Nanoformulated Zoledronic Acid Boosts the Vδ2 T Cell Immunotherapeutic Potential in Colorectal Cancer

 ,
,  , , ,
, , , 

Abstract
:1. Introduction
2. Results
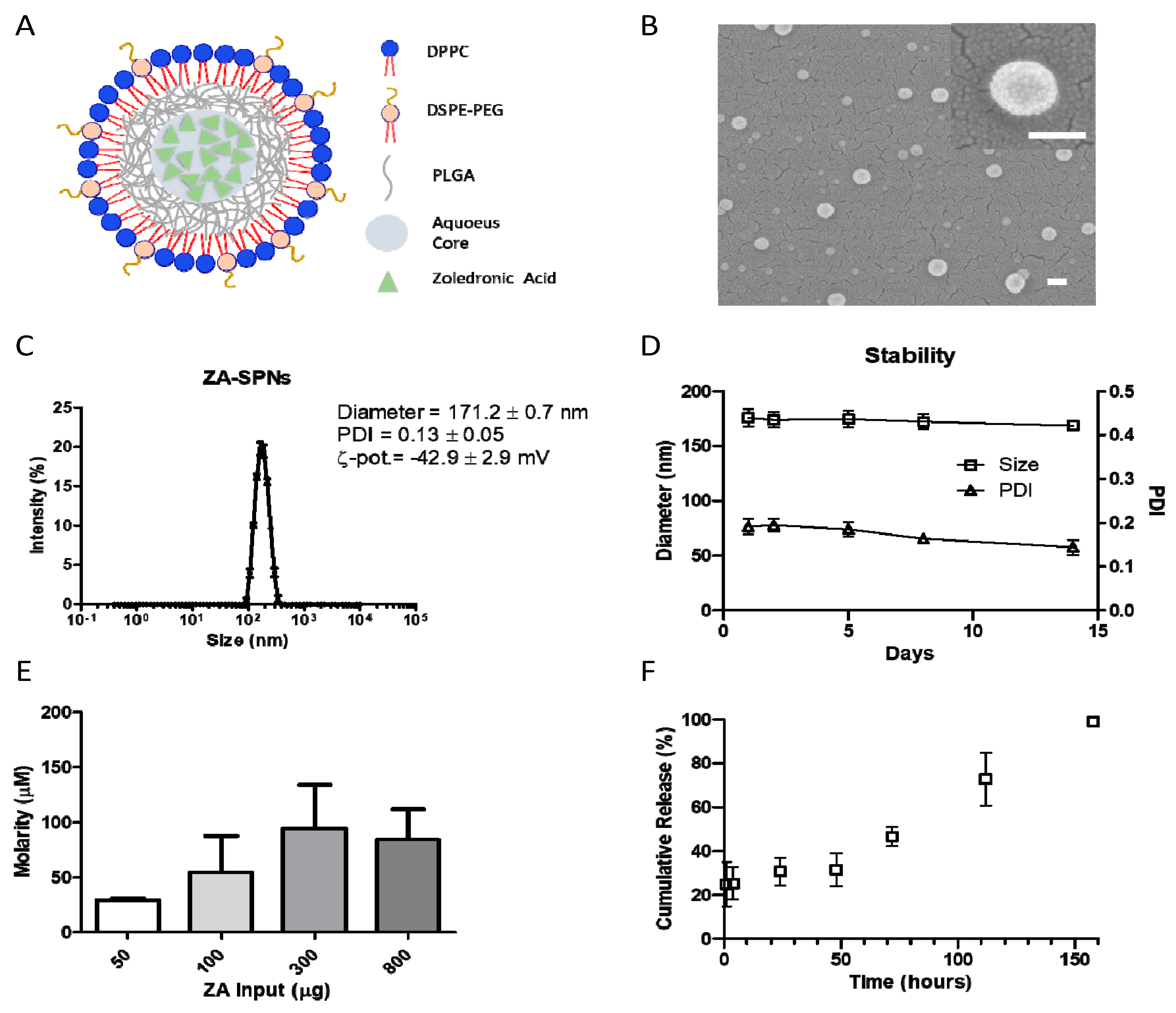
2.1. Synthesis and Characterization of ZA-SPNs
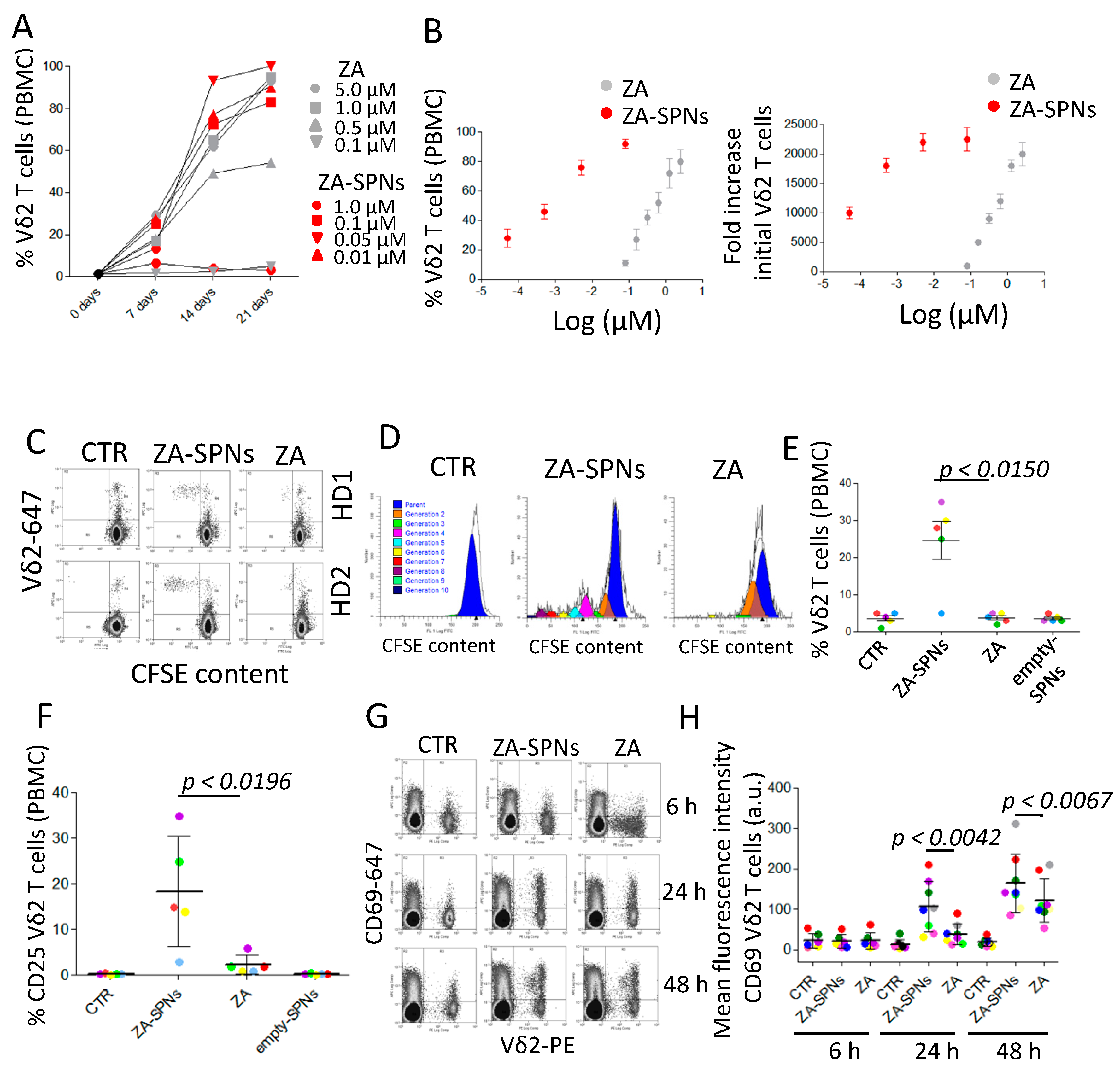
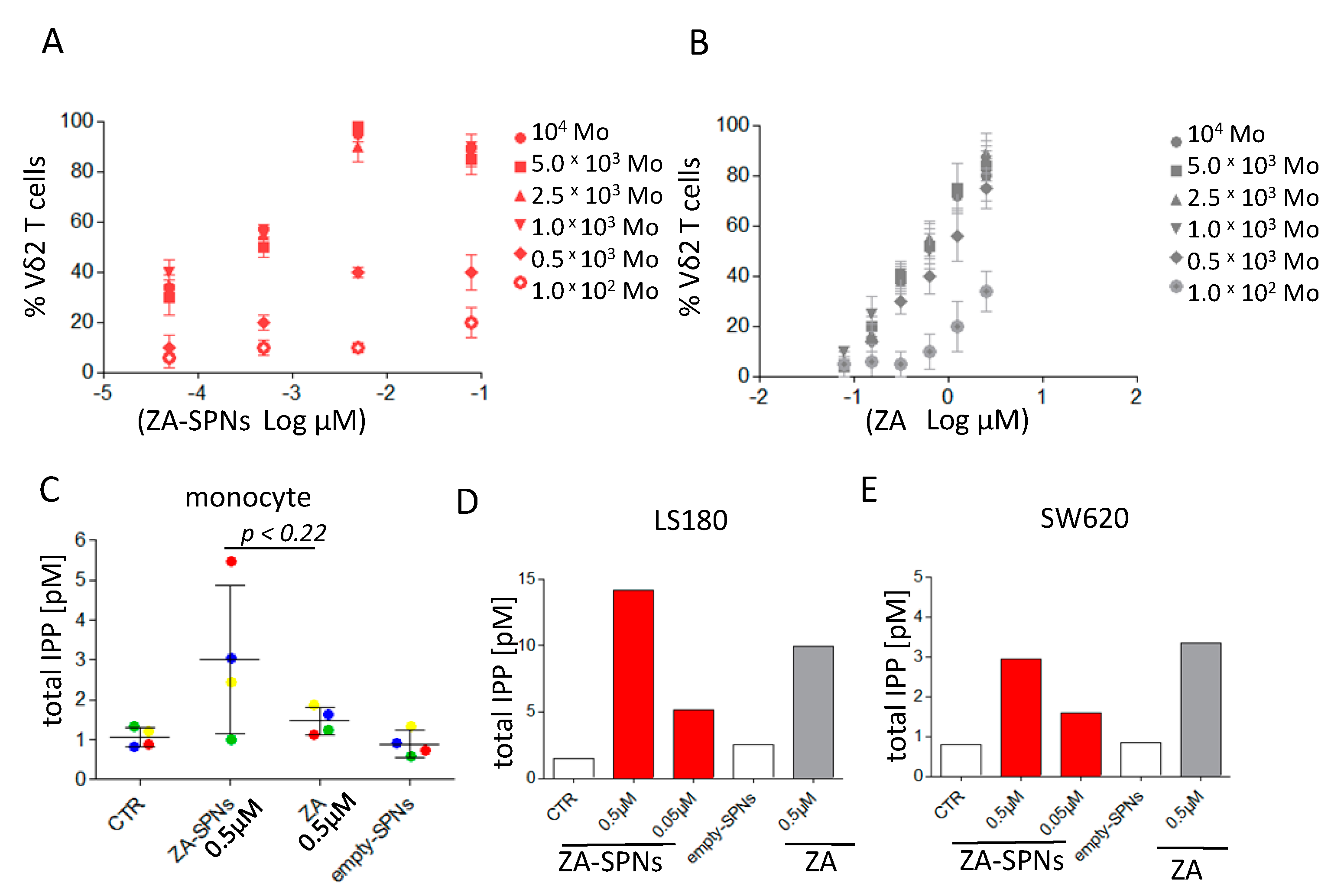
2.2. ZA-SPNs Induce the Proliferation and Activation of Vδ2 T Lymphocytes
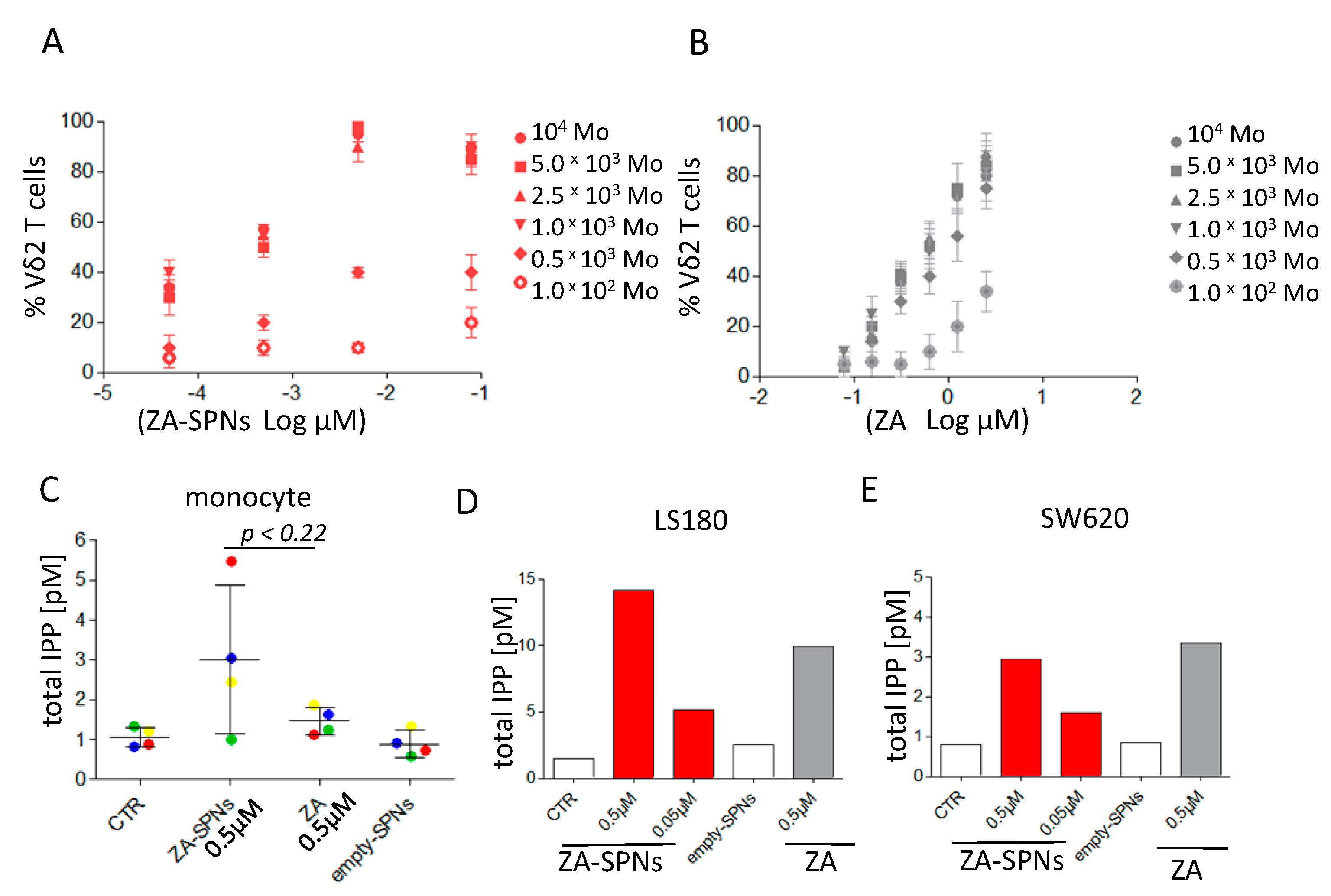
2.3. ZA-SPNs Mediated Vδ2 T Cell Expansion Is Associated with Isopentenyl-Pyrophosphate (IPP) Production
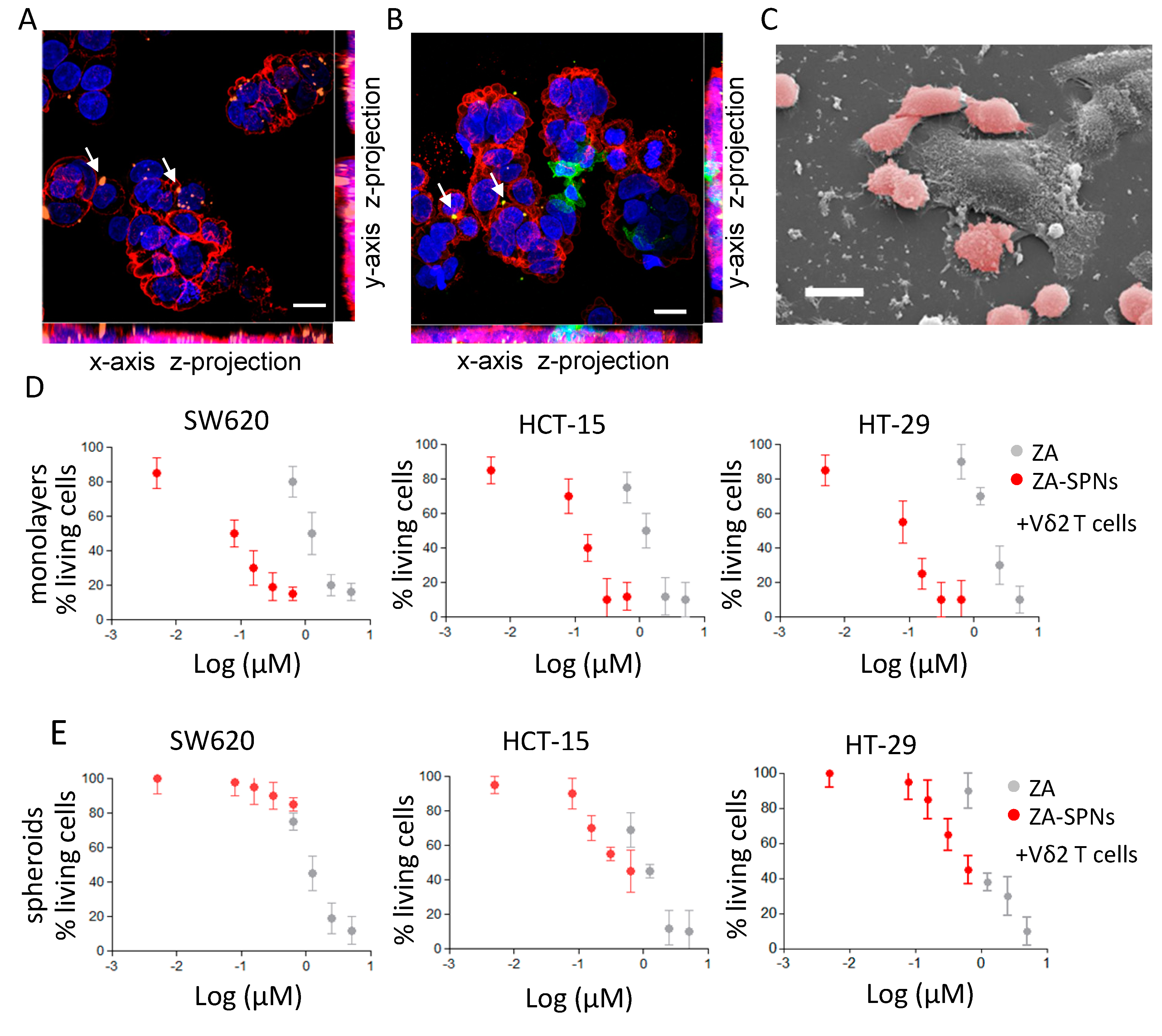
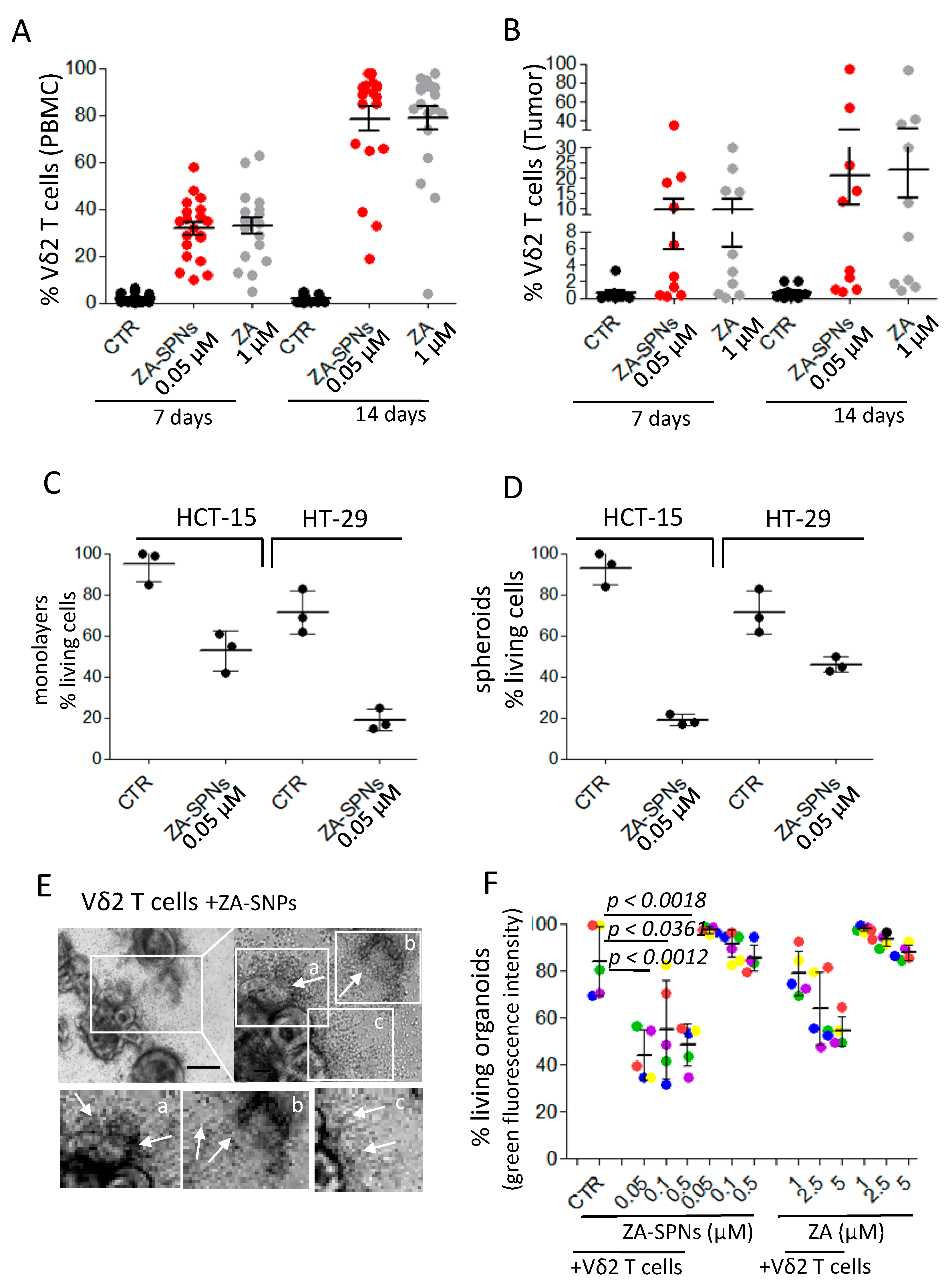
2.4. CRC Cell Death Induced by Vδ2 T Lymphocytes Stimulated with ZA-SPNs
2.5. ZA-SPNs Elicited Vδ2 T Cell Proliferation from CRC Patients and the Killing of Autologous Tumor Organoids
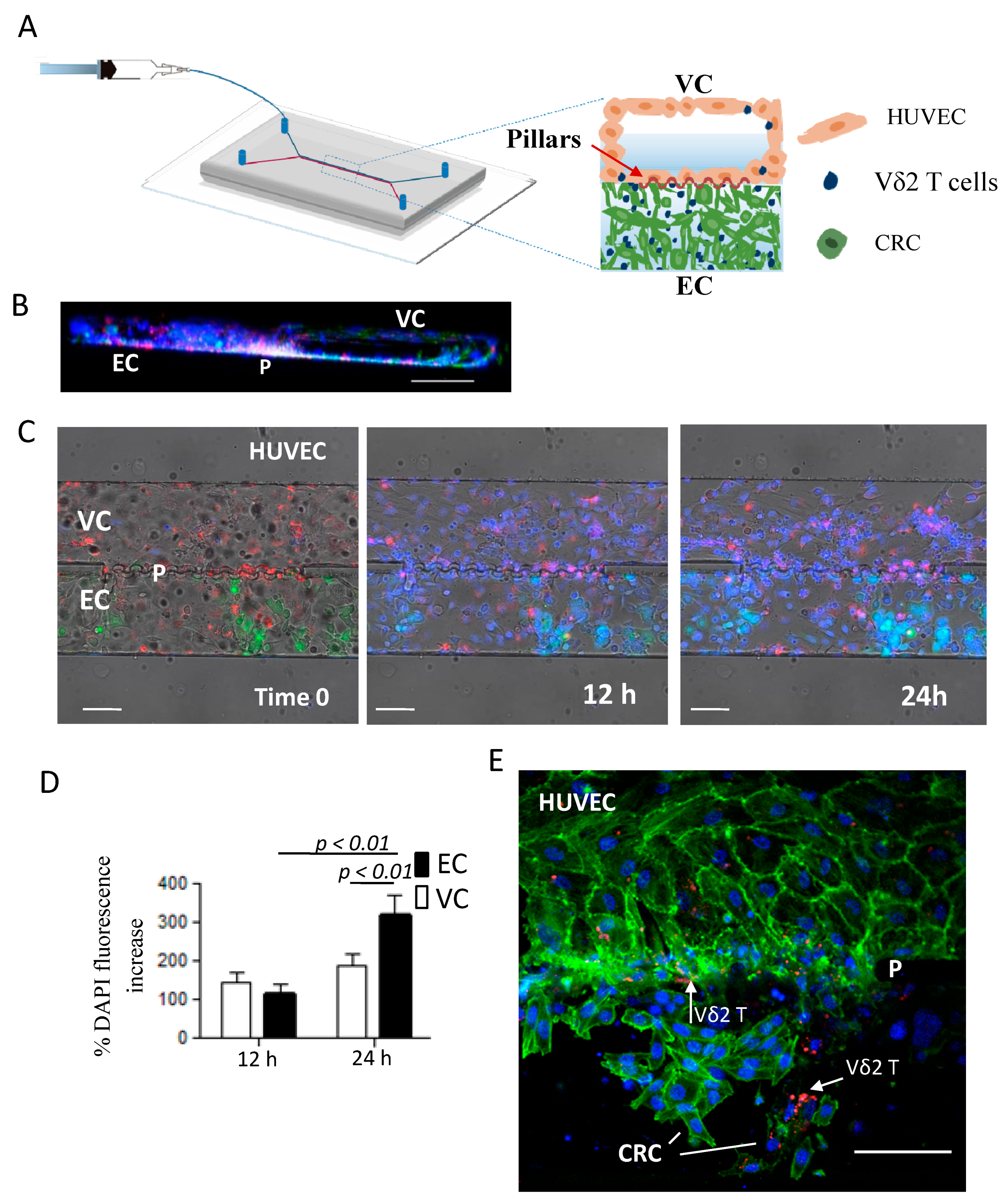
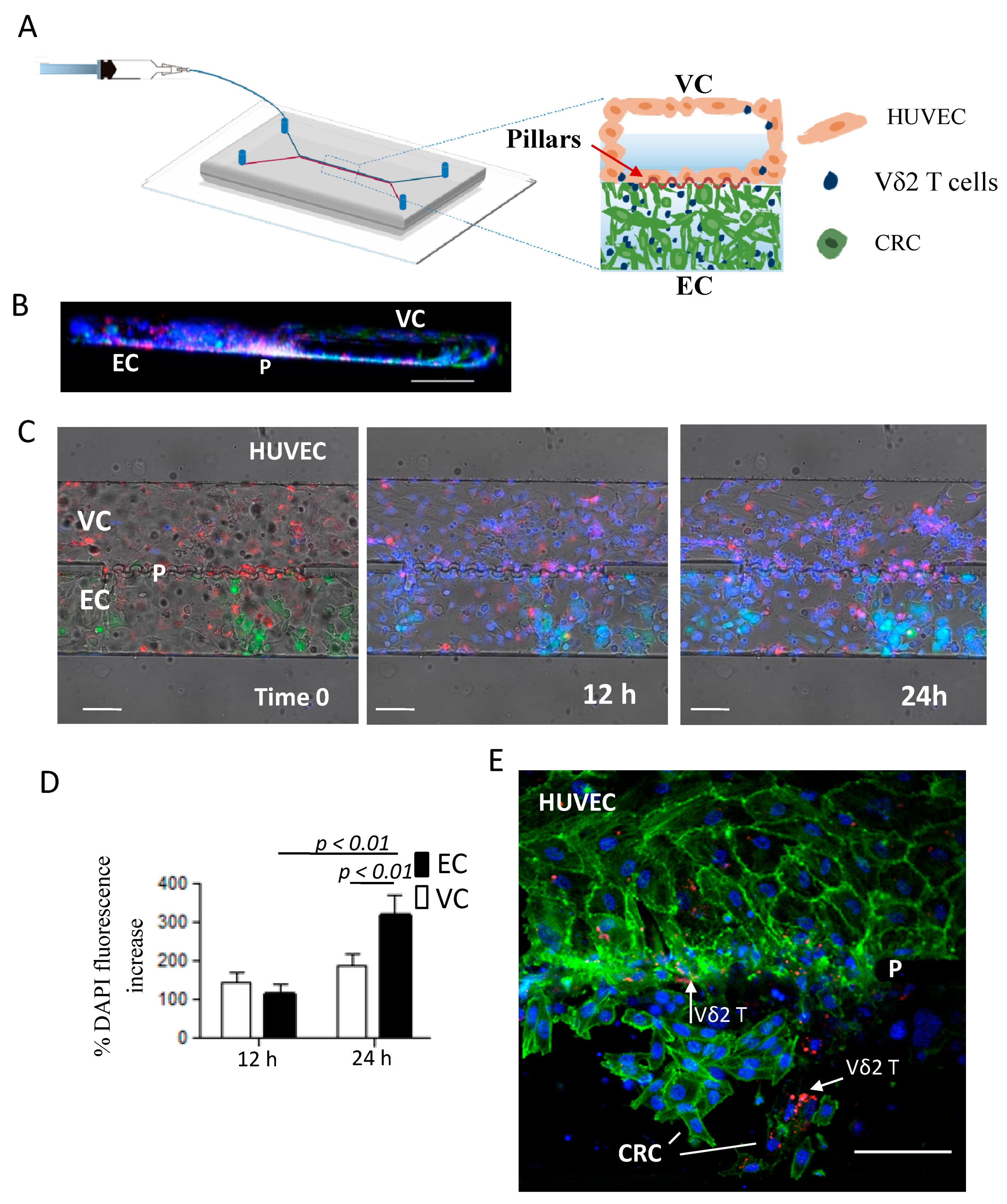
2.6. ZA-SPNs-Vδ2 T Lymphocytes Transendothelial Migration Toward CRC Tumor Cells in a Microfluidic System
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Zoledronic Acid-Loaded Spherical Polymeric Nanoparticles (ZA-SPNs) Synthesis
4.3. ZA-SPNs Physico-Chemical and Pharmacological Characterization
4.4. Patients
4.5. Ex Vivo Expansion of Vδ2 T Cells
4.6. Immunofluorescence Assay and Analysis of Lymphocyte Proliferation
4.7. CRC Cell Cultures and Spheroid Generation
4.8. CRC Cell Viability and Cytotoxicity Assay
4.9. CRC Organoid Generation and Vδ2 T Cell Cytotoxicity Elicited Using ZA-SPNs
4.10. Cell Preparation for Confocal and SEM Imaging
4.11. Microfluidic Chip Fabrication
4.12. Microfluidic Experiments
4.13. HPLC Negative-Ion Electrospray Ionization TOF-MS
4.14. Statistical Analysis
4.15. Human Subjects
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Church, S.E.; Galon, J. Tumor Microenvironment and immunotherapy: The whole picture is better than a glimpse. Immunity 2015, 43, 631–633. [Google Scholar] [CrossRef] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Meraviglia, S.; Lo Presti, E.; Tosolini, M.; La Mendola, C.; Orlando, V.; Todaro, M.; Catalano, V.; Stassi, G.; Cicero, G.; Vieni, S.; et al. Distinctive features of tumor-infiltrating γδ T lymphocytes in human colorectal cancer. Oncoimmunology 2017, 6, e1347742. [Google Scholar] [CrossRef] [Green Version]
- Hayday, A.C. Gammadelta T cells and the lymphoid stress-surveillance response. Immunity 2009, 31, 184–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonneville, M.; O’Brien, R.L.; Born, W.K. Gammadelta T cell effector functions: A blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 2010, 10, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Corvaisier, M.; Moreau-Aubry, A.; Diez, E.; Bennouna, J.; Scotet, E.; Bonneville, M.; Jotereau, F. Vgamma9 Vdelta2 T cell response to colon carcinoma cells. J. Immunol. 2005, 175, 5481–5488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabelitz, D.; Wesch, D.; He, W. Perspectives of gammadelta T lymphocytes in tumor immunology. Cancer Res. 2007, 67, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Todaro, M.; D’Asaro, M.; Caccamo, N.; Iovino, F.; Francipane, M.G.; Meraviglia, S.; Orlando, V.; La Mendola, C.; Gulotta, G.; Salerno, A.; et al. Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J. Immunol. 2009, 182, 7287–7296. [Google Scholar] [CrossRef] [Green Version]
- Gober, H.J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; De Libero, G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef]
- Wang, H.; Sarikonda, G.; Puan, K.J.; Tanaka, Y.; Feng, J.; Giner, J.L.; Cao, R.; Mönkkönen, J.; Oldfield, E.; Morita, C.T. Indirect stimulation of human Vγ2Vδ2 T cells through alterations in isoprenoid metabolism. J. Immunol. 2011, 187, 5099–5113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vavassori, S.; Kumar, A.; Wan, G.S.; Ramanjaneyulu, G.S.; Cavallari, M.; El Daker, S.; Beddoe, T.; Theodossis, A.; Williams, N.K.; Gostick, E.; et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human γδ T cells. Nat. Immunol. 2013, 14, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Burjanadzé, M.; Condomines, M.; Reme, T.; Quittet, P.; Latry, P.; Lugagne, C.; Romagne, F.; Morel, Y.; Rossi, J.F.; Klein, B.; et al. In vitro expansion of gammadelta T cells with anti-myeloma cells activity by Phosphostim and IL-2 in patients with multiple myeloma. Br. J. Haematol. 2007, 139, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertner-Dardenne, J.; Bonnafous, C.; Bezombes, C.; Capietto, A.H.; Scaglione, V.; Ingoure, S.; Cendron, D.; Gross, E.; Lepage, J.F.; Quillet-Mary, A.; et al. Bromohydrin pyrophosphate enhances antibody-dependent cell-mediated cytotoxicity induced by therapeutic antibodies. Blood 2009, 113, 4875–4884. [Google Scholar] [CrossRef] [Green Version]
- Das, H.; Wang, L.; Kamath, A.; Bukowski, J.F. Vgamma2Vdelta2 T-cell receptor-mediated recognition of aminobisphosphonates. Blood 2001, 98, 1616–1618. [Google Scholar] [CrossRef]
- Santini, D.; Vespasiani Gentilucci, U.; Vincenzi, B.; Picardi, A.; Vasaturo, F.; La Cesa, A.; Onori, N.; Scarpa, S.; Tonini, G. The antineoplastic role of bisphosphonates: From basic research to clinical evidence. Ann. Oncol. 2003, 14, 1468–1476. [Google Scholar] [CrossRef]
- Clézardin, P.; Fournier, P.; Boissier, S.; Peyruchaud, O. In vitro and in vivo anti-tumor effects of bisphopshonates. Curr. Med. Chem. 2003, 10, 173–180. [Google Scholar] [CrossRef]
- Santolaria, T.; Robard, M.; Léger, A.; Catros, V.; Bonneville, M.; Scotet, E. Repeated systemic administration of aminobisphosphonates and human Vγ9Vδ2 T cells efficiently control tumor development in vivo. J. Immunol. 2013, 191, 1993–2000. [Google Scholar] [CrossRef] [Green Version]
- Kunzmann, V.; Bauer, E.; Feurle, J.; Weissinger, F.; Tony, H.P.; Wilhelm, M. Stimulation of gammadelta T cells by aminobisphosphonates and induction of anti plasmacell activity in multiple myeloma. Blood 2000, 96, 384–392. [Google Scholar] [CrossRef]
- Fiore, F.; Castella, B.; Nuschak, B.; Bertieri, R.; Mariani, S.; Bruno, B.; Pantaleoni, F.; Foglietta, M.; Boccadoro, M.; Massaia, M. Enhanced ability of dendritic cells to stimulate innate and adaptive immunity on short-term incubation with zoledronic acid. Blood 2007, 110, 921–927. [Google Scholar] [CrossRef]
- Zocchi, M.R.; Costa, D.; Venè, R.; Tosetti, F.; Ferrari, N.; Minghelli, S.; Benelli, R.; Scabini, S.; Romairone, E.; Catellani, S.; et al. Zoledronate can induce colorectal cancer microenvironment expressing BTN3A1 to stimulate effector γδ T cells with anti-tumor activity. Oncoimmunology 2017, 6, e1278099. [Google Scholar] [CrossRef] [PubMed]
- Bennouna, J.; Levy, V.; Sicard, H.; Senellart, H.; Audrain, M.; Hiret, S.; Rolland, F.; Bruzzoni-Giovanelli, H.; Rimbert, M.; Galéa, C.; et al. Phase I study of bromohydrinpyrophosphate (BrHPP, IPH 1101) a Vgamma9 Vdelta2 T lymphocyte agonist in patients with solid tumors. Cancer Immunol. Immunother. 2010, 59, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.M.; Kaikobad, M.R.; Wallace, M.; Staab, M.J.; Horvath, D.L.; Wilding, G.; Liu, G.; Eickhoff, J.C.; McNeel, D.G.; Malkovsky, M. Pilot trial of interleukin-2 and zoledronic acid to augment γδ T cells as treatment for patients with refractory renal cell carcinoma. Cancer Immunol. Immunother. 2011, 60, 1447–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, M.; Nakajima, J.; Murakawa, T.; Fukami, T.; Yoshida, Y.; Murayama, T.; Takamoto, S.; Matsushita, H.; Kakimi, K. Adoptive immunotherapy for advanced non-small cell lung cancer using zoledronate-expanded γδT cells: A phase I clinical study. J. Immunother. 2011, 34, 202–211. [Google Scholar] [CrossRef]
- Meraviglia, S.; Eberl, M.; Vermijlen, D.; Todaro, M.; Buccheri, S.; Cicero, G.; La Mendola, C.; Guggino, G.; D’Asaro, M.; Orlando, V.; et al. In vivo manipulation of Vgamma9Vdelta2 T cells with zoledronate and low-dose interleukin-2 for immunotherapy of advanced breast cancer patients. Clin. Exp. Immunol. 2010, 161, 290–297. [Google Scholar] [CrossRef]
- Dieli, F.; Vermijlen, D.; Fulfaro, F.; Caccamo, N.; Meraviglia, S.; Cicero, G.; Roberts, A.; Buccheri, S.; D’Asaro, M.; Gebbia, N.; et al. Targeting human gammadelta T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res. 2007, 67, 7450–7457. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H. Bisphosphonates: A review of their pharmacokinetic properties. Bone 1996, 18, 75–85. [Google Scholar] [CrossRef]
- Dhillon, S. Zoledronic Acid (Reclast®, Aclasta®): A Review in Osteoporosis. Drugs 2016, 76, 1683–1697. [Google Scholar] [CrossRef]
- Tanaka, Y.; Iwasaki, M.; Murata-Hirai, K.; Matsumoto, K.; Hayashi, K.; Okamura, H.; Sugie, T.; Minato, N.; Morita, C.T.; Toi, M. Anti-tumor activity and immunotherapeutic potential of a bisphosphonate prodrug. Sci. Rep. 2017, 7, 5987. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Wang, Y.; Tiruthani, K. Tumor immune microenvironment and nano-immunotherapeutics in colorectal cancer. Nanomedicine 2019, 21, 102034. [Google Scholar] [CrossRef]
- Zeng, B.; Middelberg, A.P.; Gemiarto, A.; MacDonald, K.; Baxter, A.G.; Talekar, M.; Moi, D.; Tullett, K.M.; Caminschi, I.; Lahoud, M.H.; et al. Self-adjuvanting nanoemulsion targeting dendritic cell receptor Clec9A enables antigen-specific immunotherapy. J. Clin. Investig. 2018, 128, 1971–1984. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, D.; Seo, N.; Hayashi, T.; Tahara, Y.; Fujii, K.; Tawara, I.; Miyahara, Y.; Okamori, K.; Yagita, H.; Imoto, S.; et al. Antigen delivery targeted to tumor-associated macrophages overcomes tumor immune resistance. J. Clin. Investig. 2019, 129, 1278–1294. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Matsumura, Y. EPR effect based drug design and clinical outlook for enhanced cancer chemotherapy. Adv. Drug Deliv. Rev. 2011, 63, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Papahadjopoulos, D.; Allen, T.M.; Gabizon, A.; Mayhew, E.; Matthay, K.; Huang, S.K.; Lee, K.D.; Woodle, M.C.; Lasic, D.D.; Redemann, C.; et al. Sterically stabilized liposomes: Improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. USA 1991, 88, 11460–11464. [Google Scholar] [CrossRef] [Green Version]
- He, K.; Tang, M. Safety of novel liposomal drugs for cancer treatment: Advances and prospects. Chem. Biol. Interact. 2018, 295, 13–19. [Google Scholar] [CrossRef]
- Zang, X.; Zhang, X.; Hu, H.; Qiao, M.; Zhao, X.; Deng, Y.; Chen, D. Targeted Delivery of Zoledronate to Tumor-Associated Macrophages for Cancer Immunotherapy. Mol. Pharm. 2019, 16, 2249–2258. [Google Scholar] [CrossRef]
- Lee, A.; Di Mascolo, D.; Francardi, M.; Piccardi, F.; Bandiera, T.; Decuzzi, P. Spherical polymeric nanoconstructs for combined chemotherapeutic and anti-inflammatory therapies. Nanomedicine 2016, 12, 2139–2147. [Google Scholar] [CrossRef]
- Cibrián, D.; Sánchez-Madrid, F. CD69: From activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef]
- Abbas, A.K.; Trotta, E.R.; Simeonov, D.; Marson, A.; Bluestone, J.A. Revisiting IL-2: Biology and therapeutic prospects. Sci. Immunol. 2018, 3, eaat1482. [Google Scholar] [CrossRef] [Green Version]
- Ameruoso, A.; Palomba, R.; Palange, A.L.; Cervadoro, A.; Lee, A.; Di Mascolo, D.; Decuzzi, P. Ameliorating amyloid-βfibrils triggered inflammation via curcumin-loaded polymeric nanoconstructs. Front. Immunol. 2017, 8, 1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mascolo, D.; Basnett, P.; Palange, A.L.; Francardi, M.; Roy, I.; Decuzzi, P. Tuning core hydrophobicity of spherical polymeric nanoconstructs for docetaxel delivery. Polym. Int. 2016, 65, 741–746. [Google Scholar] [CrossRef]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3D tumor spheroid models for in vitro therapeutic screening: A systematic approach to enhance the biological relevance of data obtained. Sci. Rep. 2016, 6, 19103. [Google Scholar] [CrossRef] [PubMed]
- Varesano, S.; Zocchi, M.R.; Poggi, A. Zoledronate triggers Vδ2 T cells to destroy and kill spheroids of colon carcinoma: Quantitative image analysis of three-dimensional cultures. Front. Immunol. 2018, 9, 998. [Google Scholar] [CrossRef]
- Costa, D.; Venè, R.; Benelli, R.; Romairone, E.; Scabini, S.; Catellani, S.; Rebesco, B.; Mastracci, L.; Grillo, F.; Minghelli, S.; et al. Mesenchymal stromal cell-mediated inhibition of natural killer cell function in colorectal cancer can be regulated by epidermal growth factor receptor targeting. Front. Immunol. 2018, 9, 1150. [Google Scholar] [CrossRef]
- Dieli, F.; Poccia, F.; Lipp, M.; Sireci, G.; Caccamo, N.; Di Sano, C.; Salerno, A. Differentiation of effector/memory Vdelta2 T cells and migratory routes in lymph nodes or inflammatory sites. J. Exp. Med. 2003, 198, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Musso, A.; Catellani, S.; Canevali, P.; Tavella, S.; Venè, R.; Boero, S.; Pierri, I.; Gobbi, M.; Kunkl, A.; Ravetti, J.L.; et al. Aminobisphosphonates prevent the inhibitory effects exerted by lymph node stromal cells on anti-tumor Vδ2 T lymphocytes in non-Hodgkin lymphomas. Haematologica 2014, 99, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Nguyen, V.; Lin, Y.L.; Kamen, L.; Song, A. Thaw-and-use target cells pre-labeled with calcein AM for antibody-dependent cell-mediated cytotoxicity assays. J. Immunol. Methods 2017, 447, 37–46. [Google Scholar] [CrossRef]
- Mollica, H.; Palomba, R.; Primavera, R.; Decuzzi, P. Two-Channel Compartmentalized Microfluidic Chip for Real-Time Monitoring of the Metastatic Cascade. ACS Biomater. Sci. Eng. 2019, 5, 4834–4843. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.M.; Fidler, I.J. Finding the tumor copycat. Therapy fails, patients don’t. Nat. Med. 2010, 16, 974–975. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A. The flaws and human harms of animal experimentation. Camb. Q. Health Eth. 2015, 24, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Valdes, S.A.; Alzhrani, R.F.; Hufnagel, S.; Hursting, S.D.; Cui, Z. Zoledronic Acid-containing Nanoparticles with Minimum Premature Release Show Enhanced Activity Against Extraskeletal Tumor. ACS Appl. Mater. Interfaces 2019, 11, 7311–7319. [Google Scholar] [CrossRef] [PubMed]
- Silva Santos, B.; Serre, K.; Norell, H. γδT cells in cancer. Nat. Rev. Immunol. 2015, 15, 683–691. [Google Scholar] [CrossRef]
- Adams, E.J.; Gu, S.; Luoma, A.M. Human gamma delta T cells: Evolution and ligand recognition. Cell. Immunol. 2015, 296, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. γδ T 17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 2014, 40, 785–800. [Google Scholar] [CrossRef] [Green Version]
- Capietto, A.H.; Martinet, L.; Cendron, D.; Fruchon, S.; Pont, F.; Fournié, J.J. Phophoantigens overcome human TCRVgamma9+ gammadelta T cell immunosuppression by TGF-beta: Relevance for cancer immunotherapy. J. Immunol. 2010, 184, 6680–6687. [Google Scholar] [CrossRef] [Green Version]
- Astler, V.B.; Coller, F.A. The prognostic significance of direct extension of carcinoma of the colon and rectum. Ann. Surg. 1954, 139, 846–852. [Google Scholar] [CrossRef]
- Musso, A.; Zocchi, M.R.; Poggi, A. Relevance of the mevalonate biosynthetic pathway in the regulation of bone marrow mesenchymal stromal cell-mediated effects on T-cell proliferation and B-cell survival. Haematologica 2011, 96, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Van De Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gor, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988. [Google Scholar] [CrossRef] [Green Version]
- Jauhiainen, M.; Monkkonen, H.; Raikkonen, J.; Monkkonen, J.; Auriola, S. Analysis of endogenous ATP analogs and mevalonate pathway mebolites in cancer cell cultures using liquid chromatography-electrospray ionization mass spectrometry. J. Chromatogr. B 2009, 877, 2967–2975. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Vδ2 Expansion a | Vδ2 Fold Increase b | Vδ2 Expansion from T + Mo c |
|---|---|---|---|
| ZA-SPNs | 1.355 × 10−3 | 2.067 × 10−4 | 7.096 × 10−4 |
| Soluble ZA | 3.685 × 10−1 | 4.2 × 10−1 | 5.449 × 10−1 |
| SW620 | HCT-15 | HT-29 | ||||
|---|---|---|---|---|---|---|
| Compound | Adh a | Sph b | Adh | Sph | Adh | Sph |
| ZA-SPNs | 8 × 10−2 | >6 × 10−1 | 1 × 10−1 | 5 × 10−1 | 1 × 10−1 | 5 × 10−1 |
| ZA | 1.2 | 1.0 | 1.0 | 1.1 | 1.7 | 1.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Mascolo, D.; Varesano, S.; Benelli, R.; Mollica, H.; Salis, A.; Zocchi, M.R.; Decuzzi, P.; Poggi, A. Nanoformulated Zoledronic Acid Boosts the Vδ2 T Cell Immunotherapeutic Potential in Colorectal Cancer. Cancers 2020, 12, 104. https://doi.org/10.3390/cancers12010104
Di Mascolo D, Varesano S, Benelli R, Mollica H, Salis A, Zocchi MR, Decuzzi P, Poggi A. Nanoformulated Zoledronic Acid Boosts the Vδ2 T Cell Immunotherapeutic Potential in Colorectal Cancer. Cancers. 2020; 12(1):104. https://doi.org/10.3390/cancers12010104
Chicago/Turabian StyleDi Mascolo, Daniele, Serena Varesano, Roberto Benelli, Hilaria Mollica, Annalisa Salis, Maria Raffaella Zocchi, Paolo Decuzzi, and Alessandro Poggi. 2020. "Nanoformulated Zoledronic Acid Boosts the Vδ2 T Cell Immunotherapeutic Potential in Colorectal Cancer" Cancers 12, no. 1: 104. https://doi.org/10.3390/cancers12010104
APA StyleDi Mascolo, D., Varesano, S., Benelli, R., Mollica, H., Salis, A., Zocchi, M. R., Decuzzi, P., & Poggi, A. (2020). Nanoformulated Zoledronic Acid Boosts the Vδ2 T Cell Immunotherapeutic Potential in Colorectal Cancer. Cancers, 12(1), 104. https://doi.org/10.3390/cancers12010104