
Signals for Muscular Protein Turnover and Insulin Resistance in Critically Ill Patients: A Narrative Review

 ,
, 

 , , and
, , and
Abstract
1. Introduction
2. Methodology
3. Extracellular Signals Control Protein Turnover and Insulin Resistance in Muscle
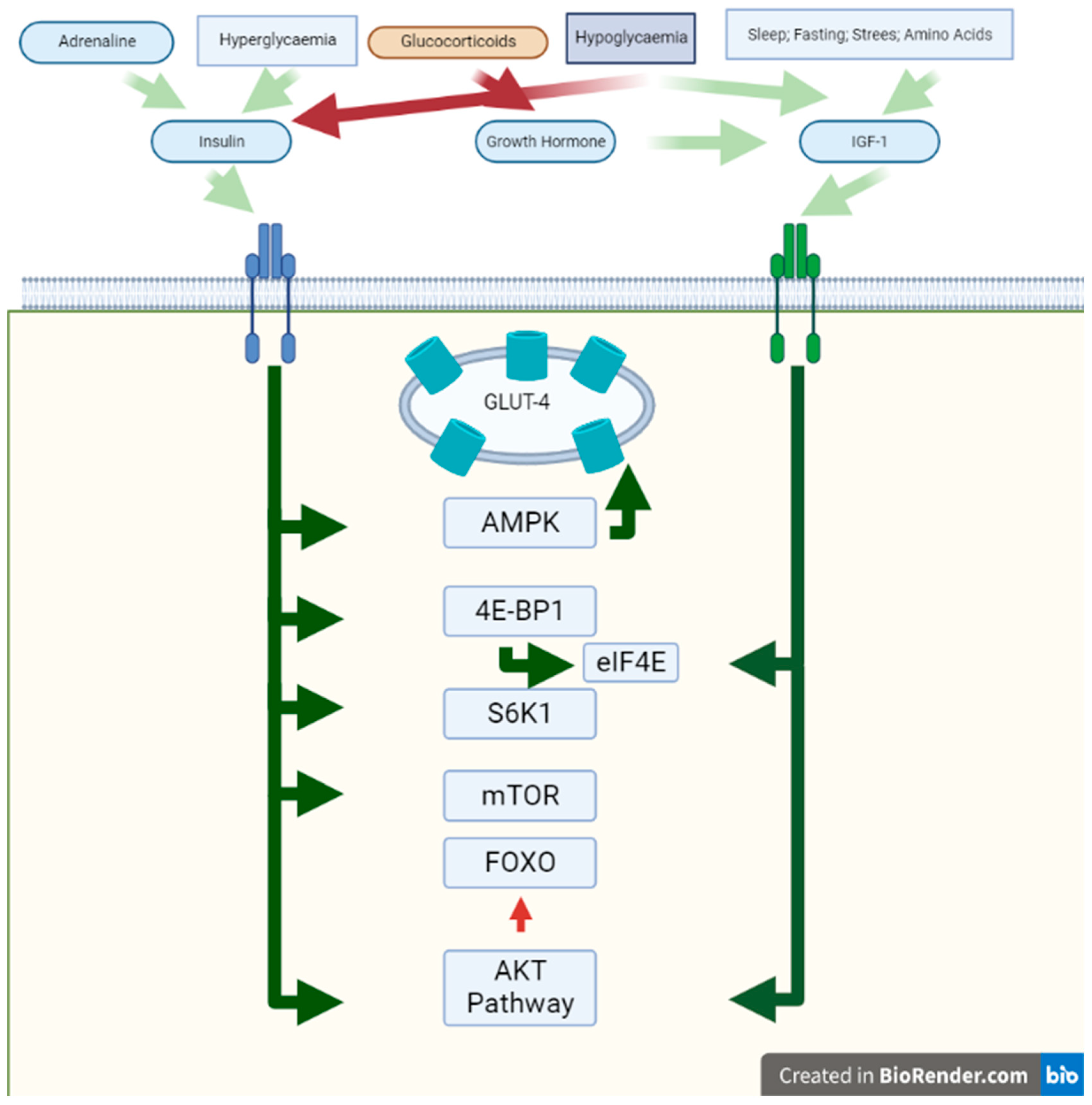
3.1. Insulin
3.2. Growth Hormone
3.3. Insulin-like Growth Factor 1 (IGF-1)
3.4. Glucocorticoids
3.5. Glucagon
3.6. TNF Alpha
3.7. Adrenaline
3.8. Hypoxia-Inducible Factor
4. Signal Translation
4.1. AMPK
4.2. AKT
4.3. GSK 3B
4.4. mTOR
4.5. Ribosomal Protein S6 Kinase 1 (P70S6k)
4.6. FOXO
5. Effectors of Protein Breakdown
5.1. Atrogin and MuRF1
5.2. Ubiquitin-Proteasome
5.3. Calpain and Caspase-3
6. Summary and Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-Activated Protein Kinase |
| eIF | eukaryotic Initiation Factor |
| FFM | Fat Free Mass |
| FM | Fat Mass |
| GC | Glucocorticoids |
| COPD | Chronic Obstructive Pulmonary Disease |
| GH | Growth Hormone |
| GLP-1 | Glucagon Like Factor-1 |
| GLUT-4 | Glucose Transporter 4 |
| GSK3B | Glycogen synthase kinase 3 β |
| HIF | Hypoxia Inducible Factor |
| IGF-1 | Insulin-like Growth Factor |
| IGFBP3 | IGF Binding Protein 3 |
| IL-1 | Interleukin 1 |
| LPS | Lipopolysaccharide |
| MAFbx1 | Muscle Atrophy Box 1 |
| mTOR | Mammalian Target of Rapamycin |
| MuRF1 | Muscle RING Finger 1 |
| NF-KB | Nuclear Factor Kappa |
| PI | Phosphatidylinositol |
| P70S6k | Ribosomal protein S6 kinase 1 |
| SOFA | Sequential Organ Failure Assessment |
| TNF | Tumour Necrosis Factor |
References
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European Consensus on Definition and Diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef]
- Peng, P.; Firoozmand, A.; Kneuertz, P.; Schulick, R.D.; Huang, D.; Makary, M.; Hirose, K.; Edil, B.; Choti, M.A.; Herman, J.; et al. Impact of Sarcopenia on Outcomes Following Resection of Pancreatic Adenocarcinoma. J. Gastrointest. Surg. 2012, 16, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, A.V.; Maddocks, M.; Martolini, D.; Polkey, M.I.; Man, W.D. Muscle Function in COPD: A Complex Interplay. Int. J. Chronic Obstr. Pulm. Dis. 2012, 7, 523–535. [Google Scholar]
- Ji, Y.; Cheng, B.; Xu, Z.; Ye, H.; Lu, W.; Luo, X.; Fu, S.; Fang, X. Impact of Sarcopenic Obesity on 30-Day Mortality in Critically Ill Patients with Intra-Abdominal Sepsis. J. Crit. Care 2018, 46, 50–54. [Google Scholar] [CrossRef]
- Masuda, T.; Shirabe, K.; Ikegami, T.; Harimoto, N.; Yoshizumi, T.; Soejima, Y.; Uchiyama, H.; Ikeda, T.; Baba, H.; Maehara, Y. Sarcopenia Is a Prognostic Factor in Living Donor Liver Transplantation. Liver Transplant. 2014, 20, 401–407. [Google Scholar] [CrossRef]
- Matsubara, Y.; Matsumoto, T.; Aoyagi, Y.; Tanaka, S. Sarcopenia Is a Prognostic Factor for Overall Survival in Patients with Critical Limb Ischemia. J. Vasc. Surg. 2015, 61, 945–950. [Google Scholar] [CrossRef]
- Dodson, R.M.; Firoozmand, A.; Hyder, O.; Tacher, V.; Cosgrove, D.P.; Bhagar, N.; Herman, J.M.; Wolfgang, C.L.; Gesschwind, J.-F.H.; Kamel, I.R.; et al. Impact of Sarcopenia on Outcomes Following Intra-Arterial Therapy of Hepatic Malignancies. J. Gastrointest. Surg. 2013, 17, 2123–2132. [Google Scholar] [CrossRef]
- DeAndrade, J.; Pedersen, M.; Garcia, L.; Nau, P. Sarcopenia Is a Risk Factor for Complications and an Independent Predictor of Hospital Length of Stay in Trauma Patients. J. Surg. Res. 2018, 221, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Englesbe, M.J.; Patel, S.P.; He, K.; Lynch, R.J.; Schaubel, D.E.; Harbaugh, C.; Holcombe, S.A.; Wang, S.C.; Segev, D.L.; Sonnenday, C.J. Sarcopenia and Post-Liver Transplant Mortality. J. Am. Coll. Surg. 2010, 211, 271–278. [Google Scholar] [CrossRef]
- Friedman, J.; Lussiez, A.; Sullivan, J.; Wang, S.; Englesbe, M. Implications of Sarcopenia in Major Surgery. Nutr. Clin. Pract. 2015, 30, 175–179. [Google Scholar] [CrossRef]
- Jones, K.I.; Doleman, B.; Scott, S.; Lund, J.N.; Williams, J.P. Simple Psoas Cross-Sectional Area Measurement Is a Quick and Easy Method to Assess Sarcopenia and Predicts Major Surgical Complications. Color. Dis. 2014, 17, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, S.; Asghar, A.; Mott, S.L.; Johnson, B.E.; Button, A.M.; Clark, E.; Mezhir, J.J. Sarcopenia Is an Independent Predictor of Complications Following Pancreatectomy for Adenocarcinoma. J. Surg. Oncol. 2015, 111, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Fairchild, B.; Webb, T.P.; Xiang, Q.; Tarima, S.; Brasel, K.J. Sarcopenia and Frailty in Elderly Trauma Patients. World J. Surg. 2015, 39, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.S. Sarcopenia and Critical Illness: A Deadly Combination in the Elderly Physiologic Derangements in Sarcopenia Age-Associated Alterations in Muscle. J. Parenter. Enter. Nutr. 2015, 39, 273–281. [Google Scholar] [CrossRef]
- Looijaard, W.G.P.M.; Dekker, I.M.; Stapel, S.N.; Girbes, A.R.J.; Twisk, J.W.R.; Straaten, H.M.O.; Weijs, P.J.M. Skeletal Muscle Quality as Assessed by CT-Derived Skeletal Muscle Density Is Associated with 6-Month Mortality in Mechanically Ventilated Critically Ill Patients. Crit. Care 2016, 20, 386. [Google Scholar] [CrossRef]
- Moisey, L.L.; Mourtzakis, M.; Cotton, B.A.; Premji, T.; Heyland, D.K.; Wade, C.E.; Bulger, E.; Kozar, R.A. Skeletal Muscle Predicts Ventilator-Free Days, ICU-Free Days, and Mortality in Elderly ICU Patients. Crit. Care 2013, 17, R206. [Google Scholar] [CrossRef]
- Weijs, P.J.M.; Looijaard, W.G.P.M.; Dekker, I.M.; Stapel, S.N.; Girbes, A.R.; Straaten, H.M.O.; Beishuizen, A. Low Skeletal Muscle Area Is a Risk Factor for Mortality in Mechanically Ventilated Critically Ill Patients. Crit. Care 2014, 18, R12. [Google Scholar] [CrossRef]
- Zusman, O.; Theilla, M.; Cohen, J.; Kagan, I.; Bendavid, I.; Singer, P. Resting Energy Expenditure, Calories and Protein Consumption in Critically Ill Patients: A Retrospective Cohort Study. Crit. Care 2016, 20, 367. [Google Scholar] [CrossRef]
- Nicolo, M.; Heyland, D.K.; Chittams, J.E.S.S.E.; Sammarco, T.; Compher, C. Clinical Outcomes Related to Protein Delivery in a Critically Ill Population: A Multicenter, Multinational Observation Study. J. Parenter. Enter. Nutr. 2016, 40, 45–51. [Google Scholar] [CrossRef]
- Compher, C.; Chittams, J.; Sammarco, T.; Nicolo, M.; Heyland, D.K. Greater Protein and Energy Intake May Be Associated with Improved Mortality in Higher Risk Critically Ill Patients: A Multicenter, Multinational Observational Study. Crit. Care Med. 2017, 45, 156–163. [Google Scholar] [CrossRef]
- Lee, Z.Y.; Sing, C.; Yap, L.; Hasan, M.S.; Engkasan, J.P.; Yusof, M.; Nisak, B.; Day, A.G.; Patel, J.J.; Heyland, D.K. The Effect of Higher versus Lower Protein Delivery in Critically Ill Patients: A Systematic Review and Meta—Analysis of Randomized Controlled Trials. Crit. Care 2021, 25, 260. [Google Scholar] [CrossRef] [PubMed]
- Compher, C.; Bingham, A.L.; McCall, M.; Patel, J.; Rice, T.W.; Braunschweig, C.; McKeever, L. Guidelines for the Provision of Nutrition Support Therapy in the Adult Critically Ill Patient: The American Society for Parenteral and Enteral Nutrition. J. Parenter. Enter. Nutr. 2022, 46, 12–41. [Google Scholar] [CrossRef] [PubMed]
- Singer, P.; Blaser, A.R.; Berger, M.M.; Alhazzani, W.; Philip, C.; Casaer, M.; Hiesmayr, M.; Mayer, K.; Montejo, J.C.; Pichard, C.; et al. ESPEN Guideline on Clinical Nutrition in the Intensive Care Unit. Clin. Nutr. 2018, 38, 48–79. [Google Scholar] [CrossRef] [PubMed]
- Preiser, J.C.; Ichai, C.; Orban, J.C.; Groeneveld, A.B.J. Metabolic Response to the Stress of Critical Illness. Br. J. Anaesth. 2014, 113, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Hollander, J.M.; Mechanick, J.I. Nutrition Support and the Chronic Critical Illness Syndrome. Nutr. Clin. Pract. 2006, 21, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Leasa, D. Chronic Critical Illness. Handb. ICU Ther. Third Ed. 2015, 21, 209–220. [Google Scholar] [CrossRef]
- Marchioni, A.; Fantini, R.; Antenora, F.; Clini, E.; Fabbri, L. Chronic Critical Illness: The Price of Survival. Eur. J. Clin. Investig. 2015, 45, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Mira, J.C.; Gentile, L.F.; Mathias, B.J.; Efron, P.A.; Brakenridge, S.C.; Mohr, A.M.; Moore, F.A.; Moldawer, L.L. Sepsis Pathophysiology, Chronic Critical Illness, and Persistent Inflammation-Immunosuppression and Catabolism Syndrome. Crit. Care Med. 2017, 45, 253–262. [Google Scholar] [CrossRef]
- Clain, J. Glucose Control in Critical Care. World J. Diabetes 2015, 6, 1082. [Google Scholar] [CrossRef]
- Tjäder, I.; Klaude, M.; Hssain, A.A.; Guillet, C.; Nennesmo, I.; Wernerman, J.; Rooyackers, O. Variability in Skeletal Muscle Protein Synthesis Rates in Critically Ill Patients. Nutrients 2022, 14, 3733. [Google Scholar] [CrossRef]
- Berg, J.; Tymoczko, J.L.; Stryer, L. Biochemistry, 7th ed.; W. H Freeman and Company: New York, NY, USA, 2012. [Google Scholar]
- Lodish, H.; Berk, A.; Matsudaira, P.; Kaiser, C.; Krieger, M.; Scott, M.; Zipursky, S.L.; Darnel, J. Biología Celular y Molecular; Panamericana: Singapore, 2013. [Google Scholar]
- Vary, T.C.; Jefferson, L.S.; Kimball, S.R. Insulin Fails to Stimulate Muscle Protein Synthesis in Sepsis despite Unimpaired Signaling to 4E-BP1 and S6K1. Am. J. Physiol.—Endocrinol. Metab. 2001, 281, 1045–1053. [Google Scholar] [CrossRef]
- Weber-Carstens, S.; Schneider, J.; Wollersheim, T.; Assmann, A.; Bierbrauer, J.; Marg, A.; Hasani, H.A.; Chadt, A.; Wenzel, K.; Koch, S.; et al. Critical Illness Myopathy and GLUT4 Significance of Insulin and Muscle Contraction. Am. J. Respir. Crit. Care Med. 2013, 187, 387–396. [Google Scholar] [CrossRef]
- Sasso, F.C.; Carbonara, O.; Cozzolino, D.; Rambaldi, P.; Mansi, L.; Torella, D.; Gentile, S.; Turco, S.; Torella, R.; Salvatore, T. Effects of Insulin-Glucose Infusion on Left Ventricular Function at Rest and during Dynamic Exercise in Healthy Subjects and Noninsulin Dependent Diabetic Patients: A Radionuclide Ventriculographic Study. J. Am. Coll. Cardiol. 2000, 36, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Dong, H.H. FoxO Integration of Insulin Signaling with Glucose and Lipid Metabolism. J. Endocrinol. 2017, 233, R67–R79. [Google Scholar] [CrossRef]
- Du, S.; Zheng, H. Role of FoxO Transcription Factors in Aging and Age-Related Metabolic and Neurodegenerative Diseases. Cell Biosci. 2021, 11, 188. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Udy, A.A.; Nichol, A.; Bellomo, R.; Deane, A.M.; El-Khawas, K.; Thummaporn, N.; Serpa Neto, A.; Bergin, H.; Short-Burchell, R.; et al. Incidence and Management of Metabolic Acidosis with Sodium Bicarbonate in the ICU: An International Observational Study. Crit. Care 2021, 25, 45. [Google Scholar] [CrossRef] [PubMed]
- De Corte, W.; Vuylsteke, S.; De Waele, J.J.; Dhondt, A.W.; Decruyenaere, J.; Vanholder, R.; Hoste, E.A.J. Severe Lactic Acidosis in Critically Ill Patients with Acute Kidney Injury Treated with Renal Replacement Therapy. J. Crit. Care 2014, 29, 650–655. [Google Scholar] [CrossRef]
- Posa, D.K.; Baba, S.P. Intracellular PH Regulation of Skeletal Muscle in the Milieu of Insulin Signaling. Nutrients 2020, 12, 2910. [Google Scholar] [CrossRef]
- Taylor, B.E.; Buchman, T.G. Is There a Role for Growth Hormone Therapy in Refractory Critical Illness? Curr. Opin. Crit. Care 2008, 14, 438–444. [Google Scholar] [CrossRef]
- Wolf, S.E.; Barrow, R.E.; Herndon, D.N. Growth Hormone and IGF-I Therapy in the Hypercatabolic Patient. Bailliere Clin. Endocrinol. Metab. 1996, 10, 447–463. [Google Scholar] [CrossRef]
- Lal, S.O.; Wolf, S.E.; Herndon, D.N. Growth Hormone, Burns and Tissue Healing. Growth Horm. IGF Res. 2000, 10, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Hammarqvist, F.; Wennström, I.; Wernerman, J. Effects of Growth Hormone and Insulin-like Growth Factor-1 on Postoperative Muscle and Substrate Metabolism. J. Nutr. Metab. 2010, 2010, 647929. [Google Scholar] [CrossRef] [PubMed]
- Voerman, H.J.; Strack van Schijndel, R.J.M.; Groeneveld, A.B.J.; De Boer, H.; Nauta, J.P.; Van der Veen, E.A.; Thijs, L.G. Effects of Recombinant Human Growth Hormone in Patients with Severe Sepsis. Ann. Surg. 1992, 216, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Takala, J.; Rouken, E.; Webster, N.R.; Nielsen, M.S.; Zandstra, D.F.; Vundelinckx, G.; Hinds, C.J. Increased Mortality Associated with Growth Hormone Treatment In Critically Ill Adults. NEJM 1999, 341, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Mallon, D.; Boyle, D.W.; Liechty, E.A. IGF-I and Insulin Regulate EIF4F Formation by Different Mechanisms in Muscle and Liver in the Ovine Fetus. Am. J. Physiol.—Endocrinol. Metab. 2002, 283, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Hajsadeghi, S.; Khamseh, M.E.; Gholami, S.; Kerman, S.R.J.; Gohardehi, G.; Moghadam, N.S.; Sabet, A.S.; Moradi, M.; Mollahoseini, R.; Najafi, M.; et al. IGF-I Concentration and Changes in Critically Ill Patients. J. Res. Med. Sci. 2011, 16, 170–178. [Google Scholar]
- Tong, J.F.; Yan, X.; Zhu, M.J.; Du, M. AMP-Activated Protein Kinase Enhances the Expression of Muscle-Specific Ubiquitin Ligases despite Its Activation of IGF-1/Akt Signaling in C2C12 Myotubes. J. Cell. Biochem. 2009, 108, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, W.; Sun, R.; Liu, J.; Hong, J.; Li, Q.; Hu, B.; Gong, F. IGF-1 May Predict the Severity and Outcome of Patients with Sepsis and Be Associated with MicroRNA-1 Level Changes. Exp. Ther. Med. 2017, 14, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Papastathi, C.; Mavrommatis, A.; Mentzelopoulos, S.; Konstandelou, E.; Alevizaki, M.; Zakynthinos, S. Insulin-like Growth Factor I and Its Binding Protein 3 in Sepsis. Growth Horm. IGF Res. 2013, 23, 98–104. [Google Scholar] [CrossRef]
- Vary, T.C. IGF-I Stimulates Protein Synthesis in Skeletal Muscle through Multiple Signaling Pathways during Sepsis. Am. J. Physiol.—Regul. Integr. Comp. Physiol. 2006, 290, 313–321. [Google Scholar] [CrossRef]
- Morandi, A.; Gunther, M.L.; Pandharipande, P.P.; Jackson, J.C.; Thompson, J.L.; Shintani, A.K.; Ely, E.W.; Girard, T.D. Insulin-like Growth Factor-1 and Delirium in Critically Ill Mechanically Ventilated Patients: A Preliminary Investigation. Int. Psychogeriatr. 2011, 23, 1175–1181. [Google Scholar] [CrossRef]
- Hunninghake, G.W.; Doerschug, K.C.; Nymon, A.B.; Schmidt, G.A.; Meyerholz, D.K.; Ashare, A. Insulin-like Growth Factor-1 Levels Contribute to the Development of Bacterial Translocation in Sepsis. Am. J. Respir. Crit. Care Med. 2010, 182, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Ashare, A.; Nymon, A.B.; Doerschug, K.C.; Morrison, J.M.; Monick, M.M.; Hunninghake, G.W. Insulin-like Growth Factor-1 Improves Survival in Sepsis via Enhanced Hepatic Bacterial Clearance. Am. J. Respir. Crit. Care Med. 2008, 178, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Frost, R.A. Differential Effect of Sepsis on Ability of Leucine and IGF-I to Stimulate Muscle Translation Initiation. Am. J. Physiol.—Endocrinol. Metab. 2004, 287, E721–E730. [Google Scholar] [CrossRef]
- Teng Chung, T.; Hinds, C.J. Treatment with GH and IGF-1 in Critical Illness. Crit. Care Clin. 2006, 22, 29–40. [Google Scholar] [CrossRef]
- Goeters, C.; Mertes, N.; Tacke, J.; Bolder, U.; Kuhmann, M.; Lawin, P.; Löhlein, D. Repeated Administration of Recombinant Human Insulin like Growth Factor I in Patients after Gastric Surgery. Effect on metabolic and hormonal patterns. Ann. Surg. 1995, 222, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Schakman, O.; Kalista, S.; Barbé, C.; Loumaye, A.; Thissen, J.P. Glucocorticoid-Induced Skeletal Muscle Atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2163–2172. [Google Scholar] [CrossRef]
- Liu, Z.; Li, G.; Kimball, S.R.; Jahn, L.A.; Barrett, E.J. Glucocorticoids Modulate Amino Acid-Induced Translation Initiation in Human Skeletal Muscle. Am. J. Physiol.—Endocrinol. Metab. 2004, 287, E275–E281. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo Transcription Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Sandoval, D.A.; D’Alessio, D.A. Physiology of Proglucagon Peptides: Role Ofglucagon and GLP-1 in Health and Disease. Physiol. Rev. 2015, 95, 513–548. [Google Scholar] [CrossRef] [PubMed]
- Thiessen, S.E.; Gunst, J.; Van Den Berghe, G. Role of Glucagon in Protein Catabolism. Curr. Opin. Crit. Care 2018, 24, 228–234. [Google Scholar] [CrossRef]
- Thiessen, S.E.; Derde, S.; Derese, I.; Dufour, T.; Vega, C.A.; Langouche, L.; Goossens, C.; Peersman, N.; Vermeersch, P.; Perre, S.V.; et al. Role of Glucagon in Catabolism and Muscle Wasting of Critical Illness and Modulation by Nutrition. Am. J. Respir. Crit. Care Med. 2017, 196, 1131–1143. [Google Scholar] [CrossRef]
- Remels, A.H.V.; Gosker, H.R.; Schrauwen, P.; Hommelberg, P.P.H.; Sliwinski, P.; Polkey, M.; Galdiz, J.; Wouters, E.F.M.; Langen, R.C.J.; Schols, A.M.W.J. TNF-α Impairs Regulation of Muscle Oxidative Phenotype: Implications for Cachexia? FASEB J. 2010, 24, 5052–5062. [Google Scholar] [CrossRef]
- Li, Y.-P.; Lecker, S.H.; Chen, Y.; Waddell, I.D.; Goldberg, A.L.; Reid, M.B. TNF-α Increases Ubiquitin-conjugating Activity in Skeletal Muscle by Up-regulating UbcH2/E2 20k. FASEB J. 2003, 17, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-neriah, Y. Phosporilation Meets the Ubiquitination: The Control of NF- Kapp B Activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Kanarek, N.; London, N.; Schueler-Furman, O.; Ben-neriah, Y. Ubiquitination and Degradation of Proteins. Methods Mol. Biol. 2011, 753, 335–357. [Google Scholar] [CrossRef]
- Moylan, J.S.; Smith, J.D.; Chambers, M.A.; McLoughlin, T.J.; Reid, M.B. TNF Induction of Atrogin-1/MAFbx MRNA Depends on Foxo4 Expression but Not AKT-Foxo1/3 Signaling. Am. J. Physiol.—Cell Physiol. 2008, 295, C986–C993. [Google Scholar] [CrossRef]
- Lang, C.H.; Pruznak, A.M.; Frost, R.A. TNFα Mediates Sepsis-Induced Impairment of Basal and Leucine-Stimulated Signaling via S6K1 and ElF4E in Cardiac Muscle. J. Cell. Biochem. 2005, 94, 419–431. [Google Scholar] [CrossRef]
- Remels, A.H.V.; Gosker, H.R.; Verhees, K.J.P.; Langen, R.C.J.; Schols, A.M.W.J. TNF-α-Induced NF-ΚB Activation Stimulates Skeletal Muscle Glycolytic Metabolism through Activation of HIF-1-α. Endocrinology 2015, 156, 1770–1781. [Google Scholar] [CrossRef]
- Wang, D.T.; Yin, Y.; Yang, Y.J.; Lv, P.J.; Shi, Y.; Lu, L.; Wei, L.B. Resveratrol Prevents TNF-α-Induced Muscle Atrophy via Regulation of Akt/MTOR/FoxO1 Signaling in C2C12 Myotubes. Int. Immunopharmacol. 2014, 19, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Gore, D.C.; Jahoor, F.; Wolfe, R.R.; Herndon, D.N. Acute Response of Human Muscle Protein to Catabolic Hormones. Ann. Surg. 1993, 218, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Fryburg, D.A.; Gelfand, R.A.; Jahn, L.A.; Sherwin, S.; Sacca, L.; Barrett, J.; David, A.; Gelfand, R.A.; Jahn, L.A.; Oliveras, D.; et al. Effects of Epinephrine on Human and Protein Metabolism Muscle Glucose. Am. J. Physiol. (Endocrinol. Metab.) 1995, 268, E55–E59. [Google Scholar] [CrossRef]
- Manfredi, L.H.; Lustrino, D.; Machado, J.; Silveira, W.A.; Zanon, N.M.; Navegantes, L.C.; Kettelhut, I.C. Adrenodemedullation Activates the Ca2+-Dependent Proteolysis in Soleus Muscles from Rats Exposed to Cold. J. Appl. Physiol. 2017, 122, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Silveira, W.A.; Gonçalves, D.A.; Graça, F.A.; Bergantin, L.B.; Zanon, N.M.; Godinho, R.O.; Kettelhut, I.C.; Navegantes, L.C.C. Activating CAMP/PKA Signaling in Skeletal Muscle Suppresses the Ubiquitin-Proteasome-Dependent Proteolysis: Implications for Sympathetic Regulation. Am. J. Physiol. Endocrinol. Metab. 2014, 117, 11–19. [Google Scholar] [CrossRef]
- Graça, F.A.; Gonçalves, D.A.P.; Silveira, W.A.; Lira, E.C.; Chaves, V.E.; Zanon, N.M.; Garófalo, M.A.R.; Kettelhut, I.C.; Navegantes, L.C.C. Epinephrine Depletion Exacerbates the Fasting-Induced Protein Breakdown in Fast-Twitch Skeletal Muscles. Am. J. Physiol. Endocrinol. Metab. 2013, 305, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Vanderhaeghen, T.; Vandewalle, J.; Libert, C. Hypoxia-Inducible Factors in Metabolic Reprogramming during Sepsis. FEBS J. 2020, 287, 1478–1495. [Google Scholar] [CrossRef]
- Bogdanovski, D.A.; DiFazio, L.T.; Bogdanovski, A.K.; Csóka, B.; Jordan, G.B.; Paul, E.R.; Antonioli, L.; Pilip, S.A.; Nemeth, Z.H. Hypoxia-Inducible-Factor-1 in Trauma and Critical Care. J. Crit. Care 2017, 42, 207–212. [Google Scholar] [CrossRef]
- Bar-Or, D.; Carrick, M.; Tanner, A.; Lieser, M.J.; Rael, L.T.; Brody, E. Overcoming the Warburg Effect: Is It the Key to Survival in Sepsis? J. Crit. Care 2018, 43, 197–201. [Google Scholar] [CrossRef]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 Regulates the Warburg Effect and Promotes HMGB1 Release in Sepsis. Nat. Commun. 2014, 5, 4436. [Google Scholar] [CrossRef]
- Van Wyngene, L.; Vandewalle, J.; Libert, C. Reprogramming of Basic Metabolic Pathways in Microbial Sepsis: Therapeutic Targets at Last? EMBO Mol. Med. 2018, 10, e8712. [Google Scholar] [CrossRef]
- Grimaldi, D.; Hraiech, S.; Boutin, E.; Lacherade, J.C.; Boissier, F.; Pham, T.; Richard, J.C.; Thille, A.W.; Ehrmann, S.; Lascarrou, J.B.; et al. Hypoxemia in the ICU: Prevalence, Treatment, and Outcome. Ann. Intensive Care 2018, 8, 82. [Google Scholar] [CrossRef]
- Wandrag, L.; Siervo, M.; Riley, H.L.; Khosravi, M.; Fernandez, B.O.; Leckstrom, C.A.; Martin, D.S.; Mitchell, K.; Levett, D.Z.H.; Montgomery, H.E.; et al. Does Hypoxia Play a Role in the Development of Sarcopenia in Humans? Mechanistic Insights from the Caudwell Xtreme Everest Expedition. Redox Biol. 2017, 13, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Textoris, J.; Beaufils, N.; Quintana, G.; Lassoud, A.B.; Zieleskiewicz, L.; Wiramus, S.; Blasco, V.; Lesavre, N.; Martin, C.; Gabert, J.; et al. Hypoxia-Inducible Factor (HIF1α) Gene Expression in Human Shock States. Crit. Care 2012, 16, 2–7. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Cacciani, N.; Paoli, A.; Reggiani, C.; Patruno, M. Hypoxia: The Third Wheel between Nerve and Muscle. Neurol. Res. 2008, 30, 149–154. [Google Scholar] [CrossRef] [PubMed]
- De Theije, C.; Costes, F.; Langen, R.C.; Pison, C.; Gosker, H.R. Hypoxia and Muscle Maintenance Regulation: Implications for Chronic Respiratory Disease. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Winkelman, C. Inactivity and Inflammation in the Critically Ill Patient. Crit. Care Clin. 2007, 23, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Taylor, C.T. Hypoxia and Inflammation. Biochemistry 2017, 39, 34–36. [Google Scholar] [CrossRef]
- Musi, N.; Goodyear, L.J. AMP-Activated Protein Kinase and Muscle Glucose Uptake. Acta Physiol. Scand. 2003, 178, 337–345. [Google Scholar] [CrossRef]
- Krawiec, B.J.; Nystrom, G.J.; Frost, R.A.; Jefferson, L.S.; Lang, C.H. AMP-Activated Protein Kinase Agonists Increase MRNA Content of the Muscle-Specific Ubiquitin Ligases MAFbx and MuRF1 in C2C12 Cells. Am. J. Physiol.—Endocrinol. Metab. 2007, 292, 1555–1567. [Google Scholar] [CrossRef]
- Sanchez, A.M.J.; Csibi, A.; Raibon, A.; Cornille, K.; Gay, S.; Bernardi, H.; Candau, R. AMPK Promotes Skeletal Muscle Autophagy through Activation of Forkhead FoxO3a and Interaction with Ulk1. J. Cell. Biochem. 2012, 113, 695–710. [Google Scholar] [CrossRef]
- Nakashima, K.; Yakabe, Y. AMPK Activation Stimulates Myofibrillar Protein Degradation and Expression of Atrophy-Related Ubiquitin Ligases by Increasing FOXO Transcription Factors in C2C12 Myotubes. Biosci. Biotechnol. Biochem. 2007, 71, 1650–1656. [Google Scholar] [CrossRef]
- Jaitovich, A.; Angulo, M.; Lecuona, E.; Dada, L.A.; Welch, L.C.; Cheng, Y.; Gusarova, G.; Ceco, E.; Liu, C.; Shigemura, M.; et al. High CO2 Levels Cause Skeletal Muscle Atrophy via AMP-Activated Kinase (AMPK), FoxO3a Protein, and Muscle-Specific Ring Finger Protein 1 (MuRF1). J. Biol. Chem. 2015, 290, 9183–9194. [Google Scholar] [CrossRef] [PubMed]
- Favier, F.B.; Costes, F.; Defour, A.; Bonnefoy, R.; Lefai, E.; Baugé, S.; Peinnequin, A.; Benoit, H.; Freyssenet, D. Downregulation of Akt/Mammalian Target of Rapamycin Pathway in Skeletal Muscle Is Associated with Increased REDD1 Expression in Response to Chronic Hypoxia. Am. J. Physiol.—Regul. Integr. Comp. Physiol. 2010, 298, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt Pathway Prevents Expression of Muscle Atrophy-Induced Ubiquitin Ligases by Inhibiting FOXO Transcription Factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Joassard, O.R.; Amirouche, A.; Gallot, Y.S.; Desgeorges, M.M.; Castells, J.; Durieux, A.C.; Berthon, P.; Freyssenet, D.G. Regulation of Akt-MTOR, Ubiquitin-Proteasome and Autophagy-Lysosome Pathways in Response to Formoterol Administration in Rat Skeletal Muscle. Int. J. Biochem. Cell Biol. 2013, 45, 2444–2455. [Google Scholar] [CrossRef] [PubMed]
- Behrens, S.; Kozeniecki, M. Nutrition Support During Prone Positioning: An Old Technique Reawakened by COVID-19. Nutr. Clin. Pract. 2020, 36, 105–109. [Google Scholar] [CrossRef]
- Plyte, S.E.; Hughes, K.; Nikolakaki, E.; Pulverer, B.J.; Woodgett, J.R. Glycogen Synthase Kinase-3: Functions in Oncogenesis and Development. BBA—Rev. Cancer 1992, 1114, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Subber, T.; Zhao, Y. GSK3β and Its Role in Sepsis. J. Allergy Ther. 2017, 8, 13–14. [Google Scholar] [CrossRef]
- Vines, A.; Cahoon, S.; Goldberg, I.; Saxena, U.; Pillarisetti, S. Novel Anti-Inflammatory Role for Glycogen Synthase Kinase-3β in the Inhibition of Tumor Necrosis Factor-α- and Interleukin-1β-Induced Inflammatory Gene Expression. J. Biol. Chem. 2006, 281, 16985–16990. [Google Scholar] [CrossRef]
- Rocha, J.; Figueira, M.E.; Barateiro, A.; Fernandes, A.; Brites, D.; Pinto, R.; Freitas, M.; Fernandes, E.; Mota-Filipe, H.; Sepodes, B. Inhibition of Glycogen Synthase Kinase-3β Attenuates Organ Injury and Dysfunction Associated with Liver Ischemia-Reperfusion and Thermal Injury in the Rat. Shock 2015, 43, 369–378. [Google Scholar] [CrossRef]
- Dugo, L.; Abdelrahman, M.; Murch, O.; Mazzon, E.; Cuzzocrea, S.; Thiemermann, C. Glycogen Synthase Kinase-3β Inhibitors Protect against the Organ Injury and Dysfunction Caused by Hemorrhage and Resuscitation. Shock 2006, 25, 485–491. [Google Scholar] [CrossRef]
- Dugo, L.; Collin, M.; Allen, D.A.; Patel, N.S.A.; Bauer, I.; Mervaala, E.M.A.; Louhelainen, M.; Foster, S.J.; Yaqoob, M.M.; Thiemermann, C. GSK-3β Inhibitors Attenuate the Organ Injury/Dysfunction Caused by Endotoxemia in the Rat. Crit. Care Med. 2005, 33, 1903–1912. [Google Scholar] [CrossRef]
- Dugo, L.; Collin, M.; Thiemermann, C. Glycogen Synthase Kinase 3β as a Target for the Therapy of Shock and Inflammation. Shock 2007, 27, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Verhees, K.J.P.; Schols, A.M.W.J.; Kelders, M.C.J.M.; den Kamp, C.M.H.O.; van der Velden, J.L.J.; Langen, R.C.J. Glycogen Synthase Kinase-3β Is Required for the Induction of Skeletal Muscle Atrophy. Am. J. Physiol.—Cell Physiol. 2011, 301, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Shen, E.; Fan, J.; Peng, T. Glycogen Synthase Kinase-3beta Suppresses Tumor Necrosis Factor-Alpha Expression in Cardiomyocytes during Lipopolysaccharide Stimulation. J. Cell. Biochem. 2008, 104, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a Protein Target of the FKBP12-Rapamycin Complex in Mammalian Cells. J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Luna, J.I.; Marusina, A.I.; Merleev, A.; Kundu-Raychaudhuri, S.; Fiorentino, D.; Raychaudhuri, S.P.; Maverakis, E. Dual MTOR Inhibition Is Required to Prevent TGF-β-Mediated Fibrosis: Implications for Scleroderma. J. Investig. Dermatol. 2015, 135, 2873–2876. [Google Scholar] [CrossRef]
- Lynch, C.J.; Halle, B.; Fujii, H.; Vary, T.C.; Wallin, R.; Damuni, Z.; Hutson, S.M. Potential Role of Leucine Metabolism in the Leucine-Signaling Pathway Involving MTOR. Am. J. Physiol.—Endocrinol. Metab. 2003, 285, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. MTOR- and HIF-1α-Mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef]
- Xiao, F.; Huang, Z.; Li, H.; Yu, J.; Wang, C.; Chen, S.; Meng, Q.; Cheng, Y.; Gao, X.; Li, J.; et al. Leucine Deprivation Increases Hepatic Insulin Sensitivity via GCN2/MTOR/S6K1 and AMPK Pathways. Diabetes 2011, 60, 746–756. [Google Scholar] [CrossRef]
- Zhang, Q.; Duplany, A.; Moncollin, V.; Mouradian, S.; Goillot, E.; Mazelin, L.; Gauthier, K.; Streichenberger, N.; Angleraux, C.; Chen, J.; et al. Lack of Muscle MTOR Kinase Activity Causes Early Onset Myopathy and Compromises Whole-Body Homeostasis. J. Cachexia Sarcopenia Muscle 2019, 10, 35–53. [Google Scholar] [CrossRef]
- Fry, C.S.; Glynn, E.L.; Drummond, M.J.; Timmerman, K.L.; Fujita, S.; Abe, T.; Dhanani, S.; Volpi, E.; Rasmussen, B.B. Blood Flow Restriction Exercise Stimulates MTORC1 Signaling and Muscle Protein Synthesis in Older Men. J. Appl. Physiol. 2010, 108, 1199–1209. [Google Scholar] [CrossRef]
- Grove, J.R.; Banerjee, P.; Balasubramanyam, A.; Coffer, P.J.; Price, D.J.; Avruch, J.; Woodgett, J.R. Cloning and Expression of Two Human P70 S6 Kinase Polypeptides Differing Only at Their Amino Termini. Mol. Cell. Biol. 1991, 11, 5541–5550. [Google Scholar] [CrossRef] [PubMed]
- Jameel Shah, O.; Iniguez-Lluhi, J.A.; Romanelli, A.; Kimball, S.R.; Jefferson, L.S. The Activated Glucocorticoid Receptor Modulates Presumptive Autoregulation of Ribosomal Protein S6 Protein Kinase, P70 S6K. J. Biol. Chem. 2002, 277, 2525–2533. [Google Scholar] [CrossRef] [PubMed]
- Datan, E.; Shirazian, A.; Benjamin, S.; Matassov, D.; Tinari, A.; Malorni, W.; Lockshin, R.A.; Garcia-Sastre, A.; Zakeri, Z. MTOR/P70S6K Signaling Distinguishes Routine, Maintenance-Level Autophagy from Autophagic Cell Death during Influenza A Infection. Virology 2014, 452–453, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Léger, B.; Cartoni, R.; Praz, M.; Lamon, S.; Dériaz, O.; Crettenand, A.; Gobelet, C.; Rohmer, P.; Konzelmann, M.; Luthi, F.; et al. Akt Signalling through GSK-3β, MTOR and Foxo1 Is Involved in Human Skeletal Muscle Hypertrophy and Atrophy. J. Physiol. 2006, 576, 923–933. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.M.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Foletta, V.C.; White, L.J.; Larsen, A.E.; Léger, B.; Russell, A.P. The Role and Regulation of MAFbx/Atrogin-1 and MuRFl in Skeletal Muscle Atrophy. Pflugers Arch. Eur. J. Physiol. 2011, 461, 325–335. [Google Scholar] [CrossRef]
- Rom, O.; Reznick, A.Z. The Role of E3 Ubiquitin-Ligases MuRF-1 and MAFbx in Loss of Skeletal Muscle Mass. Free Radic. Biol. Med. 2016, 98, 218–230. [Google Scholar] [CrossRef]
- Nakao, R.; Hirasaka, K.; Goto, J.; Ishidoh, K.; Yamada, C.; Ohno, A.; Okumura, Y.; Nonaka, I.; Yasutomo, K.; Baldwin, K.M.; et al. Ubiquitin Ligase Cbl-b Is a Negative Regulator for Insulin-Like Growth Factor 1 Signaling during Muscle Atrophy Caused by Unloading. Mol. Cell. Biol. 2009, 29, 4798–4811. [Google Scholar] [CrossRef] [PubMed]
- Wray, C.J.; Mammen, J.M.V.; Hershko, D.D.; Hasselgren, P.-O. Sepsis Upregulates the Gene Expression of Multiple Ubiquitin Ligases in Skeletal Muscle. Int. J. Biochem. Cell Biol. 2003, 35, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Frost, R.A.; Nystrom, G.J.; Jefferson, L.S.; Lang, C.H. Hormone, Cytokine, and Nutritional Regulation of Sepsis-Induced Increases in Atrogin-1 and MuRF1 in Skeletal Muscle. Am. J. Physiol.—Endocrinol. Metab. 2007, 292, E501–E512. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Protein Breakdown in Muscle Wasting: Role of Autophagy-Lysosome and Ubiquitin-Proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef]
- Ogawa, T.; Furochi, H.; Mameoka, M.; Hirasaka, K.; Onishi, Y.; Suzue, N.; Oarada, M.; Akamatsu, M.; Akima, H.; Fukunaga, T.; et al. Ubiquitin Ligase Gene Expression in Healthy Volunteers with 20-Day Bedrest. Muscle Nerve 2006, 34, 463–469. [Google Scholar] [CrossRef]
- Nelson, W.B.; Smuder, A.J.; Hudson, M.B.; Talbert, E.E.; Powers, S.K. Cross-Talk Betwen the Calpain and Caspase-3 Proteolytic Systems in the Diaphragm during Prolonged Mechanical Ventilation. Crit. Care Med. 2012, 40, 1857–1863. [Google Scholar] [CrossRef]
- Supinski, G.S.; Wang, W.; Callahan, L.A. Caspase and Calpain Activation Both Contribute to Sepsis-Induced Diaphragmatic Weakness. J. Appl. Physiol. 2009, 107, 1389–1396. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. BBA—Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Aziz, M.; Jacob, A.; Wang, P. Revisiting Caspases in Sepsis. Cell Death Dis. 2014, 5, e1526. [Google Scholar] [CrossRef]
- Llano-Diez, M.; Fury, W.; Okamoto, H.; Bai, Y.; Gromada, J.; Larsson, L. RNA-Sequencing Reveals Altered Skeletal Muscle Contraction, E3 Ligases, Autophagy, Apoptosis, and Chaperone Expression in Patients with Critical Illness Myopathy. Skelet. Muscle 2019, 9, 9. [Google Scholar] [CrossRef]
- Hammarqvist, F.; Wernerman, J.; Ali, R.; von der Decken, A.; Vinnars, E. Addition of Glutamine to Total Parenteral Nutrition after Elective Abdominal Surgery Spares Free Glutamine in Muscle, Counteracts the Fall in Muscle Protein Synthesis, and Improves Nitrogen Balance. Ann. Surg. 1989, 209, 455–461. [Google Scholar] [CrossRef]
- Lambell, K.J.; Goh, G.S.; Tierney, A.C.; Franzcr, M.; Forsyth, A.; Nanjayya, V.; King, S.J.; Nyulasi, I.; King, S.J. Marked Losses of Computed Tomography–Derived Skeletal Muscle Area and Density over the First Month of a Critical Illness Are Not Associated with Energy and Protein Delivery. Nutrition 2021, 82, 111061. [Google Scholar] [CrossRef]
- Fetterplace, K.; Deane, A.M.; Tierney, A.; Beach, L.J.; Knight, L.D.; Presneill, J.; Rechnitzer, T.; Forsyth, A.; Gill, B.M.T.; Mourtzakis, M.; et al. Targeted Full Energy and Protein Delivery in Critically Ill Patients: A Pilot Randomized Controlled Trial (FEED Trial). J. Parenter. Enter. Nutr. 2018, 42, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- McNelly, A.S.; Bear, D.E.; Connolly, B.A.; Arbane, G.; Allum, L.; Tarbhai, A.; Cooper, J.A.; Hopkins, P.A.; Wise, M.P.; Brealey, D.; et al. Effect of Intermittent or Continuous Feed on Muscle Wasting in Critical Illness: A Phase 2 Clinical Trial. Chest 2020, 158, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Yeh, D.D.; Ortiz, L.A.; Lee, J.M.; Chan, J.; McKenzie, K.; Young, B.; Chetelat, L.; Collier, B.; Benson, A.; Heyland, D.K. PEP uP (Enhanced Protein-Energy Provision via the Enteral Route Feeding Protocol) in Surgical Patients—A Multicenter Pilot Randomized Controlled Trial. J. Parenter. Enter. Nutr. 2020, 44, 197–204. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Hormone | Effects |
|---|---|
| Insulin |
|
| Growth Hormone |
|
| IGF-1 |
|
| Glucocorticoids |
|
| Glucagon |
|
| TNF-alpha |
|
| Adrenaline |
|
| HIF-1 |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chapela, S.P.; Simancas-Racines, D.; Montalvan, M.; Frias-Toral, E.; Simancas-Racines, A.; Muscogiuri, G.; Barrea, L.; Sarno, G.; Martínez, P.I.; Reberendo, M.J.; et al. Signals for Muscular Protein Turnover and Insulin Resistance in Critically Ill Patients: A Narrative Review. Nutrients 2023, 15, 1071. https://doi.org/10.3390/nu15051071
Chapela SP, Simancas-Racines D, Montalvan M, Frias-Toral E, Simancas-Racines A, Muscogiuri G, Barrea L, Sarno G, Martínez PI, Reberendo MJ, et al. Signals for Muscular Protein Turnover and Insulin Resistance in Critically Ill Patients: A Narrative Review. Nutrients. 2023; 15(5):1071. https://doi.org/10.3390/nu15051071
Chicago/Turabian StyleChapela, Sebastián P., Daniel Simancas-Racines, Martha Montalvan, Evelyn Frias-Toral, Alison Simancas-Racines, Giovanna Muscogiuri, Luigi Barrea, Gerardo Sarno, Pablo I. Martínez, María J. Reberendo, and et al. 2023. "Signals for Muscular Protein Turnover and Insulin Resistance in Critically Ill Patients: A Narrative Review" Nutrients 15, no. 5: 1071. https://doi.org/10.3390/nu15051071
APA StyleChapela, S. P., Simancas-Racines, D., Montalvan, M., Frias-Toral, E., Simancas-Racines, A., Muscogiuri, G., Barrea, L., Sarno, G., Martínez, P. I., Reberendo, M. J., Llobera, N. D., & Stella, C. A. (2023). Signals for Muscular Protein Turnover and Insulin Resistance in Critically Ill Patients: A Narrative Review. Nutrients, 15(5), 1071. https://doi.org/10.3390/nu15051071