsal-5,7-X2 (X = Cl, Br, I, L12), with a different substitution pattern, namely halo-substituted at the 5,7 positions of the aminoquinoline moiety, and their Fe(III) [Fe(L12)2]− complexes. The Cl and Br compounds are isostructural dimers, where the distorted octahedral environment of each Fe(III) metal ion consists of one N,N,O tridentate (qsal-5,7-X2)− (X = Cl and Br) ligand, one N-coordinated SCN− anion, and two bridging methanolate anions. The I derivative is obtained as another dimer with co-crystallized MeOH. The Br and I ligands form a different polymorph based on centrosymmetric tetranuclear species with two types of Fe(III): two are bound by three nitrogen atoms and three oxygen atoms (from one tridentate ligand, (qsal-5,7-X2)− (X = Br and I), one SCN−, one methanolate anion and a bridging µ3-oxo moiety); the other two are coordinated to two nitrogen and four oxygen atoms (from one tridentate ligand, (qsal-5,7-X2)− (X = Br and I), one methanolate anion and two bridging µ3-oxo moieties). Cl···Cl (3.39 Å) and Br···Br (3.51 Å) intermolecular halogen bonds, CH···S weak hydrogen bonds with the aromatic ring and the SCN− anion, and π-π stacking occur between adjacent halogen-quinoline molecules. In the second polymorph of I (tetranuclear), the iodine atoms dominate the packing interactions through the establishment of a halogen-bonding network. The binuclear and the tetranuclear complexes with the Br ligand are depicted in Figure 9.
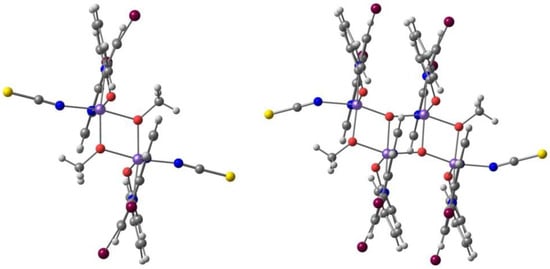
Figure 9.
The binuclear (left) and the tetranuclear (right) complexes of Fe(III) with the (qsal-5,7-X2)− ligand.
The magnetic behavior of dimers and tetramers indicate the presence of strong antiferromagnetic interactions between Fe(III) centers (S = 5/2), mediated by the alkoxy or oxo bridges. DFT calculations (mPW1PW/def2-SVP) support the experimental results. The authors highlighted the versatility that halogen substitution brings to the rational design of new molecules and crystals with a special focus on their magnetic and physical properties.
Clemente-León, Coronado and co-workers [105] inserted the compounds [Fe(III)(5-Xsal2-trien)]+ (sal2-trien = N,N-disalicylidenetriethylene-tetramine, X = Cl, Br, L13) into the 3D oxalate lattice of a coordination polymer based on [Mn(II)Cr(III)(ox)3]− resulting in formation of a 3D chiral network for both compounds. This structure contrasts with the 2D network obtained when X=NO2 and the 3D achiral one for X=MeO. The authors reported the magnetic profiles of the oxalate networks and found that the magnetic plots are dominated by the ferromagnetic behavior of the oxalate network. After isolating this contribution, they were able to determine that both complexes displayed gradual SCO before showing ferromagnetic ordering. This was also confirmed by Mössbauer spectroscopy. The authors also concluded that the halogenated substituents favored a helical structural arrangement of the complexes which enabled the growth of a 3D chiral network.
Clemente-León, Coronado et al. [106] extended their previous study to more Fe(III) complexes of R-sal2-trien ligands (L13, L14, L15, Scheme 4), functionalized with halogens in different positions of the phenolate ring, and inserted them in the same 2D bimetallic Mn(II) and Cr(III) oxalate network. The structures consist of layers of this 2D honeycomb anionic layer and an intercalated cationic layer of the Fe(III) complexes. The authors found that a change of Br by the smaller Cl in [Fe(III)(3-Br-sal2-trien)] [Mn(II)Cr(III)(ox)3]·(CH3CN)2 and [Fe(III)(3-Cl-sal2trien)][Mn(II)Cr(III)(ox)3]·(CH3OH)2 ·(CH3CN)2 can change the templating effect of the cation to afford a very different structure. The two crystallographically independent [Fe(III)(3-Cl-sal2-trien)]+ complexes form dimers, which are connected to other dimers by short Cl···Cl contacts (Cl–Cl distances of 3.304 and 3.307 Å) giving rise to double chains along the a axis. The magnetic profiles showed that the compound based on 4-Br-sal2-trien exhibited abrupt HS to LS SCO from 400 to 270 K, while the one based on 3-Br-sal2-trien continued in the HS state and the one based on 3-Cl-sal2-trien showed a very gradual and incomplete SCO.
Scheme 4.
Ligands L13–L24 (protonated form).
Clemente-León, Coronado et al. [107] also reported the syntheses, structures, and magnetic properties of a family of Fe(III) SCO cationic complexes of the H2(5-Cl-sal2-trien) ligand (L13) and tren(imid)3 (L16, Scheme 4), which were inserted in a 2D network of bimetallic Mn(II) and Cr(III) coordinated to bridging 2,5-dihydroxy-1,4-benzoquinone dianion derivatives (anilate) ligands (L17). Two neighbor Fe(III) complexes present a number of intermolecular interactions, which involve the two phenolate rings (π−π stacking interactions between the two aromatic rings and C−H···π or C−Cl···π bonds). These dimers of [Fe(III)(5-Cl-sal2-trien)]+ complexes are well separated from other Fe(III) complexes, although they display several intermolecular interactions with solvent molecules and with the anilate-based layers. Magnetic measurements revealed that the compound is mostly LS.
Takahashi, Nakamura, Ikeue and co-workers [108] reported the spectroscopic and magnetic properties of several Fe(III) complexes of porphyrins (L18, Scheme 4), one of them bearing pentafluorinated aryl groups, and monodentate ligands (pyNO, 4-ClpyNO, 4-MepyNO, 4-MeOpyNO, 4-Me2NpyNO) both in the solid state and solution. The crystal structure of some of them was also described. The authors found that some of the non-fluorinated compounds were HS and some were SCO, while the pentafluorinated compound (with 4-Me2NpyNO) exhibited SCO behavior. It was not possible to draw any conclusions from the crystal structure of this compound about the role of the fluoride substituents on the packing and SCO behavior.
Trávníček and Herchel [109] published a series of mononuclear iron(III) complexes involving 5-aminotetrazole (L19, Hatz, Scheme 4) and pentadentate Schiff-base ligands (L20) one of which N,N′-bis(5-chloro-2-hydroxybenzylidene)-1,6-diamino-3-azahexane (R=Cl), and two other N,N′-bis(5-bromo-2-hydroxybenzylidene)-1,6-diamino-3-azahexane (R=Br) and N,N′-bis(3,5-dibromo-2-hydroxybenzylidene)-1,6-diamino-3-azahexane) (R=Br). The compounds were characterized by standard techniques, and the magnetic properties studied by magnetic measurements and Mössbauer spectroscopy, and complemented by single-crystal X-ray analysis of the parent phenolate compound (R=H). While this one is HS, all the new complexes with halogen substituents show SCO with critical temperatures above 415 K. DFT calculations with ORCA (B3LYP/def2TZVP) showed that both the LS and HS geometries and the spin transition depend on the computational methodology employed, namely the solvent model, additional van der Waals corrections, and the treatment of relativistic effects.
Boca, Renz et al. reported [110] the cloro-substituted [Fe(LCl)(X)] complexes, where L is also the pentadentate Schiff-base with a Cl on the phenolate (L20, Scheme 4) but a halide or pseudo-halide occupies the remaining coordination position. They studied the effect of X on the properties. The crystal structures are triclinic (Cl, CN and NCO) or monoclinic (P21/c) (NCS and NCSe) and the compounds in each group are isostructural. The compounds in the triclinic series contain in their asymmetric units two [Fe(L)(X)] molecules that differ in ligand orientation (Λ, Δ), whereas the monoclinic ones have only one [Fe(L)(X)] symmetry-independent molecule. In the two latter compounds (NCS, NCSe), the complexes form pairs supported by the weak hydrogen bond between the amine group of one and the chloride atom from the pentadentate ligand of the adjacent [Fe(L)(X)] molecule (d(NH···Cl) = 3.418(2) Å in NCS and 3.437(2) Å in NCSe). The adjacent supramolecular dimers are interconnected by π–π stacking between phenyl groups. Magnetic data revealed that the Cl and NCO compounds are HS, the CN compound is LS and the remaining two exhibit SCO. The substitution of the thiocyanato ligand for the selenocyanato one influences the transition temperature which rises from T1/2 = 280 K to 293 K.
Pavlik et al. reported [111] similar pentadentate ligands (L21) with bromo substituents on the phenolate, which form octahedral Fe(III) complexes, [Fe(LBr)X], with addition of another ligand (X=halides or pseudo-halides). The Cl derivative was obtained as a water solvate, the N3 as a methanol solvate, and the others crystallized without solvent. Complexes with Cl and NCS remained in the HS state, while the other two displayed SCO. The profiles are gradual and incomplete and the T1/2 for the NCSe is almost 100 K higher than for the N3 compound. A detailed analysis of the crystal and molecular structures reveals that the occurrence of SCO in the series of the [Fe(LCl/Br)(L)] compounds is influenced by a number of factors. First, the compounds [Fe(LCl)(NCS)], [Fe(LCl)(NCSe)]110 are isostructural with [Fe(LBr)(NCSe)] (P21/c) and exhibit SCO. On the other hand, [Fe(LBr)(NCS)] displays a different crystal packing (Pn) with Br⋅⋅⋅S halogen bonds and it stays HS over the whole temperature range. A close analysis of the molecular structures of all the complexes shows that the differences in bond lengths or bond angles do not deviate significantly from the typical LS/HS forms of the SCO compounds, or from pure HS compounds previously reported for this type of complexes. Thus, there is no evidence of any structural change which could be unequivocally assigned to changes in the overall ligand-field strength, leading to a weaker field and concomitant stabilization of the HS state. Only a very slight difference between the ligand-field strengths of the LCl and LBr ligands can be deduced from the transition temperatures of the SCO compounds: T1/2 ([Fe(LCl)(NCSe)]) = 293 K and T1/2 ([Fe(LBr)(NCSe)]) = 320 K. Additionally, the modification of the pseudo-halide ligand from NCSe− to NCS− does not lead to a sizable decrease of the increment to the crystal-field strength. The non-occurrence of SCO for the halide compound was explained on the basis of the low ligand-field strength arising from the weak π-donor chloride ligand.
The results of our DFT calculations for complexes with LBr and LCl are shown in Table 4. They confirm certain proposals of the authors. The ligand fields created by NCS− and NCSe− are similar, though slightly higher for the selenocyanate and T1/2 also increases accordingly for the LCl derivatives. For the LBr complexes, the ligand field is higher and T1/2 (NCSe) is thus higher than for LCl. The same comparison cannot be made for the NCS− complexes because with LBr the structure is totally different, but the trend also holds for Cl (HS for LBr).
Table 4.
Calculated energy differences (kcal mol−1) between HS and LS states in the optimized (opt) geometries of complexes [Fe(LCl/Br)(X)], between MECP and LS and HS, Δoct (cm−1) and T1/2 (K).
Martinho and co-workers [112] reported the synthesis of two salts of the 3,5-Br-salEen ligand (3,5-Br-salEen = N-ethyl-N-(2-aminoethyl)-3,5-Br-salicylaldiminate) (L22, R1 = R2 = Br), [Fe(L22)2]X.sol. The perchlorate was obtained as an ethanol solvate and was found to remain LS over the temperature range measured. On the other hand, the tetraphenylborate, crystallized as a dimethylformamide (DMF) solvate, showed an incomplete and gradual SCO centered around room temperature. Only the crystal structure of the tetraphenylborate was reported and no conclusions could be taken on the effect of the halogens.
Martinho and co-workers [113] reported another similar iron(III) perchlorate compound based on the 5-Br-salEen ligand (L22, R1 = H, R2 = Br). The SCO of this compound was coupled with the thermosalient effect and crystal pulverization was observed at the abrupt SCO transition temperature (~320 K). The magnetic profile was very special owing to an abrupt transition in the heating mode and a rather gradual transition in the cooling mode resulting in a hysteresis loop of about 30 K. The LS state is dominant in the magnetic profile because the HS is never attained. The hysteresis loop is stable over several cycles and appears to be independent from the pulverization process. Single-crystal and powder X-ray diffraction studies performed at various temperatures helped to reveal that the abrupt transition is accompanied by a phase transition without symmetry breaking. The salt crystallizes in an orthorhombic group, without solvent, and the packing diagrams at low temperature reveal only N–H⋅⋅⋅O hydrogen bonding between the anion and the cation, as well as weak C–H⋅⋅⋅O hydrogen bonds linking anions in adjacent rows. No other interactions were found.
Martinho and co-workers reported [114] a second polymorph of the Fe(III) complex [Fe(5-Br-salEen)2]ClO4 (L22, R1 = H, R2 = Br) described in Reference [113]. The complex in the new polymorphic form showed an abrupt spin crossover at 172 K with a small 1 K hysteresis window and over a narrow 10 K range. The structure of the cubic crystals shows also two N–H⋅⋅⋅O hydrogen bonding between the anion and the cation, but they are bifurcated and stronger. However, a cooperative network is built from a variety of intermolecular interactions such as C–H⋯Br and C–H⋯π hydrogen bonds, π⋯π stacking, and C–Br⋯π halogen bonds. This crystal packing, not available in the other polymorph, is responsible for the spin-transition profile.
Martinho et al. [115] extended their studies to halogen-derived salEen ligands (L22, R1 = H, R2 = I) and described the new iron(III) complex [Fe(5-I-salEen)2]ClO4 containing iodine, which exhibits both spin crossover, between 304 K and 320 K, with a 16 K hysteresis loop, and thermosalient phenomena. The hysteresis loop disappears after a symmetry-breaking phase transition. The crystal structure is again based on the N–H⋅⋅⋅O hydrogen bonding between the cation and the anion. The behavior of this compound is different from that of the 5-Br-salEen analogue described above [113].
Harding et al. [116] discussed solvatomorphism and anion effects in the predominantly low spin Fe(III) Schiff-base complexes [Fe(naphEen)2]X⋅sol, where naphEen (L23) = 1-{[2-(ethylamino)-ethylimino]methyl}-2-naphtholate is an extension of the salEen ligand (L22). When X = F, sol was 0.5CH2Cl2⋅H2O, but with X = Cl and X = Br a water solvate is obtained, while no solvent co-crystallizes with X = I. The X-ray crystal structures of Cl, Br and I complexes show LS Fe(III) centers in all cases assembled by extensive π-π stacking aryl interactions involving four cations and forming supramolecular squares. The Br salt loses half an equivalent of water at room temperature and the space group changes from monoclinic P21/n to C2/c. Magnetic studies indicate that the F compound is trapped in a mixed spin state (~ 40% HS), while for Cl, B3r, and I the low-spin forms are observed up to 350 K.
Mansour and Shehab [117] reported Cr(III), Fe(III) and Ru(III) complexes with three 7-bromo-1,3-dihydro-5-(2-pyridyl)-2H-1,4-benzodiazepin-2-one ligands (L24). The properties determined for this Fe(III) compound indicated that it is in the LS form.
2.3. d6 Complexes, HS (S=2) and LS (S=0)—Fe(II)
Kao and Wei [118] studied by Mössbauer spectroscopy the effect of ligand substituent X on the SCO of complexes [(dithiocyanato)bis(N-X-phenyl-2-pyridinaldimine)iron(II)] (L25, Scheme 5, X = 4-OCH3, 4-CH3CONH, 4-C4H6, 4-CH3, 4-OH, 4-H, 4-Cl, 4-NO2). Mössbauer spectra at 78 K for the 4-substituted derivatives, except the 4-OH-substituted species, indicate that the fraction of low-spin states increases when the electron-withdrawing ability of the substituent decreases, as measured by the Hammet constant (4-OCH3<4-CH3CONH 4-C4H6<4-CH3<4-H<4-Cl<4-NO2).
Scheme 5.
Ligands L25–L33 (protonated form).
Murray et al. [119] investigated in detail a family of halogen-substituted Schiff-base iron(II) complexes, [Fe(qsal-X)2], (L3, qsal-X = 5-X-N-(8-quinolyl)salicylaldimines) with X = F, Cl, Br or I. F exhibits a temperature invariant HS state, assigned to the very weak π···π and C-H···O/F weak bonds. Cl, Br and I show abrupt SCO with T1/2 = 295 and 342 K for X=I and Br, respectively, whereas Cl shows a two-step transition at T1/2 = 308 and 316 K for the 1st and 2nd steps, respectively, before reaching the fully HS form at high temperatures. C-H···Cl/Br or I···π halogen bonds, as well as π···π stacking and P4AE interactions, are responsible for the abrupt SCO transitions in this group of Fe(II) complexes. The Fe(III) analogues have an anion and therefore the crystal packing contains stronger electrostatic interactions which determine different SCO patterns, as in the [Fe(qsal-X)2]NCS.solvent complexes [95].
DFT calculations were performed on the Fe(II) complexes and the computed parameters are in general agreement with the experimental observations, although there are discrepancies in fine details such as ground-state energies, possibly due to a lack of inclusion of an elastic model, referred to above, and/or inclusion of intermolecular interactions such as Madelung potential fields, recently used in calculations on solid-state Fe(II) spin-crossover systems. We also tested our DFT approach, the results being shown in Table 5.
Table 5.
Calculated energy differences (kcal mol−1) between HS and LS states of [Fe(qsal-X)2] in the X-ray structure (shown for X = Cl) and in the optimized (opt) geometries, between MECP and LS and HS, Δoct (cm−1) and T1/2 (K).
In these complexes, there is no correlation between the calculated parameters and T1/2. The influence of the ligand field is outweighed by the different crystal structures and the nature of intermolecular interactions.
Murray et al. [120] further examined the chloride ligand complex [Fe(qsal-Cl)2] with qsal-X = 5-X-N-(8-quinolyl)salicylaldimines (L3), which shows an abrupt two-step spin transition at 308 and 316 K and a symmetry breaking at the highest temperature. Packing reveals a chain of Fe(II) molecules organized by means of π···π stacking interactions between the salen and the quinoline rings of adjacent units, as well as C–H···O interactions along the b axis. Further intermolecular interactions, namely C–H···Cl hydrogen bonds and two different sets of parallel fourfold aryl embraces (P4AE) are observed along a and define the 3D nature of the structure. The P4AE have been associated with the abrupt SCO transition. The structure is very different from that of analogous Fe(III) complexes studied by References [95,96,97,101,102,103] and other authors [104], where the counter ions prevent a similar arrangement. The use of the planar [Ni(dmit)2]− anion chosen in some works as counter ion might have been an attempt to increase π···π stacking in structures of [Fe(III)(qsal-X)2]+ derivatives.
Kuroda-Sowa et al. [121] studied the halogen effect on [Fe(qsal-X)2] (Hqsal-X = N-(8′-quinolyl)-2-hydroxy-5-halogeno-1-salicylaldimine (L3), X = F, Cl, Br, I) complexes. Magnetic studies on these compounds revealed that the fluoride is HS, but the remaining undergo SCO with gradual profiles and T1/2 of 308 K (Cl), 341 K (Br), and 340 K (I). Based on the analysis of the crystal structures and packing diagrams of all the compounds, the authors assigned the origin of the difference in their magnetic behavior to the distortion in the iron coordination geometry, which is affected by the CH⋅⋅⋅F hydrogen-bond interactions and π···π stacking. The authors consider that these results indicate the increasing stability of the low-spin states from F to I.
Kahn and co-workers reported [122] several binuclear complexes cis-[{FeL(NCS)2}2(μ-bpim)], where the bpim ligand (bpim=2,2’-bipyrimidine) bridges two Fe(II) centers (Figure 10), each of them also coordinated to two NCS and one bidentate ligands L (L = bpim, 2,2′-bipyridine or bromazepan, L24, Scheme 4). The latter compound showed SCO (smooth transition) with T1/2 = 235 K. At 300 K, the two iron(II) ions in all the dinuclear units are in a high spin state and about 60% of them are involved in the smooth transition 235 K. Besides diamagnetic dinuclear units (52%), a small amount of dinuclear units with a high-spin ion and a low-spin ion (8%) are also formed. About 40% of the dinuclear units do not undergo the transition and behave as antiferromagnetically coupled species in the whole temperature range.
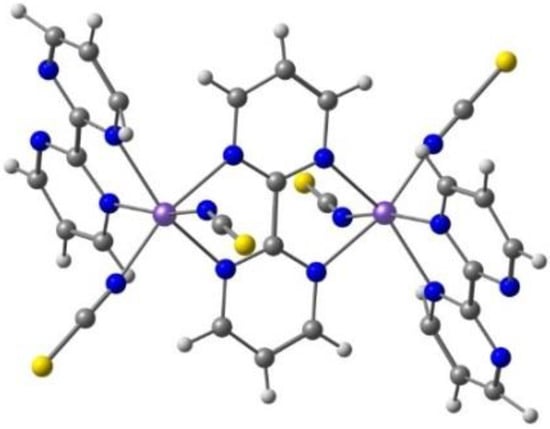
Figure 10.
A view of the molecular structure of the cis-[{Fe(bpim)(NCS)2}2(μ-bpim)] complex.
Linert et al. investigated [123] the spin-crossover behavior of cationic [Fe(L26)2]2+ complexes of X-2,6-bis(benzimidazol-2’-yl)pyridine (L26, Scheme 5, X = H, OH, Cl and CH3) using UV-visible and Fourier transform infrared spectroscopy on solid-state compounds. Variable-temperature FTIR indicated that the Cl substituted compound was predominantly in the LS state at RT but certain bands are associated with the HS state and were used to study the spin transition. Further studies were performed in solution and will be discussed later [124,125].
Paulsen and co-workers [126] analyzed the substituent effect in tris(pyrazolyl) complexes (L27) [Fe(L27)2]2+ in the perchlorate salt of the bromo substituted ligand (L27). It showed a very gradual and slow SCO with an estimated T1/2 of about 355 K. Besides susceptibility measurements, Mössbauer spectroscopy experiments were performed to study the magnetic behavior and DFT calculations complemented the experimental results.
Kaizaki et al. reported [127] the role of substituents on binuclear complexes [{Fe(NCE)(X-py)}2(μ-L28)2] (NCE = NCS or NCBH3), where each Fe(II) center binds four nitrogen atoms of L28 and one substituted pyridine and one NCE ligands coordinate to the iron centers in the axial positions. The order of T1/2 changes with the substituents of pyridine in the same way for both the NCS and the NCBH3 complexes, increasing from X = 4-Mepy < 4-Me2Npy < py < 3-Mepy < 3-Clpy ≤ 3-Brpy, and correlates with the Hammett constants. This trend shows that a higher T1/2 is observed when the X-py ligands become better π-acceptors and the stabilization of the t2g set favors the LS state.
Tuchagues et al. reported [128] the synthesis and characterization of Fe(II) complexes trans-[Fe(L29)(NCX)2] of a new dissymmetrical tetradentate ligand, two of them with R2 = Br, R1 = H or CH3, and X = S. These two compounds were in the HS state, while the others (R2 = H) underwent SCO. The X-ray structure of the methyl/bromo compound reveals a planar conformation of the L29 ligand, which is stabilized by a weak intramolecular C-H···Br hydrogen bond, with a C···Br distance of 3.434(3) Å and a C-H···Br angle of 128.8°, forming a six-membered metallacycle and distorting the octahedral environment of the Fe(II), Figure 11. The authors concluded that the HS state was more stabilized than expected by the combination of steric and electronic effects induced by the 6-bromo substituent on the pyridyl ring of the ligand. The steric effect of Br associated with the possibility of forming the C-H···Br bond seems to be more important than its electronic effects.
Figure 11.
A view of the molecular structure of the trans-[Fe(L29)(NCX)2] complex showing the C-H···Br hydrogen bond.
Halcrow and co-workers [129] described six new structurally related iron(II) complexes, two of them, [Fe(L30)2][BF4]2, containing halides (Cl, Br), and showing extremely similar abrupt thermal spin transitions. Upon cooling, ground polycrystalline samples of both the chloro and the bromo compounds revealed an abrupt thermal spin-transition. For the Cl derivative, the transition is complete and occurs at 202 K, with a narrow but reproducible hysteresis loop of 3 K, while for the Br derivative it takes place at 253 K with a 2 K hysteresis loop. This transition was only 85% complete at 250 K, with slow conversion of the remaining iron cations when the temperature was lowered further. The similarity of the spin transitions in these two halogen derivatives and in the other four complexes with non-halogen substituents is striking. Despite differences in the transition temperatures, the six compounds show highly abrupt thermal spin crossover with a small hysteresis width of 2 or 3 K. Indeed, their packing is dominated by π···π stacking interactions, C–H···π hydrogen bonds or X···π (X = Cl or Br) halogen bonds, involving pyrazole groups of adjacent molecules (Figure 12). These interactions define a three-dimensional network, which include van der Waals contacts (terpyridyl embrace) and provide the environment for the SCO.
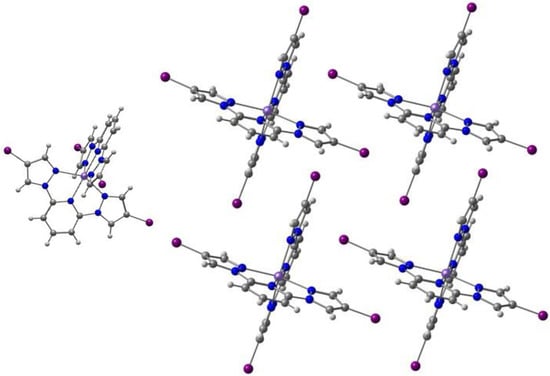
Figure 12.
A view of the [Fe(L30)2]2+ cation and a section of the packing showing the Br···π halogen bonds and the terpyridyl embrace.
Halcrow et al. described [38] with more detail the synthesis of the same Fe(II) complexes [Fe(L30)2][BF4]2, now extending the substituents to I. The Cl complex crystallizes in two different solvent-free polymorphs. The tetragonal (α) undergoes an abrupt spin-transition at 202 K and coexists with the “terpyridine embrace” structure. This packing motif is modified in the orthorhombic form (β), which displays two distinct iron sites in a 2:1 ratio. Only one-third of the molecules undergoes a very gradual thermal spin-crossover centered at 137 K. A comparison of the two structures suggests that cooperativity effects in the α-polymorph are observed in two dimensions within the extended lattice and allow for spin crossover. The Br complex is isostructural with the Cl α form and exhibits a similar abrupt spin-transition at 253 K. In contrast, the I complex is LS as a powder, at 360 K and below, and can be crystallized as two different solvates from acetone solution.
The authors reinforced their conclusions that the layered terpyridine embrace lattice is responsible for mediating a level of cooperativity in iron(II) spin-transitions, which can be disrupted by small perturbations as observed in the β polymorph, where cooperativity is less extended.
Chastanet, Halcrow and co-workers [130] extended their work to new [Fe(L31)2][BF4]2 complexes containing three new related ligands (L31), two of them with bromide and iodide substituents on the pyridine ring. Both compounds exhibited SCO behavior with T1/2 = 307 K (Br) and 332 K (I) with gradual and incomplete profiles. The two compounds are isostructural and the cation layers display an incomplete embrace structure (π–π stacking interactions, C–H···π hydrogen bonds involving the pyrazolyl rings) with more or less approximate four-fold symmetry, but the Br and I substituents at the pyridine disrupt the extended and regular network of interactions.
The DFT calculations we performed (Table 6) show that the T1/2 follows the increase of Δoct for the complexes (the ligand with R=H is added for comparison). The MECP was not calculated due to the technical difficulties of computing it in this binuclear model.
Table 6.
Calculated energy differences (kcal mol−1) between HS and LS states of [Fe(L31)2][BF4]2 in the X-ray structure and in the optimized (opt) geometries, Δoct (cm−1) and T1/2 (K).
Deeth, Halcrow et al. [132] tried to define a relationship between ligand substituents, namely Hammett parameters, and spin state (HS, LS, or SCO) in a series of the previously studied iron(II) complexes containing derivatized 2,6-di(pyrazol-1-yl)pyridine (bpp) ligands. The derivatization was either on the pyridine ring (L31, X = F, Cl, Br and I) or the pyrazole ring (L30, X = Cl, Br and I). The effect of the same electron-withdrawing substituent, for instance, could stabilize the LS or HS form depending on its nature and its position in the molecule. DFT calculations were also used and showed that the difference between the total energy of the HS and the LS states correlated well with the measured T1/2.
Halcrow and co-workers [133] reported a new series of Fe(II) complexes with similar ligands with triazine (and other azines, L32) in replacement of pyridine. The ligand bearing one halogen substituent is 2,4-di(pyrazol-1-yl)-6-chloro-1,3,5-triazine, however, as all the compounds prepared with triazine, it is HS. DFT calculations were also performed to try and find a correlation between the behavior of the different parent compounds.
Ruben and co-workers [134] reported, among others, an iodo Fe(II) derivative of the same bpp ligand L31, as the perchlorate salt. Solid-state magnetic measurements revealed that the compound shows SCO and the authors noted that this compound was one rare example of Fe(II) complexes exhibiting spin transition above room temperature with a T1/2 of 333 K. No comments were made on the nature of intermolecular interactions, namely those involving iodine.
Ishida and Kimura [135] modified the bpp ligand by replacing pyrazole by oxazoline groups (L33). This new ligand presented several substituents on the pyridine group, one of them being chloride. The crystal structure of the [Fe(L33)2][ClO4]2 compound was reported but no information on the packing of the molecules was drawn. Magnetic studies reveal that the chloride compound displays SCO, behaving in the same fashion as the complexes with other substituents (the only exception is the methoxy, which is HS). The transition is incomplete, with T1/2 = 310 K. The authors analyzed the electronic effects of the substituents on the T1/2 and concluded that the π-electron-withdrawing strength of the ligand stabilized the LS state. They also calculated distortion parameters for each substituent and found that highly distorted structures preferred to retain the HS state, while those with an intermediate distortion promoted SCO.
Murray and co-workers reported [136] polymeric and dimeric Fe(II) compounds with ligand L34 (Scheme 6). All dimeric compounds are HS.
Scheme 6.
Ligands L34–L48 (protonated form).
Hanan and co-workers [137] reported the phenyl bromo-derived bpp inspired ligands L35 (Scheme 6, R = 2-pyridyl, 2-pyrazyl, and 6-picolyl). The crystal packing of their Fe(II) complexes shows intermolecular Br⋅⋅⋅Br short contacts, of 3.46, 3.42 and 3.55 Å for [Fe(L35-2-pyridyl)2](PF6)2, [Fe(L35-2-pyrazyl)2](ClO4)2 and [Fe(L35-6-picolyl)2](ClO4)2, respectively. These Br⋅⋅⋅Br distances, shorter than the sum of the Br van der Waals radii, give rise to 1-dimensional tapes running throughout the lattice (Figure 13). In complex [Fe(L35-6-picolyl)2](ClO4)(PF6), the Br⋅⋅⋅Br contacts have lengthened to 4.26 Å. The magnetic moments of the complexes with 2-pyridyl (2.4 B.M.) and 2-pyrazyl (4.2 B.M.) substituents are indicative of spin transitions, but the 6-picolyl mixed salt (5.4 B.M.) remains in HS over the temperature range studied.
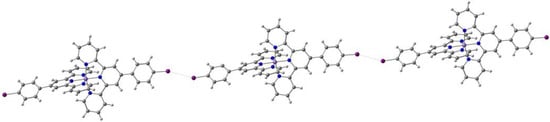
Figure 13.
A view of the 1D chain assembled by Br⋅⋅⋅Br halogen bonds between the [Fe(L35-2-pyridyl)2]2+ cations.
Murray et al. [138] reported mononuclear Fe(II) complexes containing dipyridylamino-substituted triazine ligands, one of them containing a chloride substituent on the triazine ring, DCCl = 6-chloro-N2,N2-dicyclohexyl-N4,N4-di(pyridin-2-yl)-1,3,5-triazine-2,4-diamine (L36, Scheme 6): trans-[Fe(II)(NCS)2(L36)2].2CH3OH. The temperature dependent magnetic susceptibility of this Fe(II) complex was measured below 320 K and a T1/2 of ~280 K was estimated, even though the compound was mounted on grease to prevent solvent loss from the methanol solvate. Analysis of the packing diagrams revealed intra-molecular S⋅⋅⋅π interactions between the thiocyanate ligand and the triazine ring. Lattice methanol molecules create hydrogen bonding interactions with the NCS− ligands and the parallel quadruple phenyl embrace (P4PE) packing motif is also present. Further, C–H⋅⋅⋅π hydrogen bonds are observed between the slipped π-stacked pyridyl rings and those pyridyl rings adjacent to them within the P4PE configuration.
Murray et al. [139] described the synthesis of the related ligand 6-chloro-N2,N2-diethyl-N4,N4-di(pyridin-2-yl)-1,3,5-triazine-2,4-diamine (L37) together with two other crown-ethers derivatives (no halogens) and their Fe(II) complexes, trans-[FeII(L37)2(NCX)2] (X = S, Se, BH3). The packing diagrams of the three complexes show that the diethyl ‘arms’ of the ligand are oriented perpendicular to the triazine ring in the complexes, pointing in opposite directions in S and Se and in BH3. The closest intermolecular contacts in S and Se complexes are C–H⋯Cl hydrogen bonds (C18⋯Cl1, 3.415 Å (S), 3.401 Å (Se)), but for BH3 the closest C⋯Cl distance is 3.689 Å. For the complex with S, the χMT value remains 3.49 cm3 K mol−1 between 270 K and 130 K, decreasing then to 3.04 cm3 K mol−1 at 70 K. After remaining constant until ∼10 K, a more rapid decrease occurs, χMT reaching 2.55 cm3 K mol−1 at 2 K. The complex with Se exhibits a similar behavior. On the other hand, the BH3 compound undergoes a very abrupt spin transition, which starts with a HS plateau (χMT value of 3.8 cm3 mol−1 K at 280 K), and is followed by an abrupt reduction to a χMT of around 0.9 cm3 K mol−1, typical of a mainly LS state, upon cooling to 170 K. This spin transition has a T1/2 = 210 K and shows little or no hysteresis. The authors tried to prepare a fresh powdered sample of the BH3 compound in order to gather more information on the cooperative effects taking place, but they were not able to reproduce the sample, obtaining instead a hydrated amorphous material with 1.5 water molecules. This hydrated compound displayed an abrupt SCO with hysteresis. A plateau is observed for the HS state, at room temperature, with a value of 3.2 cm3 mol−1 K, and decreases abruptly to 1.0 cm3 mol−1 K at 170 K (T1/2 ≈ 203 K). While in the heating mode, T1/2 is shifted to a higher temperature of ∼ 210 K; thus ΔT is ≈7 K.
The authors performed several types of calculations in order to detect the presence of cooperative or anticooperative interactions, determining the interaction parameter Γ or cooperativity factor C (Slichter and Drickamer thermodynamic (mean-field) or ab initio Complete Active Space Self Consistent Field, CASSCF). They studied in more detail the BH3 complex, using also DFT. Despite the broad agreement found between the methods and experiment, they were not able to pin down precise intermolecular structural reasons for the weak cooperativity detected [140].
Roubeau, Youngme, Gamez and co-worker [140] reported two ligands with a different functionalization in the triazine, 4,6-dichloro-N,N-di(pyridine-2-yl)-1,3,5-triazine-amine (Cldpat, L38) and 6-chloro-N-phenyl-N,N-di(pyridin-2-yl)-1,3,5-triazine-2,4-diamine (Cladpat, L39) and their Fe(II) complexes with two thiocyanate molecules completing the coordination sphere. The compound trans-[Fe(L38)2(NCS)2](H2O) is HS from room temperature down to 5 K, whereas trans-[Fe(L39)2(NCS)2] exhibits spin-crossover properties (fast and complete change) with T1/2 = 178 K. The [Fe(L39)2(NCSe)2] analogue also displays SCO properties, with a more gradual profile and a lower T1/2 value of 166 K. The solid-state structure of trans-[Fe(L38)2(NCS)2](H2O) shows intramolecular S(lone pair)···π(triazine) interactions (3.337(8) Å). In addition, the molecules are associated in a supramolecular 1D chain by means of intermolecular S(lone pair)···π(triazine) interactions (3.242(7) Å). The chains interact with each other through similar interactions, producing a 2D layer. These 2D layers are associated by means of OH⋅⋅⋅Cl hydrogen bonds between lattice water molecules and the chloride atoms of the ligand giving rise to a 3D network. The molecules of trans-[Fe(L39)2(NCS)2] exhibit intramolecular S(lone pair)···π(triazine)interactions, which are also altered by the SCO. These 1D supramolecular chains are linked into a 2D layer by parallel-displaced π–π interactions involving neighboring aniline rings. The authors conclude that an apparent minor modification of a ligand, namely the substitution of one of the chloride atoms of L38 by an aniline group (L39) originates a drastic change of the magnetic properties of the corresponding iron(II) complexes. The modification is not minor, however, either in terms of steric bulk, or in terms of donor/acceptor for hydrogen bonds and other weak bond formation.
Roubeau, Youngme, Gamez et al. [141] modified the previous ligand by appending two different groups, obtaining 2-(N,N-bis(2-pyridyl)amino)-4,6-bis(pentafluorophenoxy)-(1,3,5)triazine (L40) together with the non-fluorinated compound, and its complex trans-[Fe(L40)2(NCS)2]2+. The capability of this complex to participate in intermolecular interactions of different types is clear from Figure 14 (pyridine and triazine rings and fluoride atoms).
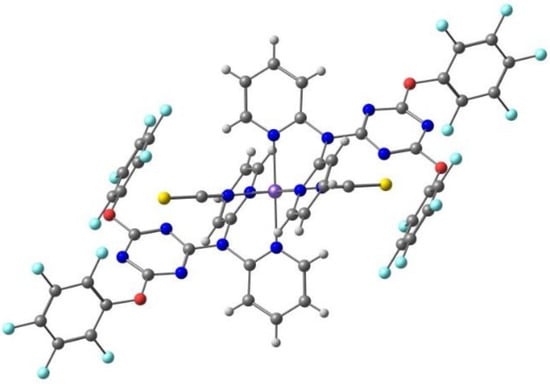
Figure 14.
A view of the molecular structure of the trans-[Fe(L40)2(NCS)2]2+ cation.
The coordination properties remain but the packing should be modified. The Fe(II) complex trans-[Fe(L40)2(NCS)2].2CH3CN shows a very abrupt SCO process, centered at T1/2 = 238 K, with a decrease of χMT from 3.10 cm3 mol−1 K at 280 K down to 0.2–0.1 below 200 K. It was explained by the cooperativity effect in the solid. Indeed, there is an intricate network of strong supramolecular bonds, forming 1D chains (π⋯π stacking between the fluorinated C6F5 rings, at 3.112 and 3.225 Å), which associate in 2D sheets through π⋯π stacking of the pyridine rings, at 3.757 Å). The final 3D assembly is achieved by F⋯F short contacts of 2.781(2) Å, a distance well below the sum of the van der Waals radii of two F atoms (2.94 Å). This strong connectivity is absent in the NCS/NCSe Fe(II) complexes with the non-fluorinated ligands and its effects have been probed with several techniques.
Masciocchi, Roubeau, Gamez [142] and co-workers describe in this work the synthesis in proprionitrile (PrCN, CH3CH2CN) of the NCSe complex [Fe(L40)2(NCSe)2]·2PrCN. The NCSe ligand is involved in the 3D packing, which is otherwise very similar to that of the NCS analogue [142]. The complex loses nitrile, forming [Fe(L40)2(NCSe)2], which can also be reproducibly obtained by thermal annealing of fresh solvated complex for 2 h at 80 °C. Both complexes display SCO, with HS to LS transitions centered at 283 and 220 K, in the solvated and non-solvated species, respectively. The authors concluded that the change of the magnetic properties is most likely due to a modification of the crystal packing of the molecules prompted by the loss of solvent, hence exemplifying the great sensitivity of the SCO phenomenon.
Zhu and co-workers [143] reported four triaryltriazole ligands two of which, 4-(p-chlorophenyl)-3,5-bis(2-pyridyl)-4H-1,2,4-triazole and 4-(p-methylphenyl)-3-(p-fluorophenyl)-5-(2-pyridyl)-4H-1,2,4-triazole (L41, Scheme 6) are halogenated, and their trans-[Fe(L42)2(NCS)2] complexes. The magnetic studies revealed that both complexes exhibit abrupt and complete SCO with T1/2 = 253 K for the chloro-compound and T1/2 = 97 K for the fluoro compound.
The packing diagrams of the chloro derivative show that each molecule of the complex is linked to another by one C–H⋯Cl and another C–H⋯π (3.627(4) Å) hydrogen bonds involving the uncoordinated pyridyl and the p-chlorophenyl group and originating 1D chain. C–H⋯N hydrogen bonds (C⋯N3 = 3.403(4) Å) between the p-chlorophenyl ring and one N of the triazole allow adjacent chains to connect and form a 2D network. π⋯π stacking interactions are responsible for the 3D network. The intermolecular interactions in the fluoro derivative are fewer, namely C–H⋯F (p-methylphenyl group and F) and C–H⋯S (pyridyl group and NCS− anion) hydrogen bonds, complemented by two types of C–H⋯π hydrogen bonds (p-methylphenyl group and pyridyl ring).
Harris et al. [144] reported a series of four binuclear complexes [(L42)2Fe2(μ-L43)]2+, where each Fe(II) coordinates the tetradentate ligand TPyA (L42) = tris(2-pyridylmethyl)amine and the two metal centers are bridged by doubly deprotonated form of 3,6-disubstituted-2,5-dianilino-1,4-benzoquinone (L43), which carries X substituents (H, Br, Cl, and F), with the aim of studying the electronic effects on SCO of these compounds. The structure of the BArF (ArF =C6H3(CF3)2) salt is shown in Figure 15.
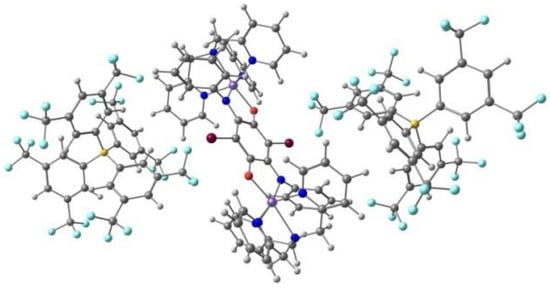
Figure 15.
A view of the molecular structure of the [(L42)2Fe2(μ-L44)][ BArF]2 (X = Br).
All four complexes showed SCO with different degrees of conversion (none of them complete). The T1/2 values were determined as 160(1), 124(1), 121(1), and 110(1) K for X = H, Br, Cl, and F, respectively, along with enthalpies, ΔH, 11.4(3), 8.5(3), 8.3(3), and 7.5(2) kJ mol−1, in the same order. The authors found a correlation between each of the two parameters and the electronegativity of the X substituent, both decreasing with increasing electronegativity (or electron-withdrawing character of X). Therefore, they proposed that the experimental trends observed resulted from the inductive effect of X and no cooperative effects in the solid were considered.
Lemaire et al. [145] also reported Fe(II) binuclear complexes, [{Fe(phen)2}(μ-L44)]4+, with substituents on the central phenyl ring of the ligand L44, one of them being a bromide. The second ligand is phen = 1,10-phenanthroline (L45). This complex shows a very slow and incomplete SCO on both sides. No crystal structures could be determined. DFT calculations (BP86/def2-TZVP) were performed to determine the molecular structures of the HS and LS states. The LS state was found to have an energy approximately 48 kcal mol−1 lower, which qualitatively agrees with the experimental data. This is a rather large energy separation, though, which probably results from the nonhybrid functional used.
Garcia et al. reported [146] mononuclear Fe(II) complexes with two phen ligands (L45) in their coordination sphere, each bearing a Br substituent, with a formula cis-[Fe(3-Br-phen)2(NCS)2]·solvent (solvent = 0.5 CH3OH (1), 2 CH2Cl2 (2), desolvation of 2 (3), 0.5 CH3COCH3 (4) and no solvent (5)). Complex 1, precipitated from MeOH, undergoes an incomplete and gradual thermally induced spin conversion upon cooling below room temperature, while 4, prepared by an extraction method, remains mostly in the low-spin state. The non-solvated 3 and 5 display a more abrupt spin crossover on cooling, with T1/2 = 175 K and T1/2 = 198 K, respectively. The crystal structure of 2 exhibits π-π stacking and Br⋅⋅⋅S halogen bonds responsible for an extended 2D network. DFT calculations (Gaussian03/B3LYP, LYP/CEP-31G) carried out by the authors for the isolated molecule reveal that the energy difference between the HS and LS forms is 7 kJ mol−1, slightly less (ca. 5 kJ mol−1) than for the parent compound [Fe(phen)2(NCS)2] (abrupt SCO, T1/2 = 175 K, polymorphs I and II), reflecting a slight destabilization of the LS isomer owing to the effect of bromine. However, the calculations cannot yet provide a comprehensive answer to the problem, owing to the impossibility of taking into account the intermolecular interactions, such as Br⋅⋅⋅S halogen bonds, which could not be present in the parent complex.
Tuczek et al. [147] analyzed four analogues of the spin-crossover complex [Fe(H2Bpz2)2(phen)] (H2Bpz2 = dihydrobis(pyrazolyl)borate), where phen represents the 1,10-phenthroline ligand (L45), with substituents 5-chloro-1,10-phenanthroline (Cl) and 4,7-dichloro-1,10-phenanthroline (Cl2). The magnetic experiments show that the Cl complex undergoes a typical steep SCO, with T1/2 = 151 K, which was determined at 163 K for the parent phen compound. This effect has been assigned to the electron-withdrawing effect of the chlorine group in lowering the transition temperature. The difunctionalized Cl2 compound, however, stays in the HS state down to 20 K.
Petzold and co-workers [148] synthesized a series of ligands of the family 2-(6-R1-pyridin-2-yl)-1,10-phenanthroline, one of them carrying a Br substituent (L46). Its Fe(II) complex, [Fe(L46)2]X2 (X = BF4− or ClO4−), undergoes SCO to the LS state on lowering the temperature. The abrupt spin transition in crystalline samples of the BF4 salt is centered at T1/2 = 202 K, pointing to cooperativity in the lattice. Indeed, it is noticed that this transition disappears in powdered samples following the loss of the solvent.
This work was continued, with the study of the behavior of the same family of complexes in solution, including the one with a Br substituent [149]. The authors also used DFT calculations to determine molecular geometries not available experimentally and to learn more about the family of compounds (most of them do not have halogens).
Renz et al. [150] concluded that the modification of ligand L47 (Scheme 6), by substitution of hydrogens at R1 and R2 positions, namely by Me and Cl, in Fe(II) complexes cis-[Fe(L47)2(NCE)2] (E= S, Se) directly influences the electronic configuration determined by Mössbauer spectroscopy both at room temperature and 80 K. Thermal SCO took place for the investigated complexes, the only exception being the complex [Fe(dppCl)2(NCS)2] which remained in HS state.
Gu and co-workers [151] reported four novel homochiral mononuclear SCO iron(II) complexes [Fe(L48)3][ClO4]2, one of them with a chloride substituent on the phenyl group (R2, L48 in Scheme 6, and R1 = iso-butenyl). The cations established C–H⋯π hydrogen bonds with their neighboring cations at a distance of 3.281 Å, forming a 1D supramolecular chain. The authors also noticed that both the imidazole and the phenyl in the ligand offered many possibilities of forming intermolecular bonds in the solid, by means of C–H⋯π and C–Cl⋯π hydrogen and halogen bonds, which should reflect the spin transition temperatures observed. Indeed, while the R2 = H complexes (R1 = n-propylenyl or n-hexenyl) exhibited reversible gradual and complete LS to HS transition with T1/2 = 257 or 282 K and no hysteresis, the Cl derivative, with C–Cl⋯π halogen bonds, showed relatively sharp transition with T1/2 = 375 K.
Gural’skiy et al. [152] studied the isothiocyanate iron(II) complexes with iodo-, bromo- and amino-substituted pyrazines (L49, pz, Scheme 7). The stoichiometries depended on the substituent, the iodo- and bromo-derivatives being [Fe(I-pz)2(SCN)2(H2O)2].2Ipz and [Fe(Br-pz)2(SCN)2(H2O)2].2Brpz, but the amino being [Fe(NH2pz)4(SCN)2]. All the complexes are HS and the 3D packing includes several kinds of hydrogen bonds, S(lone pair)⋅⋅⋅π interactions, and S⋅⋅⋅I/Br halogen bonds. Additionally, the Br complex interacts with Brpz through Br⋅⋅⋅π halogen bonds, while in the I analogue coordinated water is involved in a OH⋅⋅⋅N hydrogen bond with Ipz (Figure 16).
Scheme 7.
Ligands L49–L65 (protonated form).
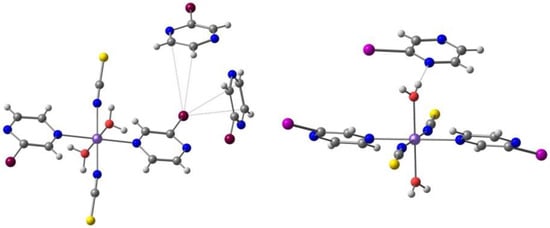
Figure 16.
A view of the structure of [Fe(X-pz)2(SCN)2(H2O)2].2Xpz, with X = Br (left) and I (right).
The same authors, Gural’skiy, Bao et al. [153], also reported three polymorphs of the complex cis-[Fe(L50)(dca)2], where L50 (Scheme 7) is N,N′-bis[(5-bromo-2-pyridyl)methyl]-ethane-1,2-diamine and dca is dicyanamide. They could prepare pure samples of each polymorph by adjusting the experimental conditions. Their magnetic properties were significantly different. The γ-polymorph remained HS in the temperature range studied, while the α-form exhibited an abrupt SCO with hysteresis and T1/2 = 134 K, and the β-form showed SCO with two steps and a plateau (T1/2 = 153 and 144 K). A wide variety of intermolecular interactions between the complexes is observed in the solid state, creating a strong network without solvent. The strongest hydrogen bond, (CN)2NH⋅⋅⋅N, links the donor NH of each dicyanamide with one N acceptor and gives rise to different motifs characterizing each polymorph and more similar for α- and β-forms. In these, pairs of NH⋅⋅⋅N hydrogen bonds between adjacent complexes with up and down orientations form chains which interconnect by means CH⋅⋅⋅π (py) and other weaker interactions. On the γ-polymorph, the NH⋅⋅⋅N hydrogen bonds form 2D sheets. This very different arrangement is reflected in the magnetic properties.
Haasnoot et al. [154] reported the synthesis of 1-(2-chloroethyl)-tetrazole (teec) (L51, n = 2, Scheme 7) and the three salts [Fe(teec)6](X)2, X = BF4, ClO4, PF6. They found that complexes obtained by slow crystallization exhibited a different magnetic behavior from those obtained by precipitation: a spin crossover of 50% (X = ClO4, crystalline), two gradual steps (X = BF4, PF6), or two steps, one of them being gradual and the other having a thermal hysteresis (X = ClO4 precipitated).
Haasnoot et al. [155] extended their studies to the fluorine, bromine, and iodine-substituted ligands 1-(2-X-ethyl)tetrazole (L51, n = 2) spin-crossover Fe(II) complexes and their BF4 and ClO4 salts. Magnetic studies on the F compound revealed for the BF4 salt a spin transition with T1/2(1) = 137 K and a second more gradual spin transition with T1/2(2) = 108 K. The magnetic susceptibility versus temperature curve for the ClO4 complex, on the other hand shows a one-step transition with T1/2 = 130 K, displaying thus a higher spin-transition temperature. The two salts of the bromide compounds undergo an almost complete and gradual spin transition (over 90%) with T1/2 = 166 K for the BF4 complex and T1/2 = 150 K for the ClO4 complex. The iodide compounds show a gradual transition for both salts and the transitions are 100% complete. The conclusion of the authors was that the substituent of the ethyl tail had practically no influence on the electron density of the tetrazole. Therefore, the differences in the spin-transition behavior are entirely assigned to the differences in crystal packing.
Müller et al. [156] designed tetrazole ligands with longer alkyl-halide substituents based on 1-(3-halopropyl)-1H-tetrazoles (F, Cl, Br and I, L51, Scheme 7, n = 3), in order to study the role of the ligand size on SCO. Their [Fe(L51)6]2+ complexes revealed very different magnetic profiles, the SCO showing different degrees of conversion to the LS state. The fluorine derivative is LS, while the iodine complex still retains 21% residual HS configuration. The T1/2 values vary from 175 K (Br), 212/160 K (Cl), 194 K (F), and 200 K (I) showing no correlation between T1/2 and the size of the halogen. All SCO take place in one step, with the exception of the Cl where two steps are observed. There is a 17 K hysteresis in the second step, which is accompanied by a structural phase transition. In contrast to the previously reported halogenated ethyltetrazoles derivatives, the authors concluded that, for the longer propyl tails analyzed in the present work, no clear correlation was found between increasing spin-transition temperature and halogen size/atomic number. DFT calculations were performed to rationalize the experimental findings. The combined experimental and quantum chemical approach confirmed that the halogen substitution only had an impact on the steric demand of the ligand, while not affecting the electronics of the coordinating tetrazole.
Tonzetich and co-workers [157] reported a Fe(II) complex [Fe(L52)(bpy)Cl], with the pincer ligand L52 (Scheme 7), 2,2′-bipyridyl (bpy) and a chloride. Magnetic studies revealed that the χMT value was 2.30 cm3 K mol−1, slightly lower than that expected for a high-spin Fe(II) ion. Upon cooling, χMT rapidly decreases before reaching a plateau near 100 K with a χMT ~ 0.4 cm3 K mol−1. This behavior reflects SCO with the high spin state being only partially populated at 340 K and rapidly depopulated as the temperature is lowered.
2.4. d7 Complexes, HS (S=3/2) and LS (S=1/2)—Co(II)
Octahedral Co(II) complexes may exist in the HS state, with three unpaired electrons (S = 3/2), or in the LS state, with only one unpaired electron in the eg set, which is therefore associated with a distortion owing to the Jahn–Teller effect. However, among the few reported examples of SCO in Co(II) complexes bearing ligands with halides, the majority does not fall in the previous situation, making this sample very different from the others analyzed before (2.1, 2.2, 2.3). In several complexes, the coordination geometry is trigonal prismatic. The d levels will be split in three groups, according with D3h symmetry, the lowest being dz2 (A’1), followed by dxy, dx2-y2 (E’), and dxz, dyz (E”). The HS state will have three unpaired electrons and the LS only one, as in the octahedral field. However, the energy differences between groups of levels are much smaller in the trigonal prism than in the octahedron, so that a different behavior is expected. The other complexes are tetracoordinate, being mostly tetrahedral for Co(II), with three unpaired electrons half occupying the t2 set. There are no low-spin tetrahedral complexes. In conditions leading to LS octahedral complexes, tetracoordinate species will in general change geometry and become square planar. In the presence of certain ligands, the energy difference between the two geometries will be very small and they may interconvert. These situations are included by some authors as SCO. Indeed, a d7 metal center will have one unpaired electron in a square planar field. Therefore, the spin change will be from 3/2 to 1/2 also in this situation. It is also possible that a distorted tetrahedral geometry occurs when temperature is changed, eventually keeping the same spin, but modifying the magnetic properties, owing to the lower symmetry and other effects.
Li et al. [158] synthesized and characterized two octahedral cobalt complexes with radical ligands, [Co(L53)2(N3)2]MeOH and [Co(L54)2(N3)2] (L53 = IM-5-Br-2Py = 2-(5-bromo-2-pyridyl)-4,4,5,5-tetramethylimidazoline-1-oxyl, L54 = NIT-5-Br-2Py = 2-(5-bromo-2-pyridyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide). For the former, the χMT values increased slowly with cooling from 300 K, reaching a maximum value (3.83 cm3 K mol−1) at 64 K, and then decreased steeply to 1.19 cm3 K mol−1 at 2.0 K. 3.66 cm3 K mol−1, the χMT value at 300 K is larger than the value 2.62 cm3 K mol−1 (with g = 2), expected for one uncoupled system containing HS Co(II) (S = 3/2) and two radical ligands (S = 1/2), the trend observed at high temperatures suggests an intramolecular ferromagnetic interaction between the cobalt(II) atom and the coordinated radical. For the second complex, the χMT value at room temperature is 2.34 cm3 K mol−1, slightly lower than the spin-only value of 2.62 cm3 K mol−1 (also HS Co(II) and two radicals). The χMT value decreases more and more rapidly upon cooling to reach 0.64 cm3 K mol−1 at 2.0 K, indicating strong antiferromagnetic interactions.
Murugesu et al. [159] studied octahedral Co(II) complexes with a terpyridine-inspired tridentate ligand, 2,6-bis(5,6-dialkyl-1,2,4-triazin-3-yl)-pyridine (L55). They obtained the mononuclear complexes [Co(L55)2]2+ or the binuclear [Co2(μ-X)2(L55)2X2] (X = Cl or Br), depending on reaction conditions (Figure 17).
Figure 17.
The structure of the dimeric complex [Co2(μ-Cl)2(L55)2Cl2].
The monomeric [CoII(BTP)2]2+ showed gradual spin-crossover properties between 1.8–370 K, with some dependence on the anion (BF4−, ClO4−, and NCS−). Ferromagnetic exchange interactions were determined with the –2J approach for the binuclear species, and values of J = +2.57 and +2.98 cm−1 were obtained for X = Cl and Br, respectively.
Voloshin and co-workers dedicated much attention to cobalt(II) clathrochelates, many of them bearing halogens, such as Cl [160] (L56, L57, 58) or Br [161] (L56) at the diimine or F at the boron (L58). The chloride derivatives were studied earlier. In the presence of apical F (L58), the complex has a μeff = 3.87 B.M. at 4 K, which is very close to the spin-only magnetic moment for the HS state, and remains HS at low temperatures. When the phenyl or nBu groups replace F (L58 and L58), there is a gradual and incomplete transition from a value close to the doublet LS (1.78 and 1.98 B.M., respectively) to the HS quartet. At 300 K the transition is not yet complete, but it is more advanced for the Ph (0.8 HS) than for the nBu derivative (0.5 HS).
The magnetic properties of isostructural cobalt hexabromoclathrochelate were expected to be similar to those of the Cl analogue. It is LS at temperatures below 100 K, and experiences than the doublet to quartet SCO, reflected in the gradual increase in the effective magnetic moment at higher temperatures and a gap caused by the structure phase transition. This spin transition is gradual and incomplete even at room temperature in solids, but in solution only the HS state is populated above 200 K. The spin transition is not complete even at 400 K owing to significant stabilization of the low spin state by intermolecular interactions.
In these complexes, Co(II) has a trigonal prismatic coordination environment. From our DFT calculations for the Br compound, it can be seen that the HS is rather symmetric, as shown in Figure 18, left, and this is particularly visible in the CoN6 core shown below in two views. The distances are very similar for all bonds (2.083-2.085 Å). The crystal structure at 290 K, containing a majority of HS but also LS species, gives three different distances, 1.949, 1.995, and 2.033 Å, which provides an acceptable comparison. The calculated geometry of the LS species can be compared with the crystal structure obtained at 60 K. The calculated bond lengths were 1.896, 2.100/2.102, and 2.015/2.018 Å, very close to the experimental 1.880, 1.990, and 2.051 Å, and the trigonal prism is much distorted. Interestingly, there is some shrinkage of the bonds, but it is negligible compared to what is usually observed for octahedral species. This is clearly shown in the experimental values [161].
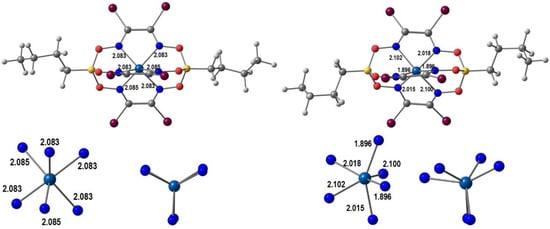
Figure 18.
DFT calculated structures of the HS (left) and LS (right) forms of the cobalt(II) hexabromoclathrochelate, showing the molecule (top) and the CoN6 core in side and top views (bottom).
At the molecular level the changes are small. Indeed, the three Cl compounds differ in the B substituent outside the cage and have different magnetic behavior. On the other hand, Cl and Br derivatives share the nBu tails and behave similarly (structure and magnetism). These aspects led the authors to a more detailed analysis of the role of the environment [162], having concluded that in Co(L56-Cl) the Cl⋅⋅⋅π halogen bonds stabilized the LS state and assigning the SCO features to an anticooperative behavior.
On the other hand, a sudden decrease in magnetic moment was observed at 273 K in the Co(L56-Br), but not in the Cl analogue. This “reverse spin transition” pattern was described previously for SCO cobalt complexes with aliphatic substituents and assigned to conformational changes in their long alkyl chains. In this Br clathrochelate, the nBu chain is much shorter and the overall effect of the phase transition is less dramatic, no hysteresis curve having been observed.
Bushuev et al. [163] reported a series of Co(II) complexes with 4-(3,5-dimethyl-1H-pyrazol-1-yl)-6-methyl-2-phenylpyrimidine (L59) of the general formula CoLX2 (X = Cl, Br, and I) and distorted tetrahedral coordination. All Co(II) complexes are found in the HS state at room temperature with magnetic moment values above the theoretical value for S = 3/2 and non-interacting Co(II) ions (μ = 3.87 µB). Antiferromagnetic interactions arising at low temperature cause a considerable decrease of the effective magnetic moment. A relationship or an effect of the halogen was not discussed.
Peters and Jenkins [164] examined a series of tetrahedral Co(II) with the tripodal L60 (Scheme 7) ligand and an aryloxide, siloxide, arylthiolate, and silylthiolate coligand. Using this tris(phosphino)borate ligands (L60, R = iPr, Ph), they obtained four halide-containing complexes. The [Co(L60)I] complexes are square planar (low-spin) for the less sterically hindered PhBP3 and tetrahedral (high-spin) for the bulkier PhBPiPr3. The other two compounds are both fluorinated, one at an oxide aryl (OC6F5) and the other at a siloxide aryl (Si(4-CF3-Ph)3). The authors found that the spin-state population at a given temperature changed significantly with the substituents at the para position on the siloxide aryl rings. Electron-withdrawing CF3 led to relevant lowering of the spin-crossover critical temperature. The authors also noticed that the fluorinated aryloxide complex [PhBP3]CoO(C6F5) is less likely to exhibit π-back donation to the aryl ring owing to its electron-withdrawing nature, although weak interactions with the ortho fluorine atoms of the aryl group could not be discarded. They proposed an equilibrium between four-coordinate and five-coordinate to explain the change in spin state that is observed in the SQUID measurements for this compound. The SQUID magnetic data was further supported by EPR experiments performed on toluene glasses. The work was also complemented by magnetic measurements in C6D6 solutions, electrochemical studies in THF solution and DFT calculations for structure optimization, single-point energy and continuous symmetry measurements.
3. Results Solution
Harding et al. [95] studied the magnetic behavior of the halogenated qsal Fe(III) [Fe(L3)]+ compounds (Scheme 1) in deuterated DMSO solutions by the Evans method. They detected a mixture of spin states varying from 22–49% HS. The authors noted that, although there was no correlation with the electronegativity or size of the halogens, it is clear that the halogen has some impact on the ligand field and consequently the preferred spin state of the complex. The Evans method was complemented by UV-vis measurements in the same solvent.
Takahashi, Nakamura, Ikeue and co-workers [108] also used the Evans method to investigate the magnetic behavior of the Fe(III) complexes of porphyrins (L18, Scheme 4), one of them bearing pentafluorinated aryl groups, and monodentate ligands (pyNO, 4-ClpyNO, 4-MepyNO, 4-MeOpyNO, 4-Me2NpyNO) in deuterated dichloromethane solutions between 298 and 183 K. The chloride compound (Cl in axial ligand) remains HS over the temperature range studied, but for the fluoro compound the contribution of the S = 5/2 HS state decreases with decreasing temperature. This is reflected in the change in χMT values from 3.7 µB at 298 K to 2.2 µB at 183 K. Complementary EPR measurements of frozen solutions confirmed that the chloro-compound is HS and the fluoro compound is LS at 15 K.
Martinho et al. studied [112] the mono-bromo and di-bromo salEen (L22, Scheme 4) Fe(III) perchlorate and tetraphenylborate salts in acetonitrile solutions by NMR, UV-vis and cyclic voltammetry. The magnetic susceptibility was determined by the Evans method in deuterated acetonitrile solutions and the χMT values were consistent with a mixture of both HS and LS states for both complexes, the di-bromo compounds having the major LS fraction. The number of electron-withdrawing groups (Br) was reflected in a red shift of the electronic absorption and a shift to more positive potentials of all redox processes.
Martinho et al. [165] further investigated the magnetic behavior of the mono-bromo salEen (L22) Fe(III) compound in deuterated DMSO by the Evans method at different concentrations (100, 50, 10 and 5 mM). The χMT values show that the magnetic behavior is concentration independent and shows a mixture of spin states at room temperature with about 64% HS.
The study by Halcrow et al. [38] of Fe(II) complexes of pyrazolylpyridines (bpp) [Fe(L30)2]2+ (Scheme 5) in CD3NO2 solution by 1H NMR revealed that they were predominantly HS at room temperature. Variable-temperature Evans method indicated that the χMT values of the chloride and the bromide compounds change from 3.1(1) cm3 mol−1 K at 338 K to around 2.0 cm3 mol−1 K at 248 K. T1/2 values of 231(2) K for the chloride and 238(2) K for bromide compound in deuterated nitromethane were obtained by extrapolating the data to lower temperature.
Deeth, Halcrow et al. [132] extended the previous studies of the spin states of the bpp (L30) Fe(II) complexes, in solutions of (CD3)2CO or CD3NO2 depending on their solubility, using the variable-temperature Evans method. The authors found that when complexes with the same halogen substituents on the pyrazolyl or the pyridyl group were considered, the stabilization of Eav(eg) by these electron-withdrawing substituents is identical and approximately 25% greater than Eav(t2g) for both sets of complexes. These trends are also supported by a computational study.
Ishida and Kimura [135] reported the solution properties of the Fe(II) complexes of oxazolylpyridines (L33, Scheme 5) with a Cl substituent in the pyridine. Results from the application of the Evans method showed SCO in acetone solution with a T1/2 at 270 K. The authors concluded, when Cl was substituted by other groups, that electron-donating groups suppress T1/2 while electron-withdrawing groups raise T1/2.
Petzold and co-workers [148,149] investigated the solution properties of Fe(II) complexes of pyridinylphenanthrolines (L46, Scheme 6) by UV-vis, cyclic voltammetry and NMR and computational studies. The bromo derivative exhibited SCO in acetone solutions and the authors identify SCO as an underlying chemical process responsible for the two-site exchange, which gives rise to the broadening in the NMR linewidths spectra.
Damrauer et al. [166] studied bis-homoleptic Fe(II) complexes of 2,2′:6′,2″-terpyridyl ligands (L61, Scheme 7), [Fe(L61)2]2+, bearing halogen substituents (X = F, Cl, Br), in order to determine how the halogen affected their spin states. The compounds were studied in both frozen and unfrozen acetonitrile solutions. A high-spin quintet ground state is observed for the Cl and Br derivatives, assigned to substituent-induced intramolecular strain, while the F compound displays SCO (T1/2 of 220 K) and a mixture of low-spin (singlet) and high-spin (quintet) populations at room temperature. UV-vis measurements show that although both molecules with X = Cl and Br appear to be HS with very similar absorption features and band energies, a modest increase in molar extinction is observed when going from Br to Cl. This is consistent with a stronger metal−ligand electronic interaction as the halogen atom size decreases from Br to Cl. A further decrease in the halogen size (F) gives rise to a complexity in the structural, optical, and magnetic data that is reflected in a significant temperature dependence. Notably, a spin-crossover equilibrium is observed where the singlet state is enthalpically favored and the quintet state is populated owing to entropic contributions to the free energy.
Linert et al. reported in several works [124,125,167] a series of Fe(II) using the 4X-substituted 2,6-bis(benzimidazol-2-yl)-pyridines (X = H, OH or Cl, L26, Scheme 5), [Fe(L26)2][ClO4]2. The spin-crossover equilibrium of the complex is reflected in the changes of the UV-vis spectrum, namely the intense charge-transfer band, in the temperature range 276-331 K. Variable-temperature UV-vis measurements in different non–aqueous solvents (methanol, nitromethane, acetone, acetonitrile, propanediolcarbonate and dimethylformamide) have shown that the substituted complexes are HS. The magnetic susceptibilities of the iron complexes in solution were measured in the temperature range 213–343 K, by 1H NMR using the Evans method. The temperature dependence is much less pronounced for the C1− and OH− substituted species than for the unsubstituted ones. Therefore the latter showed SCO in solution, while the other two did not.
Huttner and co-workers reported [168] that the four-coordinate high-spin complex [(η2-triphos)CoCl2] (triphos, L62, Scheme 6) undergoes in solution interconversion in a five-coordinate [{(η3-triphos)Co}2(μ-Cl)2]2+ complex (Figure 19). This isomerization is accompanied by a spin-state change of the Co(II) ion from a doublet state (S = 1/2) to a quartet state (S = 3/2), and can be monitored by both UV-vis spectroscopy and by magnetic measurements. The absorption spectra in THF display three isosbestic points, reflecting a single equilibrium between the two species and the absence of a long-living intermediate during the cooling. In this example, the change in spin state is triggered by the change in coordination geometry around the Co(II) center and the consequent change in electronic structure. The equilibrium is influenced by both temperature and solvent.
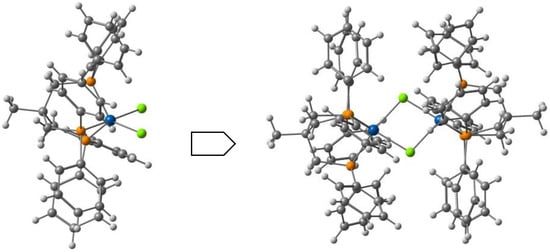
Figure 19.
The structure of [(η2-triphos)CoCl2] and the dimeric cationic complex [{(η3-triphos)Co}2(μ-Cl)2]2+.
Slattery and co-workers [169] prepared a series of iron(II) and cobalt(II) bis-terpyridine (terpy, L61, X=H, Scheme 7) complexes with the general formula [M(L61)2](PF6)2 with a variety of R groups ((C4H8)N, (C4H9)NH, HO, CH3O, CH3-phenyl, H, Cl, CH3SO, CH3SO2 in order of increasing electron-withdrawing capability), including the chloride. Their work was carried out in solution and they studied the role of these substituents in redox and spin state, using electrochemical studies in acetonitrile combined with NMR spectroscopy in d6-acetone. The Fe(II) compounds are all low-spin at room temperature, while the Co(II) compounds display different behavior depending on the nature of the substituent. This was assigned to a spin equilibrium related with the ability of undergoing spin crossover in solution. No clear relationship was found between the experimental trends and the electron-withdrawing nature of the terpyridine substituents.
Harris, Thorarinsdottir and Gaudette [170] studied the three [Fe(L)]2+ complexes (L = L63, L64, L65, Scheme 7), where the ligands are 1,4,7-triazacyclononane (tacn) with three functionalized pyridinyl pending arms, in solution by UV-vis and NMR spectroscopy. Variable-temperature UV-vis in both acetonitrile and water showed that, upon cooling, the L63 complex showed changes in the LS band, suggesting a change in spin state, the second compound (L64) remained in the HS state and the third (L65) in the LS state. In water, the LS state is stabilized and T1/2 is shifted towards higher temperatures by formation of a solvate. The magnetic moments of the compounds were studied in aqueous solutions between 5 and 60 °C by the Evans method. The L64 complex remains in the HS state. The χMT of the L63 derivative increases almost linearly with increasing temperature, from a minimum of 0.93 cm3 mol−1 K at 5 °C to a maximum of 1.99 cm3 mol−1 K at 60 °C, indicative of thermally induced spin-crossover. A linear fit gives T1/2 = 325(1) K or 52(1) °C. In acetonitrile, χMT increases almost linearly with increasing temperature, from 0.62 cm3 K mol−1 at −42 °C to 2.71 cm3 K mol−1 at 60 °C, and a linear fit affords T1/2 = 17(1) °C. The value of T1/2, which is 35 °C lower in acetonitrile than in H2O, reflects the different donor strengths of the H2O and CH3CN.
While the number and extent of solutions studies is limited and presents challenges different from those in the solid state, its study may in the future allow a more interpretation of the SCO phenomenon at the molecular level.
4. Computational Studies
In this review, we examined approximately 150 papers describing SCO in complexes containing halogens as ligands and substituents of ligands. The crystal structure for at least one spin state of the complexes studied was determined in 102 of them and the structures of both spin states were available in 14 papers. Our computational efforts focused mostly on the structures of the 27 compounds in this small subset of papers, which allowed us to compare results from experiment and from calculations performed on simpler models (see Computational Methods). The relevant parameters for the SCO phenomenon, such as ΔEHS-LS obtained from single-crystal unit cells and from DFT optimized counterparts, were calculated. The energy barrier for spin conversion (MECP) between the HS and LS isomers was also calculated. Δoct values were obtained from the previous calculations. They were estimated from the energy difference between the highest occupied t2g and the lowest unoccupied eg beta orbitals of the relevant octahedral complexes in their HS electronic configuration.
This study was aimed at calculating, for a large group of compounds, the parameters relevant for the SCO with a methodology which had been used for isolated examples and is described in Computational Methods (Section 6). The first question was whether an optimized structure can reproduce the features of an experimental one, considering that the models included cations and anions and the remaining interactions in the solid are weak (hydrogen and halogen bonds, van der Waals interactions). How important is the cooperative effect of many weak interactions? This is an important issue because experimentally determined structures are not available for many systems. In order to probe for any correlation between calculated data for SCO compounds, 27 compounds with the crystal structures available for both spin states were selected for further analysis. The difference between the energy of the LS and HS optimized structures was plotted against the same energy difference calculated from a single-point calculation performed on the experimental geometry, usually cation and anion (Supplementary materials Figure S2). Not unexpectedly, there was no correlation.
This indicates that better models are needed, e.g., incorporating periodic boundary conditions. Recently the approaches followed by Vela et al. [171,172] [have shown interesting insights, namely that the most significant role that intermolecular interactions play in the dynamics of spin interchange within a crystal lattice involve the complex and another neighboring unit and the counter ion. The degree to which these magnetic cooperative effects and the chemistry behind SCO complexes determine the energetics of spin crossover will certainly remain a significant challenge for some time to come. Without this broader insight, only minor variations on ligand composition can make rational design a viable option. Knowledge of how each factor contributes to SCO will lay the groundwork for establishing design principles. Certainly, further work is required in the field.
The attempt to correlate kinetics was based on the minimum energy crossing point (MECP), which is associated with the barrier to change between spin states. However, no correlation was observed when plotting the energy difference between MECP and the LS or HS state vs. the HS-LS energy difference (Figure S3 in SI). We should point out again that the quality of the computational approach must be improved. Still, how to predict SCO?
5. Conclusions
The thermal spin-crossover phenomenon for 3d4-3d7 transition metal mono- or binuclear complexes, namely Mn(III), Fe(III), Fe(II) and Co(II), with halogenated ligands was reviewed. SCO usually is observed in solids but there are a few solution studies. The complexes have in general octahedral geometries, the few exceptions being some Co(II) complexes. Despite the relatively large size of the sample, trends were not easily observed. Some authors studied series of complexes with different halogen substituents (F, Cl, Br, I), varying also the counter ions and the solvents (often cocrystalized) and showed how SCO depended on all these effects, reflecting the role of the nature of intermolecular interactions on SCO. The situation was made worse in some situations where polymorphs were observed. Halogens may participate in halogen and hydrogen bonds, as well as other intermolecular interactions, so that often a small change in the molecular structure has dramatic consequences. Only in few examples did the authors state that packing effects could not be held responsible for the SCO patterns and proposed a ligand field effect. In these examples we calculated, using a DFT approach, the energy difference between the HS and the LS states, both based on single points carried out on the experimental structure and on the DFT optimized geometry, as well as the MECPs (minimum energy crossing points), as defined in Figure 2. The T1/2 did not correlate with ΔEHS-LS (X-ray), ΔEHS-LS (opt), ΔEMECP-LS, or ΔEMECP-HS, but increased in general with the values of Δoct obtained from the calculations. In tetracoordinate Co(II) complexes, SCO was triggered by a geometry change. Solution studies are small in number and even more inconclusive. With this review, we hope to have shown that SCO rationalization, even for a small subset of the available molecules—such as those that bear halogens—is still an extremely intricate problem and suggest readers look beyond the HS-LS energy difference in order to try to explain these phenomena.
6. Computational Methods
Density Functional Theory [66] (DFT) calculations were performed using the ORCA program package [173]. Geometry optimizations, without symmetry constraints, were performed using the B3LYP* hybrid density functional [174,175]. Spin unrestricted calculations were performed for the relevant spin states (open shell). The split valence basis set [def2-SV(P)] [176] of Ahlrichs was used, with polarization functions on all atoms except hydrogen with the Coulomb fitting basis set by Weigend [177]. The quality of the basis set was limited by the number of systems and calculations performed. Besides, some systems had a large number of atoms. The resolution of identity with chain of spheres approximation (RIJCOSX) [178] was employed with default grid settings. Dispersion effects were calculated with the damped [179] third generation Grimme corrections (D3BJ) [180,181]. The structures were modelled after the CIF files available on the original papers (see Supporting Information). Solvation effects, when relevant, were taken into account with the conductor-like polarizable continuum model (CPCM) [182]. The minimum energy crossing points (MECPs) for the potential energy surfaces of both spin states were also obtained using the ORCA program with the keyword SurfCrossOpt. The procedure to compute the MECP was laid out by Harvey et al. [183] and will be summarized below. For two spin state potential energy Surfaces 1 and 2 with energies E1 and E2 the MECP is calculated by taking the following sum of gradients of nuclear coordinates q and equalling it to zero (Equation (1)) to fulfil the condition of a stationary point at the crossing seam:
These two components are perpendicular and parallel respectively to the hyperline in the 3N-7 coordinate space of Surface 1, where each term is:
The DFT obtained structures were graphically analyzed with Chemcraft [184] and Mercury [185].
Supplementary Materials
The following are available online at https://www.mdpi.com/2071-1050/12/6/2512/s1. Correlations and calculated energy differences. Table S1: Dataset for the correlation analysis displayed in Figures S3 and S4 of main text, Figure S1: A representation of the ligands presented in Table S1, Figure S2: Energy difference calculated between the optimized structures of HS and LS states against the same energy difference between the crystal structures for both spin states (single point calculation) for Fe(II), Mn(III), and Co(II) complexes, Figure S3: Energy difference between the MECP and the LS (top) or HS (bottom) energy against the energy difference between the HS and LS optimized structures.
Author Contributions
Literature search and data collection (P.N.M., F.F.M., N.A.G.B., M.J.C.); Computational methods (F.F.M., N.A.G.B., M.J.C.); Drafting the work (P.N.M., M.J.C.); Critical revision (P.N.M., F.F.M., N.A.G.B., M.J.C.); Supervision (P.N.M., M.J.C.). All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This work was supported by the Fundacão para a Ciência e a Tecnologia (FCT), Portugal [projects PTDC/QEQ-QIN/3414/2014, UID/MULTI/00612/2019 and UID/MULTI/04046/2019]. PNM thanks FCT for the program CEECIND/00509/2017.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cambi, L.; Szegö, L. Über die Magnetische Susceptibilität der Komplexen Verbindungen. Ber. Dtsch. Chem. Ges. 1931, 64, 2591–2598. [Google Scholar] [CrossRef]
- Gütlich, P.; Garcia, Y.; Goodwin, H.A. Spin Crossover Phenomena in Fe(II) Complexes. Chem. Soc. Rev. 2000, 29, 419–427. [Google Scholar] [CrossRef]
- Garcia, Y.; Niel, V.; Muñoz, M.C.; Real, J.A. Spin Crossover in 1D, 2D and 3D Polymeric Fe(II) Networks. Top. Curr. Chem. 2004, 233, 229–257. [Google Scholar] [CrossRef]
- Real, J.A.; Gaspar, A.B.; Muñoz, M.C.; Gütlich, P.; Ksenofontov, V.; Spiering, H. Bipyrimidine-Bridged Dinuclear Iron(II) Spin Crossover Compounds. Top. Curr. Chem. 2004, 233, 167–193. [Google Scholar] [CrossRef]
- Toftlund, H.; McGarvey, J.J. Iron(II) Spin Crossover Systems with Multidentate Ligands. Top. Curr. Chem. 2004, 233, 151–166. [Google Scholar] [CrossRef]
- Goodwin, H.A. Spin Crossover in Iron(II) Tris(diimine) and Bis(terimine) Systems. Top. Curr. Chem. 2004, 233, 59–90. [Google Scholar] [CrossRef]
- Van Koningsbruggen, P.J. Special Classes of Iron(II) Azole Spin Crossover Compounds. Top. Curr. Chem. 2004, 233, 123–149. [Google Scholar] [CrossRef]
- Thompson, A.L.; Money, V.A.; Goeta, A.E.; Howard, J.A.K. Structural Studies of Thermal- and Light-Induced Transitions in Iron(II) gSpin-Crossover Complexes. Comptes Rendus Chim. 2005, 8, 1365–1373. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, F.; Liu, T.; Yuan, M.; Wang, Z.-M.; Gao, S. Spin Crossover in a Series of Iron(II) Complexes of 2-(2-Alkyl-2H-tetrazol-5-yl)-1,10-phenanthroline: Effects of Alkyl Side Chain, Solvent, and Anion. Inorg. Chem. 2007, 46, 2541–2555. [Google Scholar] [CrossRef]
- Halcrow, M.A. The Spin-States and Spin-Transitions of Mononuclear Iron(II) Complexes of Nitrogen-Donor Ligands. Polyhedron 2007, 26, 3523–3576. [Google Scholar] [CrossRef]
- Larionov, S.V. Spin Transition in Iron(III) and Iron(II) Complexes. Russ. J. Coord. Chem. 2008, 34, 237–250. [Google Scholar] [CrossRef]
- Murray, K.S. Advances in Polynuclear Iron(II), Iron(III) and Cobalt(II) Spin-Crossover Compounds. Eur. J. Inorg. Chem. 2008, 2008, 3101–3121. [Google Scholar] [CrossRef]
- Kitchen, J.A.; Brooker, S. Spin Crossover in Iron(II) Complexes of 3,5-Di(2-pyridyl)-1,2,4-triazoles and 3,5-Di(2-pyridyl)-1,2,4-triazolates. Coord. Chem. Rev. 2008, 252, 2072–2092. [Google Scholar] [CrossRef]
- Salitros, I.; Madhu, N.T.; Boca, R.; Pavlik, J.; Ruben, M. Room-Temperature Spin-Transition Iron Compounds. Monatsh. Chem. 2009, 140, 695–733. [Google Scholar] [CrossRef]
- Wolny, J.A.; Paulsen, H.; Trautwein, A.X.; Schünemann, V. Density Functional Theory Calculations and Vibrational Spectroscopy on Iron Spin-Crossover Compounds. Coord. Chem. Rev. 2009, 253, 2423–2431. [Google Scholar] [CrossRef]
- Halcrow, M.A. Iron(II) Complexes of 2,6-Di(pyrazol-1-yl)pyridines-A Versatile System for Spin-Crossover Research. Coord. Chem. Rev. 2009, 253, 2493–2514. [Google Scholar] [CrossRef]
- Olguín, J.; Brooker, S. Spin Crossover Active Iron(II) Complexes of Selected Pyrazole-Pyridine/Pyrazine Ligands. Coord. Chem. Rev. 2011, 255, 203–240. [Google Scholar] [CrossRef]
- Gütlich, P.; Gaspar, A.B.; Garcia, Y. Spin State Switching in Iron Coordination Compounds. Beilstein J. Org. Chem. 2013, 9, 342–391. [Google Scholar] [CrossRef]
- Craig, G.A.; Roubeau, O.; Aromí, G. Spin State Switching in 2,6-Bis(pyrazol-3-yl)pyridine (3-bpp) based Fe(II) Complexes. Coord. Chem. Rev. 2014, 269, 13–31. [Google Scholar] [CrossRef]
- Levchenko, G.; Khristov, A.V.; Varyukhin, V.N. Spin Crossover in Iron(II)-Containing Complex Compounds under a Pressure. Low. Temp. Phys. 2014, 40, 571–585. [Google Scholar] [CrossRef]
- Ortega-Villar, N.; Muñoz, M.C.; Real, J.A. Symmetry Breaking in Iron(II) Spin-Crossover Molecular Crystals. Magnetochemistry 2016, 2, 16. [Google Scholar] [CrossRef]
- Scott, H.S.; Staniland, R.W.; Kruger, P.E. Spin Crossover in Homoleptic Fe(II) Imidazolylimine Complexes. Coord. Chem. Rev. 2018, 362, 24–43. [Google Scholar] [CrossRef]
- Boillot, M.-L.; Weber, B. Mononuclear Ferrous and Ferric Complexes. Comptes Rendus Chim. 2018, 21, 1196–1208. [Google Scholar] [CrossRef]
- Hogue, R.W.; Singh, S.; Brooker, S. Spin Crossover in Discrete Polynuclear Iron(II) Complexes. Chem. Soc. Rev. 2018, 47, 7303–7338. [Google Scholar] [CrossRef]
- Van Koningsbruggen, P.J.; Maeda, Y.; Oshio, H. Iron(III) Spin Crossover Compounds. Top. Curr. Chem. 2004, 233, 259–324. [Google Scholar]
- Nihei, M.; Shiga, T.; Maeda, Y.; Oshio, H. Spin Crossover Iron(III) Complexes. Coord. Chem. Rev. 2007, 251, 2606–2621. [Google Scholar] [CrossRef]
- Harding, D.J.; Harding, P.; Phonsri, W. Spin Crossover in Iron(III) Complexes. Coord. Chem. Rev. 2016, 313, 38–61. [Google Scholar] [CrossRef]
- Goodwin, H.A. Spin Crossover in Cobalt(II) Systems. Top. Curr. Chem. 2004, 234, 23–47. [Google Scholar] [CrossRef]
- Krivokapic, I.; Zerara, M.; Lawson Daku, L.M.; Vargas, A.; Enachescu, C.; Ambrus, C.; Tregenna-Piggott, P.; Amstutz, N.; Krausz, E.; Hauser, A. Spin-Crossover in Cobalt(II) Imine Complexes. Coord. Chem. Rev. 2007, 251, 364–378. [Google Scholar] [CrossRef]
- Garcia, Y.; Gütlich, P. Thermal Spin Crossover in Mn(II), Mn(III), Cr(II) and Co(III) Coordination Compounds. Top. Curr. Chem. 2004, 234, 49–62. [Google Scholar] [CrossRef]
- Halcrow, M.A. Spin-Crossover Materials: Properties and Applications; Wiley & Sons, Ltd.: West Sussex, UK, 2013. [Google Scholar]
- Borshch, S.A. Comprehensive Inorganic Chemistry II, 2nd ed.; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 469–480. [Google Scholar]
- Gudyma, I.; Maksymov, A.; Bobák, A. Nanophysics, Nanomaterials, Interface Studies, and Applications; NANO 2016, Springer Proceedings in Physics; Fesenko, O., Yatsenko, L., Eds.; Springer: Berlin, Germany, 2017; Volume 195. [Google Scholar]
- Pavlik, J.; Linares, J. Microscopic Models of Spin Crossover. Comptes Rendus Chim. 2018, 21, 1170–1178. [Google Scholar] [CrossRef]
- Calhorda, M.J.; Costa, P.J. Comprehensive Inorganic Chemistry II; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 801–809. [Google Scholar]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Jeon, I.-R.; Jeannin, O.; Clérac, R.; Rouzières, M.; Fourmigué, M. Spin-State Modulation of Molecular FeIII Complexes via Inclusion in Halogen-Bonded Supramolecular Networks. Chem. Commun. 2017, 53, 4989–4992. [Google Scholar] [CrossRef]
- Pritchard, R.; Lazar, H.; Barrett, S.A.; Kilner, C.A.; Asthana, S.; Carbonera, C.; Létard, J.F.; Halcrow, M.A.; Sheu, H.S.; Yasuda, N.; et al. Thermal and Light-Induced Spin-Transitions in Iron(II) Complexes of 2,6-Bis(4-halopyrazolyl)pyridines: The Influence of Polymorphism on a Spin-Crossover Compound. Dalton Trans. 2009, 125, 6656–6666. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Walker, F.A. Solution NMR Studies of Iron(II) Spin-Crossover Complexes. Inorg. Chem. 2007, 46, 6794–6803. [Google Scholar] [CrossRef]
- Van der Meer, M.; Rechkemmer, Y.; Breitgoff, F.D.; Dechert, S.; Marx, R.; Dörfel, M.; Neugebauer, P.; van Slageren, J.; Sarkar, B. Probing Bistability in FeII and CoII Complexes with an Unsymmetrically Substituted Quinonoid Ligand. Dalton Trans. 2016, 45, 8394–8403. [Google Scholar] [CrossRef]
- Petzold, H.; Djomgoue, P.; Hörner, G.; Speck, J.M.; Rüffer, T.; Schaarschmidt, D. 1H NMR Spectroscopic Elucidation in Solution of the Kinetics and Thermodynamics of Spin Crossover for an Exceptionally Robust Fe2+ Complex. Dalton Trans. 2016, 45, 13798–13809. [Google Scholar] [CrossRef]
- Luo, Y.H.; Nihei, M.; Wen, G.J.; Sun, B.W.; Oshio, H. Ambient-Temperature Spin-State Switching Achieved by Protonation of the Amino Group in [Fe(H2Bpz2)2(bipy-NH2)]. Inorg. Chem. 2016, 55, 8147–8152. [Google Scholar] [CrossRef]
- Rodríguez-Jiménez, S.; Yang, M.; Stewart, I.; Garden, A.L.; Brooker, S. A Simple Method of Predicting Spin State in Solution. J. Am. Chem. Soc. 2017, 139, 18392–18396. [Google Scholar] [CrossRef]
- Devid, E.J.; Martinho, P.N.; Kamalakar, M.V.; Šalitroš, I.; Prendergast, Ú.; Dayen, J.-F.; Meded, V.; Lemma, T.; González-Prieto, R.; Evers, F.; et al. Spin Transition in Arrays of Gold Nanoparticles and Spin Crossover Molecules. ACS Nano 2015, 9, 4496–4507. [Google Scholar] [CrossRef]
- Meded, V.; Bagrets, A.; Fink, K.; Chandrasekar, R.; Ruben, M.; Evers, F.; Bernand-Mantel, A.; Seldenthuis, J.S.; Beukman, A.; van der Zant, H.S.J. Electrical Control over the Fe(II) Spin Crossover in a Single Molecule: Theory and Experiment. Phys. Rev. B 2011, 83, 245415. [Google Scholar] [CrossRef]
- Miyamachi, T.; Gruber, M.; Davesne, V.; Bowen, M.; Boukari, S.; Joly, L.; Scheurer, F.; Rogez, G.; Yamada, T.K.; Ohresser, P.; et al. Robust Spin Crossover and Memristance Across a Single Molecule. Nat. Commun. 2012, 3, 938. [Google Scholar] [CrossRef] [PubMed]
- Gopakumar, T.G.; Matino, F.; Naggert, H.; Bannwarth, A.; Tuczek, F.; Berndt, R. Electron-Induced Spin Crossover of Single Molecules in a Bilayer on Gold. Angew. Chem. Int. Ed. 2012, 51, 6262–6266. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, A.C.; Aravena, D.; Cerdá, J.I.; Acís-Castillo, Z.; Li, H.; Real, J.A.; Sanz, F.; Hihath, J.; Ruiz, E.; Díez-Pérez, I. Large Conductance Switching in a Single-Molecule Device through Room Temperature Spin-Dependent Transport. Nano Lett. 2016, 16, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Lefter, C.; Rat, S.; Costa, J.S.; Manrique-Juárez, M.D.; Quintero, C.M.; Salmon, L.; Séguy, I.; Leichle, T.; Nicu, L.; Demont, P.; et al. Current Switching Coupled to Molecular Spin-States in Large-Area Junctions. Adv. Mater. 2016, 28, 7508–7514. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-H.; Liu, Q.-L.; Yang, L.-J.; Sun, Y.; Wang, J.-W.; You, C.-Q.; Sun, B.-W. Magnetic Observation of Above Room-Temperature Spin Transition in Vesicular Nano-Spheres. J. Mater. Chem. C 2016, 4, 8061–8069. [Google Scholar] [CrossRef]
- Martinho, P.N.; Ortin, Y.; Gildea, B.; Gandolfi, C.; McKerr, G.; O’Hagan, B.; Albrecht, M.; Morgan, G.G. Inducing Hysteretic Spin Crossover in Solution. Dalton Trans. 2012, 41, 7461–7463. [Google Scholar] [CrossRef]
- Gandolfi, C.; Morgan, G.G.; Albrecht, M. A Magnetic Iron(III) Switch with Controlled and Adjustable Thermal Response for Solution Processing. Dalton Trans. 2012, 41, 3726–3730. [Google Scholar] [CrossRef]
- White, N.G.; Feltham, H.L.C.; Gandolfi, C.; Albrecht, M.; Brooker, S. Towards Langmuir–Blodgett Films of Magnetically Interesting Materials: Solution Equilibria in Amphiphilic Iron(II) Complexes of a Triazole-Containing Ligand. Dalton Trans. 2010, 39, 3751–3758. [Google Scholar] [CrossRef]
- Schlamp, S.; Thoma, P.; Weber, B. Influence of the Alkyl Chain Length on the Self-Assembly of Amphiphilic Iron Complexes: An Analysis of X-ray Structures. Chem. Eur. J. 2014, 20, 6462–6473. [Google Scholar] [CrossRef]
- Rosario-Amorin, D.; Dechambenoit, P.; Bentaleb, A.; Rouzières, M.; Mathonière, C.; Clérac, R. Multistability at Room Temperature in a Bent-Shaped Spin-Crossover Complex Decorated with Long Alkyl Chains. J. Am. Chem. Soc. 2018, 140, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Aravena, D.; Ruiz, E. Coherent Transport through Spin-Crossover Single Molecules. J. Am. Chem. Soc. 2012, 134, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Baadji, N.; Piacenza, M.; Tugsuz, T.; Sala, F.D.; Maruccio, G.; Sanvito, S. Electrostatic Spin Crossover Effect in Polar Magnetic Molecules. Nat. Mater. 2009, 8, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Osorio, E.A.; Moth-Poulsen, K.; van der Zant, H.S.J.; Paaske, J.; Hedegård, P.; Flensberg, K.; Bendix, J.; Bjørnholm, T. Electrical Manipulation of Spin States in a Single Electrostatically Gated Transition-Metal Complex. Nano Lett. 2010, 10, 105–110. [Google Scholar] [CrossRef]
- Harzmann, G.D.; Frisenda, R.; van der Zant, H.S.J.; Mayor, M. Single-Molecule Spin Switch Based on Voltage-Triggered Distortion of the Coordination Sphere. Angew. Chem. Int. Ed. 2015, 54, 13425–13430. [Google Scholar] [CrossRef]
- Frisenda, R.; Harzmann, G.D.; Celis Gil, J.A.; Thijssen, J.M.; Mayor, M.; van der Zant, H.S.J. Stretching-Induced Conductance Increase in a Spin-Crossover Molecule. Nano Lett. 2016, 16, 4733–4737. [Google Scholar] [CrossRef]
- Kahn, O. Spin-Crossover Molecular Materials. Curr. Opin. Solid State Mater. Sci. 1996, 1, 547–554. [Google Scholar] [CrossRef]
- Létard, J.-F.; Guionneau, P.; Goux-Capes, L. Towards Spin Crossover Applications. Top. Curr. Chem. 2004, 235, 221–249. [Google Scholar] [CrossRef]
- Kumar, K.S.; Ruben, M. Emerging Trends in Spin Crossover (SCO) Based Functional Materials and Devices. Coord. Chem. Rev. 2017, 346, 176–205. [Google Scholar] [CrossRef]
- Li, H.; Peng, H. Recent Advances in Self-Assembly of Spin Crossover Materials and their Applications. Curr. Opin. Colloid Interface Sci. 2018, 35, 9–16. [Google Scholar] [CrossRef]
- Linares, J.; Codjovi, E.; Garcia, Y. Pressure and Temperature Spin Crossover Sensors with Optical Detection. Sensors 2012, 12, 4479–4492. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Harvey, J.N. Spin-Forbidden Reactions: Computational Insight into Mechanisms and Kinetics. WIREs Comput. Mol. Sci. 2014, 4, 1–14. [Google Scholar] [CrossRef]
- Wang, S.; Ferbinteanu, M.; Marinescu, C.; Dobrinescu, A.; Ling, Q.-D.; Huang, W. Case Study on a Rare Effect: The Experimental and Theoretical Analysis of a Manganese(III) Spin-Crossover System. Inorg. Chem. 2010, 49, 9839–9851. [Google Scholar] [CrossRef] [PubMed]
- Pandurangan, K.; Gildea, B.; Murray, C.; Harding, C.J.; Müller-Bunz, H.; Morgan, G.G. Lattice Effects on the Spin-Crossover Profile of a Mononuclear Manganese(III) Cation. Chem. Eur. J. 2012, 18, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- Gildea, B.; Gavin, L.C.; Murray, C.A.; Müller-Bunz, H.; Harding, C.J.; Morgan, G.G. Supramolecular Modulation of Spin Crossover Profile in Manganese(III). Supramol. Chem. 2012, 24, 641–653. [Google Scholar] [CrossRef]
- Fitzpatrick, A.J.; Trzop, E.; Müller-Bunz, H.; Dîrtu, M.M.; Garcia, Y.; Collet, E.; Morgan, G.G. Electronic vs. Structural Ordering in a Manganese(III) Spin Crossover Complex. Chem. Commun. 2015, 51, 17540–17543. [Google Scholar] [CrossRef]
- Sirirak, J.; Harding, D.J.; Harding, P.; Murray, K.S.; Moubaraki, B.; Liu, L.; Telfer, S.G. Spin Crossover in cis Manganese(III) Quinolylsalicylaldiminates. Eur. J. Inorg. Chem. 2015, 2015, 2534–2542. [Google Scholar] [CrossRef]
- Morgan, G.G.; Murnaghan, K.D.; Müller-Bunz, H.; McKee, V.; Harding, C.J. A Manganese(III) Complex that Exhibits Spin Crossover Triggered by Geometric Tuning. Angew. Chem. Int. Ed. 2006, 45, 7192–7195. [Google Scholar] [CrossRef]
- Fitzpatrick, A.J.; Stepanovic, S.; Müller-Bunz, H.; Gruden-Pavlović, M.A.; García-Fernández, P.; Morgan, G.G. Challenges in Assignment of Orbital Populations in a High Spin Manganese(III) Complex. Dalton Trans. 2016, 45, 6702–6708. [Google Scholar] [CrossRef]
- Brewer, C.T.; Brewer, G.; May, L.; Sitar, J.; Wang, R. Spin Crossover in Heterodinuclear Iron(III) Schiff-Base Complexes. J. Chem. Soc. Dalton Trans. 1993, 151–155. [Google Scholar] [CrossRef]
- Fallon, G.D.; McLachlan, G.A.; Moubaraki, B.; Murray, K.S.; O’Brien, L.; Spiccia, L. Mononuclear Chromium(III), Manganese(II) and Iron(III) Complexes of the Pentadentate Ligand 1;4-Bis(2-pyridylmethyl)-1;4;7-triazacyclononane. J. Chem. Soc. Dalton Trans. 1997, 2765–2769. [Google Scholar] [CrossRef]
- Hernández-Molina, R.; Mederos, A.; Dominguez, S.; Gili, P.; Ruiz-Pérez, C.; Castiñeiras, A.; Solans, X.; Lloret, F.; Real, J.A. Different Ground Spin States in Iron(III) Complexes with Quadridentate Schiff Bases: Synthesis; Crystal Structures; and Magnetic Properties. Inorg. Chem. 1998, 37, 5102–5108. [Google Scholar] [CrossRef]
- Floquet, S.; Boillot, M.-L.; Rivière, E.; Varret, F.; Boukheddaden, K.; Morineau, D.; Négrier, P. Spin Transition with a Large Thermal Hysteresis Near Room Temperature in a Water Solvate of an Iron(III) Thiosemicarbazone Complex. New J. Chem. 2003, 27, 341–348. [Google Scholar] [CrossRef]
- Zelentsov, V.V.; Bogdanova, L.G.; Ablov, A.V.; Gerbeleuand, N.V.; Dyatlova, C.V. Themomagnetic Studies of Fe(III) Complexes with Thiosemicarbazones of Substituted Ortho hydroxybenzaldehydes. Russ. J. Inorg. Chem. 1973, 18, 2654–2657. [Google Scholar]
- Zelentsov, V.V.; Mokshin, V.M.; Sobolev, S.S.; Shipilov, V.I. The Influence of Crystallization Water upon Magnetic Properties of Fe(III) Thiosemicarbazones. Dokl. Akad. Nauk. SSSR 1984, 277, 900. [Google Scholar]
- Floquet, S.; Guillou, N.; Négrier, P.; Rivière, E.; Boillot, M.-L. The Crystallographic Phase Transition for a Ferric Thiosemicarbazone Spin Crossover Complex Studied by X-ray Powder Diffraction. New J. Chem. 2006, 30, 1621–1627. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Dai, J.-W.; Gagnon, K.J.; Cai, H.-L.; Yamamoto, T.; Einaga, Y.; Zhao, H.-H.; Kanegawa, S.; Sato, O.; Dunbar, K.R.; et al. A Neutral Fe(III) Compound Exhibiting a Two-Step Spin Transition and Dielectric Anomalies. Dalton Trans. 2013, 42, 14685–14688. [Google Scholar] [CrossRef]
- Krupska, A.; Augustyniak-Jablokow, M.A.; Yablokov, Y.V.; Zelentsov, V.V.; Ulanov, V.A.; Mrozinski, J. EPR Discovery of a New Pressure-Induced Low-Spin Phase in (2Me–5Et–PyH)[Fe(Th–5Cl–Sa)2]. Acta Phys. Pol. A 2006, 110, 81–95. [Google Scholar] [CrossRef]
- Phonsri, W.; Macedo, D.S.; Davies, C.G.; Jameson, G.N.L.; Moubaraki, B.; Murray, K.S. Heteroleptic Iron(III) Schiff Base Spin Crossover Complexes: Halogen Substitution; Solvent Loss and Crystallite Size Effects. Dalton Trans. 2017, 46, 7020–7029. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Dai, J.-W.; Shiota, Y.; Yoshizawa, K.; Kanegawa, S.; Sato, O. Multi-Step Spin Crossover Accompanied by Symmetry Breaking in an FeIII Complex: Crystallographic Evidence and DFT Studies. Chem. Eur. J. 2013, 19, 12948–12952. [Google Scholar] [CrossRef]
- Floquet, S.; Rivière, E.; Boukheddaden, K.; Morineau, D.; Boillot, M.-L. Neutral Ferric Complexes of Salicylaldehyde Thiosemicarbazone Ligands: An Exceptional Family of Complexes Exhibiting Discontinuous Spin Transition Behavior. Polyhedron 2014, 80, 60–68. [Google Scholar] [CrossRef]
- Kang, S.; Shiota, Y.; Kariyazaki, A.; Kanegawa, S.; Yoshizawa, K.; Sato, O. Heterometallic FeIII/K Coordination Polymer with a Wide Thermal Hysteretic Spin Transition at Room Temperature. Chem. Eur. J. 2016, 22, 532–538. [Google Scholar] [CrossRef]
- Nakanishi, T.; Okazawa, A.; Sato, O. Halogen Substituent Effect on the Spin-Transition Temperature in Spin-Crossover Fe(III) Compounds Bearing Salicylaldehyde 2-Pyridyl Hydrazone-Type Ligands and Dicarboxylic Acids. Inorganics 2017, 5, 53. [Google Scholar] [CrossRef]
- Mukherjee, S.; Weyhermüller, T.; Eckhard, B.; Wieghardt, K.; Chaudhuri, P. Tuning of Spin Transition in Radical-Containing Iron(III) Complexes by Remote Ligand Substituents. Inorg. Chem. 2005, 44, 7099–7108. [Google Scholar] [CrossRef] [PubMed]
- Enachescu, C.; Hauser, A.; Girerd, J.; Boillot, M. Photoexcitation and Relaxation Dynamics of Catecholato–Iron(III) Spin-Crossover Complexes. ChemPhysChem 2006, 7, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Simaan, A.J.; Boillot, M.-L.; Rivière, E.; Boussac, A.; Girerd, J.-J. A Two-Step Spin Crossover in [(TPA)FeIII(cat)]BPh4. Angew. Chem. Int. Ed. 2000, 39, 196–198. [Google Scholar] [CrossRef]
- Collet, E.; Boillot, M.-L.; Hébert, J.; Moisan, N.; Servol, M.; Lorenc, M.; Toupet, L.; Cointe, B.L.; Tissot, A.; Sainton, J. Polymorphism in the Spin-Crossover Ferric Complexes [(TPA)FeIII(TCC)]PF6. Acta Crystallogr. Sect. B 2009, 65, 474–480. [Google Scholar] [CrossRef]
- Tissot, A.; Shepherd, H.J.; Toupet, L.; Collet, E.; Sainton, J.; Molnár, G.; Guionneau, P.; Boillot, M.-L. Temperature- and Pressure-Induced Switching of the Molecular Spin State of an Orthorhombic Iron(III) Spin-Crossover Salt. Eur. J. Inorg. Chem. 2013, 2013, 1001–1008. [Google Scholar] [CrossRef]
- Neves, A.I.S.; Dias, J.C.; Vieira, B.J.C.; Santos, I.C.; Branco, M.B.C.; Pereira, L.C.K.; Waerenborgh, J.C.; Almeida, M.; Belo, D.; Gama, V. A New Hybrid Material Exhibiting Room Temperature Spin-Crossover and Ferromagnetic Cluster-Glass Behavior. CrystEngComm 2009, 11, 2160–2168. [Google Scholar] [CrossRef]
- Phonsri, W.; Harding, D.J.; Harding, P.; Murray, K.S.; Moubaraki, B.; Gass, I.A.; Cashion, J.D.; Jameson, G.N.L.; Adams, H. Stepped Spin Crossover in Fe(III) Halogen Substituted Quinolylsalicylaldimine Complexes. Dalton Trans. 2014, 43, 17509–17518. [Google Scholar] [CrossRef]
- Harding, D.J.; Phonsri, W.; Harding, P.; Gass, I.A.; Murray, K.S.; Moubaraki, B.; Cashion, J.D.; Liu, L.; Telfer, S.G. Abrupt Spin Crossover in an Iron(III) Quinolylsalicylaldimine Complex: Structural Insights and Solvent Effects. Chem. Commun. 2013, 49, 6340–6342. [Google Scholar] [CrossRef] [PubMed]
- Harding, D.J.; Phonsri, W.; Harding, P.; Murray, K.S.; Moubaraki, B.; Jameson, G.N.L. Abrupt Two-Step and Symmetry Breaking Spin Crossover in an Iron(III) Complex: An Exceptionally Wide [LS–HS] Plateau. Dalton Trans. 2015, 44, 15079–15082. [Google Scholar] [CrossRef] [PubMed]
- Fukuroi, K.; Takahashi, K.; Mochida, T.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y.; Mori, H. Synergistic Spin Transition between Spin Crossover and Spin-Peierls-like Singlet Formation in the Halogen-Bonded Molecular Hybrid System: [Fe(Iqsal)2][Ni(dmit)2]⋅CH3CN⋅H2O. Angew. Chem. Int. Ed. 2014, 53, 1983–1986. [Google Scholar] [CrossRef] [PubMed]
- Vieira, B.J.C.; Dias, J.C.; Santos, I.C.; Pereira, L.C.J.; Gama, V.; Waerenborgh, J.C. Thermal Hysteresis in a Spin-Crossover FeIII Quinolylsalicylaldimine Complex, FeIII(5-Br-qsal)2Ni(dmit)2·solv: Solvent Effects. Inorg. Chem. 2015, 54, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Vieira, B.J.C.; Coutinho, J.T.; Dias, J.C.; Nunes, J.C.; Santos, I.C.; Pereira, L.C.J.; Gama, V.; Waerenborgh, J.C. Crystal Structure and Spin Crossover Behavior of the [Fe(5-Cl-qsal)2][Ni(dmit)2]·2CH3CN Complex. Polyhedron 2015, 85, 643–651. [Google Scholar] [CrossRef]
- Phukkaphan, N.; Cruickshank, D.L.; Murray, K.S.; Phonsri, W.; Harding, P.; Harding, D.J. Hysteretic Spin Crossover Driven by Anion Conformational Change. Chem. Commun. 2017, 53, 9801–9804. [Google Scholar] [CrossRef]
- Phonsri, W.; Harding, P.; Murray, K.S.; Moubaraki, B.; Harding, D.J. Spin Crossover in Mixed Ligand Iron(III) Complexes. New J. Chem. 2017, 41, 13747–13753. [Google Scholar] [CrossRef]
- Phonsri, W.; Harding, P.; Liu, L.; Telfer, S.G.; Murray, K.S.; Moubaraki, B.; Ross, T.M.; Jameson, G.N.L.; Harding, D.J. Solvent Modified Spin Crossover in an Iron(III) Complex: Phase Changes and an Exceptionally Wide Hysteresis. Chem. Sci. 2017, 8, 3949–3959. [Google Scholar] [CrossRef]
- Sadehavan, S.A.; Cadoni, E.; Monni, N.; Pipaón, C.S.; Mascarós, J.-R.G.; Abhervé, A.; Avarvi, N.; Marchiò, L.; Arca, M.; Mercuri, M.L. Structural Diversity in a New Series of Halogenated Quinolyl Salicylaldimides-Based FeIII Complexes Showing Solid-State Halogen-Bonding/Halogen···Halogen Interactions. Cryst. Growth Des. 2018, 18, 4187–4199. [Google Scholar] [CrossRef]
- Clemente-Léon, M.; Coronado, E.; López-Jordà, M.; Waerenborgh, J.C. Multifunctional Magnetic Materials Obtained by Insertion of Spin-Crossover FeIII Complexes into Chiral 3D Bimetallic Oxalate-Based Ferromagnets. Inorg. Chem. 2011, 50, 9122–9130. [Google Scholar] [CrossRef]
- Clemente-León, M.; Coronado, E.; López-Jordà, M. 2D Bimetallic Oxalate-Based Ferromagnets with Inserted [Fe(4-Br-sal2-trien)]+ and [Fe(3-R-sal2-trien)]+ (R = Br, Cl and CH3O) FeIII Spin-Crossover Complexes. Eur. J. Inorg. Chem. 2013, 5–6, 753–762. [Google Scholar] [CrossRef]
- Abhervé, A.; Clemente-León, M.; Coronado, E.; Gómez-García, C.J.; Verneret, M. Luminescent Dinuclear Cu(I) Complexes Containing Rigid Tetraphosphine Ligands. Inorg. Chem. 2014, 53, 12014–12026. [Google Scholar] [CrossRef]
- Ide, Y.; Murai, N.; Ishimae, H.; Suzuki, M.; Mori, S.; Takahashi, M.; Nakamura, M.; Toshino, K.; Ikeue, T. Spin-Crossover Between High-Spin (S = 5/2) and Low-Spin (S = 1/2) States In Six-Coordinate Iron(III) Porphyrin Complexes Having Two Pyridine-N Oxide Derivatives. Dalton Trans. 2017, 46, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Herchel, R.; Trávnícek, Z. 5-Aminotetrazole Induces Spin Crossover In Iron(III) Pentadentate Schiff Base Complexes: Experimental and Theoretical Investigations. Dalton Trans. 2013, 42, 16279–16288. [Google Scholar] [CrossRef] [PubMed]
- Krüger, C.; Augustín, P.; Nemec, I.; Trávníček, Z.; Oshio, H.; Boča, R.; Renz, F. Spin Crossover in Iron(III) Complexes with Pentadentate Schiff Base Ligands and Pseudohalido Coligands. Eur. J. Inorg. Chem. 2013, 5–6, 902–915. [Google Scholar] [CrossRef]
- Krüger, C.; Augustín, P.; Dlhán, L.; Pavlik, J.; Moncol, J.; Nemec, I.; Boča, R.; Renz, F. Iron(III) Complexes with Pentadentate Schiff-Base Ligands: Influence of Crystal Packing Change and Pseudohalido Coligand Variations On Spin. Polyhedron 2015, 87, 194–201. [Google Scholar] [CrossRef]
- Martinho, P.N.; Vicente, A.I.; Realista, S.; Saraiva, M.S.; Melato, A.I.; Brandão, P.; Ferreira, L.P.; Carvalho, M.D. Solution and Solid State Properties of Fe(III) Complexes Bearing N-Ethyl-N-(2-Aminoethyl)Salicylaldiminate Ligands. J. Organomet. Chem. 2014, 760, 48–54. [Google Scholar] [CrossRef]
- Vicente, A.I.; Joseph, A.; Ferreira, L.P.; Carvalho, M.D.; Rodrigues, V.H.N.; Duttine, M.; Diogo, H.P.; da Piedade, M.E.M.; Calhorda, M.J.; Martinho, P.N. Dynamic Spin Interchange In A Tridentate Fe(III) Schiff-Base Compound. Chem. Sci. 2016, 7, 4251–4258. [Google Scholar] [CrossRef]
- Vicente, A.I.; Ferreira, L.P.; Carvalho, M.D.; Rodrigues, V.H.N.; Dîrtu, M.; Garcia, Y.; Calhorda, M.J.; Martinho, P.N. Selecting The Spin Crossover Profile with Controlled Crystallization of Mononuclear Fe(III) Polymorphs. Dalton Trans. 2018, 47, 7013–7019. [Google Scholar] [CrossRef]
- Martins, F.F.; Joseph, A.; Diogo, H.P.; da Piedade, M.E.M.; Ferreira, L.P.; Carvalho, M.D.; Barroso, S.; Romão, M.J.; Calhorda, M.J.; Martinho, P.N. Irreversible Magnetic Behaviour Caused by the Thermosalient Phenomenon in an Iron(III) Spin Crossover Complex. Eur. J. Inorg. Chem. 2018, 25, 2976–2983. [Google Scholar] [CrossRef]
- Boonprab, T.; Harding, P.; Murray, K.S.; Phonsri, W.; Telfer, S.G.; Alkas, A.; Ketkaew, R.; Tantirungrotechai, Y.; Jameson, G.N.L.; Harding, D.J. Solvatomorphism and Anion Effects in Predominantly Low Spin Iron(III) Schiff Base Complexes. Dalton Trans. 2018, 47, 12449–12458. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.M.; Shehab, O.R. Structural Studies and Quantum Chemical Calculations of Cr(III), Fe(III) and Ru(III) Bromazepam Complexes. Appl. Organomet. Chem. 2017, 31, 3635. [Google Scholar] [CrossRef]
- Wei, H.H.; Kao, S.P. Mossbauer Study of the Effect of Intraligand Substituents on the Spin Crossover in Solid (Dithiocyanato)BIS(N-Substituted-Phenyl-2-Pyridinaldimine) Iron (II). J. Chin. Chem. Soc. 1985, 32, 399–403. [Google Scholar] [CrossRef]
- Phonsri, W.; Macedo, D.S.; Vignesh, K.R.; Rajaraman, G.; Davies, C.G.; Jameson, G.N.L.; Moubaraki, B.; Ward, J.S.; Kruger, P.E.; Chastanet, G.; et al. Halogen Substitution Effects on N2O Schiff Base Ligands in Unprecedented Abrupt FeII Spin Crossover Complexes. Chem. Eur. J. 2017, 23, 7052–7065. [Google Scholar] [CrossRef]
- Phonsri, W.; Davies, C.G.; Jameson, G.N.L.; Moubaraki, B.; Ward, J.S.; Kruger, P.E.; Chastanet, G.; Murray, K.S. Symmetry Breaking Above Room Temperature In An Fe(II) Spin Crossover Complex with An N4O2 Donor Set. Chem. Commun. 2017, 53, 1374–1377. [Google Scholar] [CrossRef]
- Kuroda-Sowa, T.; Isobe, R.; Yamao, N.; Fukumasu, T.; Okubo, T.; Maekawa, M. Variety of Spin Transition Temperatures of Iron(II) Spin Crossover Complexes with Halogen Substituted Schiff-Base Ligands, HqsalX (X = F, Cl, Br, and I). Polyhedron 2017, 136, 74–78. [Google Scholar] [CrossRef]
- Real, A.; Zarembowitch, J.; Kahn, O.; Solans, X. Magnetic Interaction and Spin Transition In Iron(II) Dinuclear Compounds. Crystal Structure of (μ-2,2’-Bipyrimidine)Bis[(2,2’-Bipyrimidine)Bis(Thiocyanato)Iron(II)]. Inorg. Chem. 1987, 26, 2939–2943. [Google Scholar] [CrossRef]
- Enamullah, M.; Linert, W.; Gutmann, V. Vibrational Spectroscopy On Iron(II) Spin-Crossover Complexes with 4-Substituted 2,6-Bis(Benzimidazol-2′-Yl)Pyridine. Vib. Spectrosc. 1995, 9, 265–271. [Google Scholar] [CrossRef]
- Linert, W.; Konecny, M.; Renz, F. Spin-State Equilibria In Non-Aqueous Solution and Quantum-Mechanical Investigations of Iron(II) and Nickel(II) Complexes with 4-Substituted 2,6-Bis(Benzimidazol-2-Yl)Pyridines. J. Chem. Soc. Dalton Trans. 1994, 1523–1531. [Google Scholar] [CrossRef]
- Linert, W.; Enamullah, M.; Gutmann, V.; Jameson, R.F. Spin-Crossover Complexes in Solution. III. Substituent- and Solvent Effects on the Spin-Equilibrium of 4-Substituted Iron(II)-2,6-Bis-(Benzimidazol-2′-Yl)-Pyridine Systems. Monatsh. Chem. 1994, 125, 661–670. [Google Scholar] [CrossRef]
- Paulsen, H.; Duelund, L.; Zimmermann, A.; Averseng, F.; Gerdan, M.; Winkler, H.; Toftlund, H.; Trautwein, A.X. Substituent Effects on the Spin-Transition Temperature in Complexes with Tris(pyrazolyl) Ligands. Monatsh. Chem. 2003, 134, 295–306. [Google Scholar] [CrossRef]
- Nakano, K.; Suemura, N.; Yoneda, K.; Kawata, S.; Kaizaki, S. Substituent Effect of The Coordinated Pyridine In A Series of Pyrazolato Bridged Dinuclear Diiron(II) Complexes On The Spin-Crossover Behavior. Dalton Trans. 2005, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Bréfuel, N.; Shova, S.; Lipkowski, J.; Tuchagues, J.-P. FeII Bi-Stable Materials Based on Dissymmetrical Ligands: N4 Schiff Bases Including 2-Pyridyl and 5-Methylimidazol-4-yl Rings Yield Various FeII Spin-Crossover Phenomena around 300 K. Chem. Mater. 2006, 18, 5467–5479. [Google Scholar] [CrossRef]
- Pritchard, R.; Kilner, C.A.; Halcrow, M.A. Iron(II) Complexes with A Terpyridine Embrace Packing Motif Show Remarkably Consistent Cooperative Spin-Transitions. Chem. Commun. 2007, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.J.K.; Shepherd, H.J.; Comyn, T.P.; Beldé, C.; Cespedes, O.; Chastanet, G.; Halcrow, M.A. Decoupled Spin Crossover and Structural Phase Transition in a Molecular Iron(II) Complex. Chem. Eur. J. 2015, 21, 4805–4816. [Google Scholar] [CrossRef] [PubMed]
- Carbonera, C.; Kilner, C.A.; Letard, J.-F.; Halcrow, M.A. Anion Doping As A Probe of Cooperativity In The Molecular Spin-Crossover Compound [FeL2][BF4]2 (L = 2,6-di{pyrazol-1-yl}pyridine). Dalton Trans. 2007, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.J.K.; Kulmaczewski, R.; Mohammed, R.; Dudley, S.; Barrett, S.A.; Little, M.A.; Deeth, R.J.; Halcrow, M.A. A Unified Treatment of the Relationship Between Ligand Substituents and Spin State in a Family of Iron(II) Complexes. Angew. Chem. Int. Ed. 2016, 55, 4327–4331. [Google Scholar] [CrossRef]
- Berdiell, I.C.; Kulmaczewski, R.; Halcrow, M.A. Iron(II) Complexes of 2,4-Dipyrazolyl-1,3,5-triazine Derivatives—The Influence of Ligand Geometry on Metal Ion Spin State. Inorg. Chem. 2017, 56, 8817–8828. [Google Scholar] [CrossRef]
- Madhu, N.T.; Salitros, I.; Schramm, F.; Klyatskaya, S.; Fhur, O.; Ruben, M. Above Room Temperature Spin Transition In A Series of Iron(II) Bis(Pyrazolyl)Pyridine Compounds. Comptes Rendus Chim. 2008, 11, 1166–1174. [Google Scholar] [CrossRef]
- Kimura, A.; Ishida, T. Pybox-Iron(II) Spin-Crossover Complexes with Substituent Effects from the 4-Position of the Pyridine Ring (Pybox = 2,6-Bis(oxazolin-2-yl)pyridine). Inorganics 2017, 5, 52. [Google Scholar] [CrossRef]
- Neville, S.M.; Leita, B.A.; Offermann, D.A.; Duriska, M.B.; Moubaraki, B.; Chapman, K.W.; Halder, G.J.; Murray, K.S. Spin-Crossover Studies on a Series of 1D Chain and Dinuclear Iron(II) Triazine-Dipyridylamine Compounds. Eur. J. Inorg. Chem. 2007, 2007, 1073–1085. [Google Scholar] [CrossRef]
- Medlycott, E.A.; Hanan, G.S.; Abedin, T.S.M.; Thompson, L.K. The Effect of Steric Hindrance On The Fe(II) Complexes of Triazine-Containing Ligands. Polyhedron 2008, 27, 493–501. [Google Scholar] [CrossRef]
- Ross, T.M.; Moubaraki, B.; Neville, S.M.; Batten, S.R.; Murray, K.S. Polymorphism and Spin Crossover In Mononuclear FeII Species Containing New Dipyridylamino-Substituted S-Triazine Ligands. Dalton Trans. 2012, 41, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.S.; Ross, T.M.; Chilton, N.F.; Gass, I.A.; Moubaraki, B.; Chastanet, G.; Paradis, N.; Lètard, J.-F.; Vignesh, K.R.; Rajaraman, G.; et al. Crown-Linked Dipyridylamino-Triazine Ligands and Their Spin-Crossover Iron(II) Derivatives: Magnetism, Photomagnetism and Cooperativity. Dalton Trans. 2013, 42, 16494–16509. [Google Scholar] [CrossRef] [PubMed]
- Wannarit, N.; Roubeau, O.; Youngme, S.; Gamez, P. Subtlety of the Spin-Crossover Phenomenon Observed with Dipyridylamino-Substituted Triazine Ligands. Eur. J. Inorg. Chem. 2013, 2013, 730–737. [Google Scholar] [CrossRef]
- Wannarit, N.; Roubeau, O.; Youngme, S.; Teat, S.J.; Gamez, P. Influence of Supramolecular Bonding Contacts on the Spin Crossover Behaviour of Iron(II) Complexes from 2,2′-Dipyridylamino/s-triazine Ligands. Dalton Trans. 2013, 42, 7120–7130. [Google Scholar] [CrossRef]
- Wannarit, N.; Nassirinia, N.; Amani, S.; Masciocchi, N.; Youngme, S.; Roubeau, O.; Teat, S.J.; Gamez, P. Drastic Effect of Lattice Propionitrile Molecules on the Spin-Transition Temperature of a 2,2′-Dipyridylamino/s-triazine-Based Iron(II) Complex. Inorg. Chem. 2014, 53, 9827–9836. [Google Scholar] [CrossRef]
- Shen, G.-P.; Qi, L.; Wang, L.; Xu, Y.; Jiang, J.-J.; Zhu, D.; Liu, X.-Q.; You, X. Spin-Crossover in a Trans-[FeL2(NCS)2] Family (L = Triaryltriazole): Remote Substituent Effects on Spin Transition Modes and Temperature. Dalton Trans. 2013, 42, 10144–10152. [Google Scholar] [CrossRef]
- Park, J.G.; Jeon, I.-R.; Harris, T.D. Electronic Effects of Ligand Substitution on Spin Crossover in a Series of Diiminoquinonoid-Bridged FeII2 Complexes. Inorg. Chem. 2015, 54, 359–369. [Google Scholar] [CrossRef]
- Wilson, D.; Djukic, B.; Lemaire, M.T. Synthesis of Bromine- or Aryl-Substituted Ditopic Schiff Base Ligands and their Bimetallic Iron(II) Complexes: Electronic and Magnetic Properties. Transit. Metal Chem. 2014, 39, 17–24. [Google Scholar] [CrossRef]
- Naik, A.D.; Tinant, B.; Muffler, K.; Wolny, J.A.; Schünemann, V.; Garcia, Y. Relevance of Supramolecular Interactions, Texture and Lattice Occupancy in the Designer Iron(II) Spin Crossover Complexes. J. Solid State Chem. 2009, 182, 1365–1376. [Google Scholar] [CrossRef]
- Naggert, H.; Rudnik, J.; Kipgen, J.; Bernien, M.; Nickel, F.; Arruda, L.M.; Kuch, W.; Näther, C.; Tuczek, F. Vacuum-Evaporable Spin-Crossover Complexes: Physicochemical Properties in the Crystalline Bulk and in Thin Films Deposited from the Gas Phase. J. Mater. Chem. C 2015, 3, 7870–7877. [Google Scholar] [CrossRef]
- Petzold, H.; Djomgoue, P.; Hörner, G.; Heider, S.; Lochenie, C.; Weber, B.; Rüffner, T.; Schaarschmidt, D. Spin State Variability in Fe2+ Complexes of Substituted (2-(Pyridin-2-yl)-1,10-phenanthroline) Ligands as Versatile Terpyridine Analogues. Dalton Trans. 2017, 46, 6218–6229. [Google Scholar] [CrossRef] [PubMed]
- Petzold, H.; Djomgoue, P.; Hörner, G.; Lochenie, C.; Weber, B.; Rüffer, T. Bis-Meridional Fe2+ Spin crossover Complexes of Phenyl and Pyridyl Substituted 2-(Pyridin-2-yl)-1,10-Phenanthrolines. Dalton Trans. 2018, 47, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Natke, D.; Unruh, D.; Dreyer, B.; Klimke, S.; Jahns, M.; Preiss, A.; Sindelar, R.; Klingelhöfer, G.; Renz, F. Tuning Spin Transitions of Iron(II)-dpp Systems. Hyperfine Interact. 2018, 239, 12. [Google Scholar] [CrossRef]
- Ren, D.-H.; Sun, X.-L.; Gu, L.; Qiu, D.; Li, Z.; Gu, Z.-G. A Family of Homochiral Spin-Crossover Iron(II) Imidazole Schiff-Base Complexes. Inorg. Chem. Commun. 2015, 51, 50–54. [Google Scholar] [CrossRef]
- Shylin, S.I.; Gural’skiy, I.A.; Bykov, D.; Demeshko, S.; Dechert, S.; Meyer, F.; Hauka, M.; Fritsky, I.O. Iron(II) Isothiocyanate Complexes with Substituted Pyrazines: Experimental and Theoretical Views on their Electronic Structure. Polyhedron 2015, 87, 147–155. [Google Scholar] [CrossRef]
- Hang, H.; Fei, B.; Chen, X.Q.; Tong, M.L.; Ksenofontov, V.; Gural’skiy, I.A.; Bao, X. Multiple Spin Phases in a Switchable Fe(II) Complex: Polymorphism and Symmetry Breaking Effects. J. Mater. Chem. C 2018, 6, 3352–3361. [Google Scholar] [CrossRef]
- Stassen, A.F.; Dova, E.; Ensling, J.; Schenk, H.; Gütlich, P.; Haasnoot, J.G.; Reedijk, J. Spin Crossover in Hexakis(1-(2-chloroethyl)-tetrazole)iron(II) Complexes, Synthesis and Magnetic Properties. Inorg. Chim. Acta 2002, 335, 61–68. [Google Scholar] [CrossRef]
- Stassen, A.F.; Grunert, M.; Dova, E.; Müller, M.; Weinberger, P.; Wiesinger, G.; Schenk, H.; Linert, W.; Haasnoot, J.G.; Reedijk, J. Magnetic, 57Fe Mössbauer, and IR Monitoring of the Thermal Spin Transition in a New Family of Iron(II) Spin-Transition Complexes. Eur. J. Inorg. Chem. 2003, 2003, 2273–2282. [Google Scholar] [CrossRef]
- Müller, D.; Knoll, C.; Seifried, M.; Welch, J.M.; Giester, G.; Reissner, M.; Weinberger, P. Halogenated Alkyltetrazoles for the Rational Design of FeII Spin-Crossover Materials: Fine-Tuning of the Ligand Size. Chem. Eur. J. 2018, 24, 5271–5280. [Google Scholar] [CrossRef]
- Thompson, C.V.; Davis, I.; DeGayner, J.A.; Arman, H.D.; Tonzetich, Z.J. Iron Pincer Complexes Incorporating Bipyridine: A Strategy for Stabilization of Reactive Species. Organometallics 2017, 36, 4928–4935. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Li, L. Synthesis; Crystal Structure and Magnetism of Two Cobalt(II). Complexes with Imino and Nitronyl Nitroxides. Transit. Met. Chem. 2015, 40, 631–636. [Google Scholar] [CrossRef]
- Zhang, Y.; Harriman, K.L.M.; Brunet, G.; Pialat, A.; Gabidullin, B.; Murugesu, M. Reversible Redox, Spin Crossover, and Superexchange Coupling in 3d Transition-Metal Complexes of Bis-Azinyl Analogues of 2,2′:6′,2′′-Terpyridine. Eur. J. Inorg. Chem. 2018, 2018, 1212–1223. [Google Scholar] [CrossRef]
- Voloshin, Y.Z.; Varzatskii, O.A.; Novikov, V.V.; Strizhakova, N.G.; Vorontsov, I.I.; Vologzhanina, A.V.; Lyssenko, K.A.; Romanenko, G.V.; Fedin, M.V.; Ovcharenko, V.I.; et al. Tris-Dioximate Cobalt(I,II,III) Clathrochelates: Stabilization of Different Oxidation and Spin States of an Encapsulated Metal Ion by Ribbed Functionalization. Eur. J. Inorg. Chem. 2010, 2010, 5401–5415. [Google Scholar] [CrossRef]
- Dolganov, A.V.; Belov, A.S.; Novikov, V.V.; Vologzhanina, A.V.; Romanenko, G.V.; Budnikova, Y.G.; Zelinskii, G.E.; Buzin, M.I.; Voloshin, Y.Z. First Iron and Cobalt(II) Hexabromoclathrochelates: Structural, Magnetic, Redox, and Electrocatalytic Behavior. Dalton Trans. 2015, 44, 2476–2487. [Google Scholar] [CrossRef]
- Novikov, V.V.; Ananyev, I.V.; Pavlov, A.A.; Fedin, M.V.; Lyssenko, K.A.; Voloshin, Y.Z. Spin-Crossover Anticooperativity Induced by Weak Intermolecular Interactions. J. Phys. Chem. Lett. 2014, 5, 496–500. [Google Scholar] [CrossRef]
- Bushuev, M.B.; Krivopalov, V.P.; Peresypkina, E.V.; Virovets, A.V.; Shvedenkov, Y.G.; Sheludyakova, L.A.; Semikolenova, N.V.; Zakharov, V.A.; Larionov, S.V. Complexes of Copper(II) and Cobalt(II) Halides with 4-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-methyl-2-phenylpyrimidine: Synthesis, Structures, and Properties. Russ. J. Coord. Chem. 2009, 35, 597–608. [Google Scholar] [CrossRef]
- Jenkins, D.M.; Peters, J.C. Spin-State Tuning at Pseudotetrahedral d7 Ions: Examining the Structural and Magnetic Phenomena of Four-Coordinate [BP3]CoII-X Systems. J. Am. Chem. Soc. 2005, 127, 7148–7165. [Google Scholar] [CrossRef]
- Ren, D.-H.; Qiu, D.; Pang, C.-Y.; Li, Z.; Gu, Z.-G. Chiral Tetrahedral Iron(II) Cages: Diastereoselective Subcomponent Self-assembly, Structure Interconversion and Spin-Crossover Properties. Chem. Commun. 2015, 51, 788. [Google Scholar] [CrossRef]
- Fatur, S.M.; Shepard, S.G.; Higgins, R.F.; Shores, M.P.; Damrauer, N.H. A Synthetically Tunable System To Control MLCT Excited-State Lifetimes and Spin States in Iron(II) Polypyridines. J. Am. Chem. Soc. 2017, 139, 4493–4505. [Google Scholar] [CrossRef] [PubMed]
- Enamullah, M.; Linert, W. Substituent- and Solvent-Effects on the Stability of Iron(II)-4-X-2,6-bis-(benzimidazol-2′-yl)pyrdine Complexes Showing Spin-Crossover in Soltution. J. Coord. Chem. 1996, 40, 193–201. [Google Scholar] [CrossRef]
- Heinze, K.; Huttner, G.; Zsolnai, L.; Schober, P. Complexes of Cobalt(II) Chloride with the Tripodal Trisphosphane triphos: Solution Dynamics, Spin-Crossover, Reactivity, and Redox Activity. Inorg. Chem. 1997, 36, 5457–5469. [Google Scholar] [CrossRef]
- Chambers, J.; Eaves, B.; Parker, D.; Claxton, R.; Ray, P.S.; Slattery, S.J. Inductive Influence of 4′-Terpyridyl Substituents on Redox and Spin State Properties of Iron(II) and Cobalt(II) Bis-terpyridyl Complexes. Inorg. Chim. Acta 2006, 359, 2400–2406. [Google Scholar] [CrossRef]
- Thorarinsdottir, A.E.; Gaudette, A.I.; Harris, T.D. Spin-Crossover and High-Spin Iron(II) Complexes as Chemical Shift 19F Magnetic Resonance Thermometers. Chem. Sci. 2017, 8, 2448–2456. [Google Scholar] [CrossRef]
- Vela, S.; Paulsen, H. Cooperativity in Spin Crossover Systems. An Atomistic Perspective on the Devil’s Staircase. Inorg. Chem. 2018, 57, 9478–9488. [Google Scholar] [CrossRef]
- Vela, S.; Paulsen, H. Deciphering Crystal Packing Effects in the Spin Crossover of Six [FeII(2-pic)3]Cl2 Solvatomorphs. Dalton Trans. 2019, 48, 1237–1245. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Reiher, M.; Salomon, O.; Hess, B.A. Reparameterization of Hybrid Functionals Based on Energy Differences of States of Different Multiplicity. Theor. Chem. Acc. 2001, 107, 48–55. [Google Scholar] [CrossRef]
- Salomon, O.; Reiher, M.; Hess, B.A. Assertion and Validation of the Performance of the B3LYP⋆ Functional for the First Transition Metal Row and the G2 Test Set. J. Chem. Phys. 2002, 117, 4729–4737. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Kossmann, S.; Neese, F. Comparison of Two Efficient Approximate Hartee–Fock approaches. Chem. Phys. Lett. 2009, 481, 240–243. [Google Scholar] [CrossRef]
- Becke, A.D.; Johnson, E.R. A Density-Functional Model of the Dispersion Interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Harvey, J.N.; Aschi, M.; Schwarz, H.; Koch, W. The Singlet and Triplet States of Phenyl Cation. A Hybrid Approach for Locating Minimum Energy Crossing Points Between Non-Interacting Potential Energy Surfaces. Theor. Chem. Acc. 1999, 99, 95–99. [Google Scholar] [CrossRef]
- Chemcraft Program. Available online: http://www.chemcraftprog.com/index.html (accessed on 31 August 2019).
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 20-New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]



























© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).