Abstract
Liver cancer is the fourth leading cause of cancer-related death globally, accounting for approximately 800,000 deaths annually. Hepatocellular carcinoma (HCC) is the most common type of liver cancer, comprising approximately 80% of cases. Murine models of HCC, such as chemically-induced models, xenograft models, and genetically engineered mouse (GEM) models, are valuable tools to reproduce human HCC biopathology and biochemistry. These models can be used to identify potential biomarkers, evaluate potential novel therapeutic drugs in pre-clinical trials, and develop molecular target therapies. Considering molecular target therapies, a novel approach has been developed to create genetically engineered murine models for HCC, employing hydrodynamics-based transfection (HT). The HT method, coupled with the Sleeping Beauty transposon system or the CRISPR/Cas9 genome editing tool, has been used to rapidly and cost-effectively produce a variety of HCC models containing diverse oncogenes or inactivated tumor suppressor genes. The versatility of these models is expected to broaden our knowledge of the genetic mechanisms underlying human hepatocarcinogenesis, allowing the study of premalignant and malignant liver lesions and the evaluation of new therapeutic strategies. Here, we review recent advances in GEM models of HCC with an emphasis on new technologies.
1. Introduction
Liver cancer is a major health concern worldwide; it is the second leading cause of cancer-related deaths in East Asia and sub-Saharan Africa and the sixth in Western countries [1,2]. The main risk factors for liver cancer are well known and include hepatitis B virus (HBV) infection, hepatitis C virus (HCV) infection, alcohol intake, liver cirrhosis, metabolic syndrome, or a combination of the above [3]. Hepatocellular carcinoma (HCC) develops through a series of genetic and epigenetic changes in proto-oncogenes and tumor suppressor genes in the liver environment, wherein hepatic fibrosis or cirrhosis occurs after sustained liver damage [4].
Therapeutic options for patients with chronic liver disease and cirrhosis exist, such as potent antiviral therapies for hepatitis B and C, alcohol abstinence programs, and exercise routines for metabolic syndrome. However, HCC patients who are diagnosed at an advanced stage have limited medical options; moreover, these may only increase the mean overall survival by a few months [5]. Furthermore, the process of anti-HCC drug discovery has been difficult and inefficient, as reflected by the high attrition rate of drugs that enter preclinical testing but fail to attain FDA (Food and Drug Administration) approval [6]. Therefore, understanding the molecular signaling pathways or protein–protein interactions that play crucial roles in hepatocarcinogenesis should provide important information for the treatment of HCC.
The mouse (Mus musculus) is considered the best animal model for cancer research due to its physiological and molecular similarities with human biology, in addition to its advantages in terms of size, reproductive capacity, and lifespan. Mouse models for HCC have been developed through the introduction of genetic changes that contribute to the pathogenesis of the disease; however, with regard to the modeling of spontaneous HCC arising from a chronic inflammatory environment, there are considerable challenges that need to be overcome. Moreover, as it is becoming increasingly clear that there is no unique molecular pathway underlying the pathogenesis of HCC, various models are needed to mimic the different types of liver tumorigenesis.
This review aimed to provide a blueprint to understand the pathogenesis of HCC and optimize the preclinical models used in drug efficacy testing.
2. Non-Genetically Engineered Mouse (GEM) Models
2.1. Chemically Induced Models
Many carcinogens that induce HCC have been identified. It has been reported that both synthetic chemicals (such as carbon tetrachloride (CCl4), diethylnitrosamine (DENA), 2-acetylaminofluorene (AAF), N-nitrosodimethylamine, arsenic, o-aminoazotoluene, N-nitrosomorpholine (NMOR), and 1,2-dichloroethane) and natural substances (such as xanthosine, pyrrolizidine alkaloids, and safrole) can be liver carcinogens [7,8]. These carcinogens are administered to the animal through food, drinking water, gas inhalation, or intraperitoneal or subcutaneous injections. Chemically induced HCC models are sometimes combined with liver resection to induce cell proliferation, which increases the frequency of mutations induced by the chemical. The characteristics of each carcinogenic chemical are summarized in Table 1.

Table 1.
Chemically induced models.
Hepatic cirrhosis and multifocal HCC developed 50 weeks after intraperitoneal injection of DENA [9,10,11]. When using NMOR as a carcinogen, HCC and lung metastasis were observed after approximately 12 weeks of carcinogen supply in drinking water at a concentration of 120 ppm [12]. A choline-deficient diet with a low intake of methionine over 2 weeks can lead to rapid death of liver cells, and their combination with DENA, azaserine, or AAF treatments can accelerate the development of liver cancer cells [8,13,14,15,16,17]. This model, however, has the limitation of a large variation in the susceptibility to choline deficiency.
Metastasis can be induced by treatment with carcinogens; however, the low long-term survival rate of the treated animals makes it difficult to assess metastasis. In 2005, Yoshino et al. established an animal metastasis model that showed a significantly higher survival rate than previous models [18]. It was developed by treatment with DENA at a concentration of 120 ppm, followed by the supply of water containing NMOR at a low concentration (40 or 80 ppm) for 14 weeks. The 40 ppm NMOR-treated model is considered to be suitable to study the mechanism of metastasis. Here, HCC develops without lung metastasis until 22 weeks and frequent lung metastasis is found after 40 weeks.
2.2. Transplantation Models
Since 1969, when it was first identified that tumor cells implanted subcutaneously into immunodeficient mice gave rise to cancer, many types of cell lines and tissues have been implanted in mice for cancer research [19,20]. The transplantation model provides a suitable niche for the survival of tumor cells in vivo. Transplantation models that use human tumor sources are classified by the type of sample (tumor cell culture or tumor tissue obtained from surgery) and the anatomical location of transplantation (ectopic or orthotopic) [21]. The ectopic xenograft model has the advantage of allowing easy measurement of tumor size, thus facilitating antitumor drug efficacy testing in vivo [22]. For the exploration of immunotherapies for HCC, syngeneic models established with immunocompetent mouse strains are preferable; such models, however, cannot recapitulate the histology, natural carcinogenesis characteristics, or microenvironment of human HCC [23,24,25]. In addition, a vaccination effect may occur due to differences in the human and mouse immune systems [26]. Humanized mouse models established with patient-derived xenografts and human peripheral-blood mononuclear cells (PBMCs) can recapitulate human HCC and the human immune system, although they are expensive and require long periods of time [27].
Ma et al. isolated CD133-expressing HCC cells from human cell lines and used them for transplantation into immunodeficient mice to test their characteristics and resistance to chemical drugs. These CD133-expressing tumor cells survived at a higher rate than those that did not express CD133 [28,29]. Researchers at the University Hospital Bonn injected alcohol and thioacetamide into C3H mice to induce liver fibrosis and, then, transplanted Hepa129 cells into the liver [30], showing that tumor growth in the fibrotic liver is faster than that in the normal liver. In addition, Kornek et al. found that the expression levels of vascular endothelial growth factor (VEGF), vascular endothelial growth factor receptor (VEGFR), and matrix metalloproteinase-2 (MMP-2) and MMP-9 were higher in tumors derived from fibrotic livers. This xenograft model has been a useful tool in drug efficacy testing in the context of liver fibrosis. The various transplantation models are summarized in Table 2.
Table 2.
Transplantation models.
3. GEM Models
3.1. Traditional GEM Model
The best model for human cancer should resemble the pathological and molecular characteristics of human malignancy [31]. The model should also allow the investigation of the interactions between the tumor microenvironment (TME) and tumor cells and provide information on the molecular signaling pathways leading to cancer [32]. Transgenic mice were first created in the 1980s and enabled the study and characterization of molecular signaling pathways in human malignant tumors [33,34,35].
Transgenic models have been engineered to express oncogenes or inactivate tumor suppressor genes; when performed in the liver, these genetic manipulations have been shown to induce HCC [31,32]. Such gene expression can be restricted to liver cells using liver-specific promoters such as the albumin promoter. Genes that are used to develop GEM models for HCC include those related to the hepatitis virus, cell proliferation, and apoptosis.
3.2. Viral Genes
Chronic viral hepatitis is the most common etiology of HCC and accounts for 80% of all HCC worldwide [36]. Hepatitis B is an endemic disease in China, Southeast Asia, and Sub-Saharan Africa, where liver cancer shows a high incidence rate. HCV is more widely distributed in the United States and Europe than HBV or the human immunodeficiency virus (HIV) [37].
3.2.1. Hepatitis B Virus (HBV)
HBV, a circular DNA virus that has four open reading frames, is difficult to propagate in vitro using cell culture. The HBV X protein (HBx) is commonly used to induce HCC in murine models. In 1994, Koike et al. showed that HCC developed in transgenic mice with high HBx expression within 13 to 24 months in 84% of cases [38]. DNA analyses of these mice revealed that persistent HBx expression induced DNA synthesis and secondary mutations in many hepatocytes. Moreover, HBx transgenic mice showed differences in hepatocarcinogenesis depending on the HBV genotypes. HCC occurred after the expression of HBx from HBV genotype C in transgenic mice, whereas HBx of other genotypes hardly induced HCC [39].
In addition to HBx, Chisari et al. developed a transgenic model overexpressing large envelope polypeptides of HBV [40]. This model demonstrated that the expression of a single structural viral gene is sufficient to induce malignant transformation. The hepatocyte injury due to the accumulation of a viral product led to HCC via an inflammatory response, regeneration, transcriptional deregulation, and aneuploidy. This model supported the hypothesis that sustained hepatocyte injury can induce secondary genetic events that cause unrestricted proliferation [41].
3.2.2. Hepatitis C Virus (HCV)
HCV is an RNA virus that is not integrated into the host genome but bears proteins that exert a wide range of biological effects by interacting with many host cell factors [42]. Several models have been developed to understand the tumorigenesis of HCC by HCV.
Experiments using cell culture systems have shown that the core protein of HCV itself can regulate various cell functions, which may be directly associated with the development of HCV-related HCC [43]. The HCV core protein cooperates with the Hras oncogene in rat embryo fibroblasts, inhibits apoptosis associated with c-Myc, and represses transcription of Tp53 [44,45]. Moreover, it allows the peroxisome proliferator-activated receptor (PPAR) to interact with various proteins leading to hepatic carcinogenesis [46,47,48].
To determine the contribution of HCV structural proteins to hepatocarcinogenesis, Kamegaya et al. developed transgenic mice expressing only the HCV core protein or both the HCV core protein and E1/E2 proteins [49]. HCC cell proliferation was not significantly different between the two groups; however, HCCs in HCV core-E1/E2 transgenic mice showed significantly lower cell death rates than those in HCV core transgenic mice. In addition, tumor size in HCV core-E1/E2 transgenic mice was larger than that in HCV core transgenic mice.
3.2.3. Woodchuck Hepatitis Virus (WHV)
WHV belongs to the hepadnavirus family. In woodchucks, it induces hepatitis and hepatocellular carcinoma that are remarkably similar to those associated with HBV infection in humans. Ultimately, almost all WHV infections develop into well-differentiated HCCs. HCC is generally caused by the overexpression of the c-Myc and N-Myc genes when the WHV DNA is integrated at a specific location of the genome [50,51].
Viral-derived HCC models are valuable for long-term testing of chemoprevention strategies and evaluating targeted therapies for established HCCs. However, these models have severe limitations, such as a long latency of HCC development (usually over 2 years).
3.3. Manipulation of Host Proto-Oncogenes and Tumor Suppressor Genes
To induce HCC, liver-specific promoters are employed to drive the expression of oncogenes. Several promoters have been used for hepatic expression, such as albumin, metallothionein, transtthyretin, and liver activator protein (LAP) [49,52,53,54,55].
Sandgren et al. developed transgenic mice that specifically expressed c-Myc in the liver using an albumin enhancer/promoter. In this experiment, c-Myc expression resulted in mild to severe levels of hepatic dysfunction in young mice and hepatoblastoma in old mice after 15 months [56]. Furthermore, it was shown that c-Myc could lead to a mutation in the β-catenin gene that led to changes in β-catenin signaling transduction, which eventually led to HCC [57].
To investigate the interaction between c-Myc and transforming growth factor alpha (TGF-α) in HCC development, a double transgenic mouse model was developed. It expressed c-Myc through an albumin enhancer/promoter and TGF-α through a metallothionein 1 promoter [58,59,60,61]. This model significantly reduced the HCC onset time compared to transgenic mice expressing c-Myc or TGF-α individually. The mouse model co-expressing c-Myc and TGF-α induced continuous hepatocyte proliferation, followed by tumor development 2 months later [59]. Compared to lesions caused by c-Myc expression alone at 10 weeks, the simultaneous expression of c-Myc and TGF-α significantly increased the production of reactive oxygen species (ROS), genetic instability, and loss of heterozygosity [62]. Other double transgenic mice models expressing c-Myc plus E2F1 or c-Myc plus EGF were also developed [61,63,64].
β-Catenin, the key downstream effector of the Wnt signaling pathway, plays an important role in the liver. Activation of the Wnt/β-catenin pathway in humans can occur via an activating mutation within the β-catenin gene or the reduced expression of adenomatous polyposis coli (APC), a negative regulator of β-catenin [57]. GEM models expressing an activated form of β-catenin or with a liver-specific Apc knockout showed hepatomegaly or HCC with a long latency [65]. However, the coexpression of activated β-catenin with an activated Ras led to HCC as early as 8 weeks [66].
The phosphatase and tensin homolog (PTEN) is a tumor suppressor that negatively regulates the PI3K-Akt signaling pathway, which, in turn, regulates cell survival, proliferation, and energy metabolism. GEM models with a Pten deletion in the liver exhibited HCC after 44 weeks [67]. The characteristics of each mouse model using proto-oncogenes or tumor suppressor genes are summarized in Table 3.
Table 3.
HCC mouse models with proto-oncogenes or tumor suppressor genes.
3.4. Inducible Gene Expression Models
Employing a liver-specific promoter allows an oncogene to be specifically expressed in cells of the hepatic lineage from embryogenesis [32]. Considering HCC usually develops in adults via somatic mutations, the expression of an oncogene since early embryonic development might cause embryonic lethality or unexpected abnormal characteristics that could make the HCC model deviate from human HCC [68]. Additionally, to investigate the so-called oncogenic addiction, one might want to inactivate the driver oncogene in transgenic mice after establishing liver cancer. Thus, systems that exert temporal control of target gene expression, such as the tamoxifen-regulated Cre-loxP and the tetracycline (Tet) regulatory systems, are sometimes favored.
In the tamoxifen-regulated Cre-loxP system, the expression of a gene of interest can be induced with tamoxifen treatment. In the absence of tamoxifen, a Cre-estrogen receptor fusion protein (Cre-ER) remains in the cytosol. However, in response to tamoxifen introduced via an intraperitoneal injection, Cre-ER is translocated into the nucleus and induces recombination between loxP sites, resulting in the removal of transcriptional stop DNA elements (Figure 1A). This allows the target gene to be expressed in the liver by a liver-specific promoter (LSP) [69,70,71]. Virus-mediated Cre delivery systems are another option for temporal control of target gene expression in the liver. For example, a recombinant adenovirus expressing the Cre gene can be used to remove a floxed target gene (Figure 1B) [72,73]. Colnot et al. created a transgenic mouse in which the tumor suppressor gene Apc was located between two loxP sites. Intravenously injected Cre-adenovirus induced the deletion of Apc in a liver-specific manner. After 8 months, 67% of the mice developed liver cancer and the signaling pathway of β-catenin was strongly upregulated in the Apc-inactivated HCC [65].
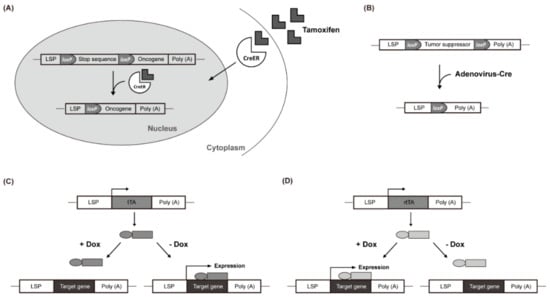
Figure 1.
Liver-Specific Genetic Modification Models. (A) Cre-ER translocates into the nucleus and induces recombination between loxP sites. (B) recombinant adenovirus removes a floxed target gene. (C) doxycycline (Dox) suppresses the transcription of the gene of interest. (D) Dox activates rtTA by attaching it to the promoter and promotes the transcription of the target gene.
The Tet-Off system uses the tetracycline transactivator (tTA), whereas the Tet-On system uses the reverse tetracycline-controlled transactivator (rtTA). In the Tet-Off system, doxycycline (Dox) suppresses the transcription of the gene of interest by preventing tTA from attaching to the promoter (Figure 1C). Conversely, in the Tet-On system, Dox activates rtTA by attaching it to the promoter, which, in turn, promotes the transcription of the target gene (Figure 1D). These inducible gene expression systems have been used in c-Myc-induced liver cancer studies. Shachaf et al. developed a mouse model in which c-Myc was regulated by the tetracycline promoter and tTA was regulated by the liver-specific LAP promoter [55]. The mouse model expressed c-Myc in the liver but not when treated with Dox (Tet-Off c-Myc model). Liver cancer occurred in all transgenic mice with upregulated c-Myc after approximately 12 weeks. After 4 days of Dox treatment, liver cancer differentiated into normal hepatocytes with apoptosis and almost all of it was eliminated within 2 weeks.
3.5. Hydrodynamics-Based Transfection and Sleeping Beauty (SB) Transposon
Recently, a simple liver-specific transgenic approach that employs the SB transposase system and the hydrodynamics-based transfection (HT) method was developed to create a mouse model for liver cancer (Figure 2A, Table 4). HT is a simple physical method to deliver naked DNA plasmids to liver cells [74]. As episomal plasmids only allow the transient expression of a target gene, chromosomal integration is required to sustain gene expression. The SB transposase mediates chromosomal integration of transposons; thus, the gene to be expressed is placed in the plasmid between specific repeating sequences (IRs). The transposase recognizes this position and integrates the gene into the chromosome [75,76,77]. A variety of transgenic mouse models have been developed using this method, through which the oncogenic signaling pathway is activated or the tumor suppressor pathway is inactivated. A selection of representative models using HT is summarized in Table 4 with emphasis on the most recent findings.
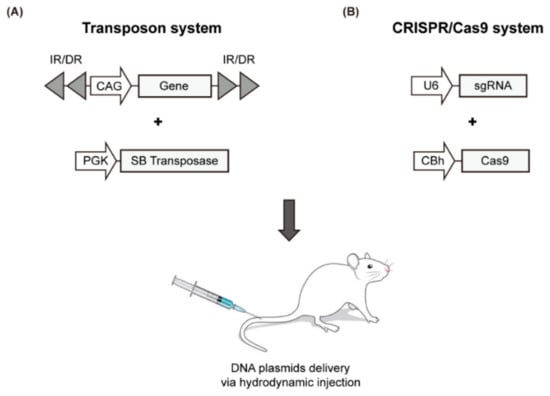
Figure 2.
Schematic illustration of the HT-based mouse model using the SB transposase system (A) and CRISPR/Cas9 genome editing tool (B).
Table 4.
Mouse models of HCC generated via hydrodynamics-based transfection.
3.5.1. Combination of Genetic Modifications and Disease-Specific Injury
Transgenic mice expressing an activated form of the Akt proto-oncogene, which were created using HT and the SB transposon, developed HCC after 6 months [78]. This model showed high similarity to a transgenic mouse model in which Pten was knocked out in the liver via traditional genetic manipulation [79].
Studies showed that transgenic mouse models overexpressing only the c-Myc proto-oncogene develop cancer in 60% to 70% of cases [80,81]. The tumors of c-Myc-induced models display high heterogeneity and mimic alcohol-induced HCCs, based on genomic changes. Additionally, a mouse model expressing c-Myc in the liver and developed using HT showed liver cancer in 5 to 8 weeks, exhibiting histological features similar to those seen in human hepatoblastoma [82]. Based on these findings, Chung et al. used a chemical induction methodology (CCl4) combined with HT to express c-Myc and a short hairpin RNA downregulating p53 expression (shp53) in mice [83]. The tumor incidence with liver fibrosis was significantly higher in transgenic mice treated with CCl4 than in those treated with vehicle.
Recent studies also showed that the overexpression of more than two oncogenes or the combination of an activated oncogene and an inactivated tumor suppressor gene could effectively contribute in the development of HCC models to obtain shorter latency and increased tumor induction. For instance, activated Met and the mutation of the β-catenin gene are considered to be more frequently present in human HCCs [84,85]. To investigate their cooperation in tumorigenesis, Tao et al. co-expressed Met and β-catenin point mutants (S33Y or S45Y) in hepatocytes, leading to HCC development as early as 7 weeks after HT [86]. Additionally, the hepatic expression of active β-catenin and Yap1 using HT and the SB transposon resulted in the development of hepatoblastoma (HB) in mice [87]. SPRY2, a gene with reduced function in half of human HCCs, is known to act as a tumor suppressor in the development of liver cancer via inhibition of the Ras signaling pathway [88]. The potential tumor suppressive role of Spry2 in HCC was investigated by expressing a dominant negative form of Spry2 (Spry2Y55F) and an activated β-catenin in mouse liver through hydrodynamic injection and the SB transposon system.
The HT method coupled with the SB transposon system could be useful in studies of specific signaling roles in tumorigenesis. HCC mouse models were developed via hydrodynamic delivery of activated Ras combined with either Tp53 downregulation or Taz activation. In these models, TGF-β signaling played a pro-tumorigenic role during the early stages of hepatocarcinogenesis, upregulating Snai1 [89]. In addition, Wang et al. generated intrahepatic cholangiocarcinoma (ICCA) mouse models using this system. The overexpression of Fbxw7ΔF, a dominant negative form of Fbxw7, did not cause significant abnormalities in the mouse liver, whereas its co-expression with an activated form of Akt resulted in ICCA [90].
3.5.2. The HT Model as a Tool for Preclinical Treatment
HCC models developed using HT and transposon methods are also very useful for evaluating the efficacy of anticancer drugs in the liver. Zhang et al. evaluated the therapeutic potential of mTOR inhibitor MLN0128 vs. gemcitabine/oxaliplatin using ICCA mouse models containing activated forms of Akt (myr-Akt) and Yap (YapS127A). The study showed the antitumor potential of MLN0128 in ICCA, suggesting that it may be superior to gemcitabine/oxaliplatin-based chemotherapy, especially in tumors exhibiting activated Akt/mTOR signaling [91].
Liu et al. established an HCC preclinical mouse model co-expressing Akt and c-Met proto-oncogenes. In this model, the therapeutic efficacies of sorafenib, regorafenib, the MEK inhibitor PD901, and the pan-mTOR inhibitor MLN0128 were investigated. Treatment with PD901 or MLN0128 alone suppressed HCC growth. Simultaneous administration of both drugs showed a stronger inhibition effect on cell cycle and tumor growth. Thus, the combination of MEK and mTOR inhibitors may represent an effective therapeutic approach in human HCCs [92].
HT and transposon-based HCC models can also be used for testing a potential anti-HCC drug. Chen et al. identified niclosamide ethanolamine (NEN) as a potential antitumor agent through a bioinformatics-based search. In an HCC mouse model produced by hydrodynamic delivery of transposons expressing activated forms of Ras and β-catenin (N90), the oral administration of NEN significantly reduced HCC growth, with a more potent anticancer effect when combined with sorafenib [93].
The HT method coupled with the SB transposon system is an ideal approach to study the roles of diverse novel genes in hepatocarcinogenesis due to the simplicity and cost-effectiveness of these techniques. The HT and SB transposon method is expected to provide insights into the biology of hepatic tumorigenesis and allow us to test novel therapies towards precision medicine.
3.6. HT and CRISPR/Cas9
Murine models developed via HT have been widely used for genetic studies of liver cancer. Although the transgenic methodology has considerable advantages over the traditional GEM approach, which requires a series of time-consuming and resource-demanding steps, the transposon-based liver cancer models also present disadvantages. First, transposons encoding oncogenes are randomly integrated into chromosomes, affecting endogenous genes at or near the integration sites. This raises the possibility of tumorigenesis induced by oncogenes influenced by the genomic integration location. Second, the oncogenes in transposons are generally placed under the control of an ectopic and strong promoter; thus, genes are expressed at an extremely high level. These shortcomings might under or overrepresent the true tumorigenic potential of an oncogene at the endogenous locus.
To overcome these limitations, direct manipulation of endogenous genes in the liver has been attempted using the CRISPR/Cas9 genome editing tool, which has been successfully applied in many organisms. This genome editing technique utilizes an RNA-guided DNA endonuclease (Cas9 nuclease) and a single guide RNA (sgRNA) that guides the Cas9 to a complementary DNA sequence where it cleaves both DNA strands. During repair processes of double-stranded DNA breaks generated by Cas9, insertions and deletions are created (via non-homologous end joining) or a specific nucleotide sequence is introduced at the target site (via homology-directed repair). To induce genome editing by the CRISPR/Cas system in the liver, plasmids encoding Cas9 and sgRNA are delivered to the liver via the HT method (Figure 2B and Table 4).
The first successful application of the CRISPR/Cas9 system in a liver cancer model was reported in 2014 by Tyler Jacks’ research group [94]. Using HT for the hepatic delivery of Cas9 and sgRNAs targeting Pten and Tp53, they successfully and simultaneously induced somatic gene disruption in the murine liver tumor suppressor genes, in a manner similar to liver tumors induced by Pten and Tp53 double knock-outs built via the traditional GEM approach. In a similar study, Liu et al. used HT and the CRISPR/Cas9 system to inactivate Tp53 and Pten simultaneously in the livers of adult transgenic mice that expressed the HBV large envelope polypeptide [40,95]. The hepatic gene disruption of Tp53 and Pten in the HBV transgenic mice significantly accelerated tumorigenesis in the liver, resulting in tumors as early as 4 months after HT.
Recently, Engelholm et al. employed HT and the CRISPR/Cas9 system to investigate the genetic aspects of the initiation of fibrolamellar hepatocellular carcinoma (FL-HCC), a liver cancer that predominantly affects children [96]. A 400 kb deletion on chromosome 19, which leads to gene fusion between DnaJ heat shock protein family member B1 gene (DNAJB1) and the protein kinase cAMP-activated catalytic subunit alpha gene (PRKACA), is repeatedly found in FL-HCC patients. Therefore, this study attempted to investigate the role of this gene fusion event in FL-HCC using HT and the CRISPR/Cas9 system. They designed sgRNA targeting intron 1 of murine Dnajb1 and intron 1 of murine Prkaca to induce DNA double-strand cuts in these regions. When the sgRNA and Cas9 were delivered to wild-type mice via HT, DNA double-strand breaks were introduced at the two genetic loci as expected; the subsequent cellular repair led to DNA end joining that created the Dnajb1–Prkaca gene fusion, as observed in human FL-HCC. They found that mice with this gene fusion in the liver developed tumors that have many features of human FL-HCC.
Thus, the HT and the CRISPR/Cas9 system can be used to faithfully mimic the genetic features found in patients with liver cancer and applied to investigate the roles of genetic alterations in liver carcinogenesis. However, CRISPR/Cas9 technology should be applied on a liver fibrosis or cirrhosis background to correctly mirror human HCC.
4. Conclusions
A successful experimental model for HCC should reflect the biological characteristics of human HCCs; it should also be reliable, highly reproducible, and technically simple. To date, GEM models for liver cancer have provided invaluable information on HCC, such as the function of oncogenes and tumor suppressor genes, the interaction between tumor and host cells, cellular responses to chemotherapeutic agents, and the role of stem cells in tumor progression. Traditional GEM methodology requires a series of time-consuming and resource-demanding procedures that delay the development of a variety of liver cancer models. HT methodology, combined with either the SB transposon system or the CRISPR/Cas9 technique, is a promising alternative to the traditional approach for generating GEM models for liver cancer (Table 5). Further development and refinement of the HT-based GEM models are expected to broaden our knowledge of the genetic mechanisms underlying hepatocarcinogenesis and provide a novel therapeutic strategy targeting genes that maintain and promote liver cancer.
Table 5.
Methodologies for creating genetically engineered mouse models for liver cancer.
Author Contributions
Conception: D.Y.K. and S.U.K.; Contribution to the literature review and manuscript preparation: K.C., S.W.R.; S.H.S., Y.J., H.M., D.Y.K., and S.U.K.; Guarantors: D.Y.K. and S.U.K. All authors reviewed the paper and approved the final version. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT & Future Planning (2019R1A2C4070136 and 2017R1C1B2007770). The funding sources had no role in study design, data collection and analysis, decision to publish, or manuscript preparation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.K.; Kim, D.Y.; Han, K.H.; Seong, J. Changes in real-life practice for hepatocellular carcinoma patients in the Republic of Korea over a 12-year period: A nationwide random sample study. PLoS ONE 2019, 14, e0223678. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Disease Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Chan, S.L. Drug development for hepatocellular carcinoma: Knowing the past helps to understand the future. Oncologist 2014, 19, 1115–1117. [Google Scholar] [CrossRef][Green Version]
- Gold, L.S.; Slone, T.H.; Manley, N.B.; Bernstein, L. Target organs in chronic bioassays of 533 chemical carcinogens. Environ. Health Perspect. 1991, 93, 233–246. [Google Scholar] [CrossRef]
- Newell, P.; Villanueva, A.; Friedman, S.L.; Koike, K.; Llovet, J.M. Experimental models of hepatocellular carcinoma. J. Hepatol. 2008, 48, 858–879. [Google Scholar] [CrossRef]
- Goldfarb, S.; Pugh, T.D.; Koen, H.; He, Y.Z. Preneoplastic and neoplastic progression during hepatocarcinogenesis in mice injected with diethylnitrosamine in infancy. Environ. Health Perspect. 1983, 50, 149–161. [Google Scholar] [CrossRef]
- Koen, H.; Pugh, T.D.; Goldfarb, S. Centrilobular distribution of diethylnitrosamine-induced hepatocellular foci in the mouse. Lab. Investig. 1983, 49, 78–81. [Google Scholar]
- Tsujiuchi, T.; Tsutsumi, M.; Sasaki, Y.; Takahama, M.; Konishi, Y. Different frequencies and patterns of beta-catenin mutations in hepatocellular carcinomas induced by N-nitrosodiethylamine and a choline-deficient L-amino acid-defined diet in rats. Cancer Res. 1999, 59, 3904–3907. [Google Scholar] [PubMed]
- Futakuchi, M.; Hirose, M.; Ogiso, T.; Kato, K.; Sano, M.; Ogawa, K.; Shirai, T. Establishment of an in vivo highly metastatic rat hepatocellular carcinoma model. Jpn. J. Cancer Res. 1999, 90, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Solt, D.B.; Medline, A.; Farber, E. Rapid emergence of carcinogen-induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am. J. Pathol. 1977, 88, 595–618. [Google Scholar] [PubMed]
- Yaswen, P.; Goyette, M.; Shank, P.R.; Fausto, N. Expression of c-Ki-ras, c-Ha-ras, and c-myc in specific cell types during hepatocarcinogenesis. Mol. Cell. Biol. 1985, 5, 780–786. [Google Scholar] [CrossRef]
- Chandar, N.; Lombardi, B.; Locker, J. c-myc gene amplification during hepatocarcinogenesis by a choline-devoid diet. Proc. Natl. Acad. Sci. USA 1989, 86, 2703–2707. [Google Scholar] [CrossRef]
- Groos, J.; Bannasch, P.; Schwarz, M.; Kopp-Schneider, A. Comparison of mode of action of four hepatocarcinogens: A model-based approach. Toxicol. Sci. 2007, 99, 446–454. [Google Scholar] [CrossRef]
- Williams, G.M.; Iatropoulos, M.J.; Wang, C.X.; Jeffrey, A.M.; Thompson, S.; Pittman, B.; Palasch, M.; Gebhardt, R. Nonlinearities in 2-acetylaminofluorene exposure responses for genotoxic and epigenetic effects leading to initiation of carcinogenesis in rat liver. Toxicol. Sci. 1998, 45, 152–161. [Google Scholar] [CrossRef][Green Version]
- Yoshino, H.; Futakuchi, M.; Cho, Y.M.; Ogawa, K.; Takeshita, F.; Imai, N.; Tamano, S.; Shirai, T. Modification of an in vivo lung metastasis model of hepatocellular carcinoma by low dose N-nitrosomorpholine and diethylnitrosamine. Clin. Exp. Metastasis 2005, 22, 441–447. [Google Scholar] [CrossRef]
- Rygaard, J.; Povlsen, C.O. Heterotransplantation of a human malignant tumour to “Nude” mice. APMIS 1969, 77, 758–760. [Google Scholar] [CrossRef]
- Cespedes, M.V.; Casanova, I.; Parreno, M.; Mangues, R. Mouse models in oncogenesis and cancer therapy. Clin. Transl. Oncol. 2006, 8, 318–329. [Google Scholar] [CrossRef]
- Killion, J.J.; Radinsky, R.; Fidler, I.J. Orthotopic models are necessary to predict therapy of transplantable tumors in mice. Cancer Metastasis Rev. 1998, 17, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. Orthotopic metastatic mouse models for anticancer drug discovery and evaluation: A bridge to the clinic. Investig. New Drugs 1999, 17, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy Research. Cancer Dis. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Pitt, J.M.; Daillere, R.; Smyth, M.J.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat. Rev. 2018, 15, 536–554. [Google Scholar] [CrossRef]
- Lee, J.; Liao, R.; Wang, G.; Yang, B.H.; Luo, X.; Varki, N.M.; Qiu, S.J.; Ren, B.; Fu, W.; Feng, G.S. Preventive Inhibition of Liver Tumorigenesis by Systemic Activation of Innate Immune Functions. Cell Rep. 2017, 21, 1870–1882. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Ann. Rev. Pathol. 2017, 12, 187–215. [Google Scholar] [CrossRef]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.; Wo, J.Y.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef]
- Kornek, M.; Raskopf, E.; Tolba, R.; Becker, U.; Klockner, M.; Sauerbruch, T.; Schmitz, V. Accelerated orthotopic hepatocellular carcinomas growth is linked to increased expression of pro-angiogenic and prometastatic factors in murine liver fibrosis. Liver Int. 2008, 28, 509–518. [Google Scholar] [CrossRef]
- Frese, K.K.; Tuveson, D.A. Maximizing mouse cancer models. Nat. Rev. Cancer 2007, 7, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Jacks, T. Technologically advanced cancer modeling in mice. Curr. Opin. Genet. Dev. 2002, 12, 105–110. [Google Scholar] [CrossRef]
- Gordon, J.W.; Scangos, G.A.; Plotkin, D.J.; Barbosa, J.A.; Ruddle, F.H. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 7380–7384. [Google Scholar] [CrossRef] [PubMed]
- Pattengale, P.K.; Stewart, T.A.; Leder, A.; Sinn, E.; Muller, W.; Tepler, I.; Schmidt, E.; Leder, P. Animal models of human disease. Pathology and molecular biology of spontaneous neoplasms occurring in transgenic mice carrying and expressing activated cellular oncogenes. Am. J. Pathol. 1989, 135, 39–61. [Google Scholar] [PubMed]
- Shuldiner, A.R. Transgenic animals. N. Engl. J. Med. 1996, 334, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.N.; Hwang, S.G.; Kim, B.K.; Park, J.Y.; Kim, D.Y.; Han, K.H.; Kim, S.U.; Ahn, S.H. Liver Cirrhosis, Not Antiviral Therapy, Predicts Clinical Outcome in Cohorts with Heterogeneous Hepatitis B Viral Status. Gut Liver 2019, 13, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Rustgi, V.K. The epidemiology of hepatitis C infection in the United States. J. Gastroenterol. 2007, 42, 513–521. [Google Scholar] [CrossRef]
- Koike, K.; Moriya, K.; Iino, S.; Yotsuyanagi, H.; Endo, Y.; Miyamura, T.; Kurokawa, K. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 1994, 19, 810–819. [Google Scholar] [CrossRef]
- Koike, K. Hepatocarcinogenesis in hepatitis viral infection: Lessons from transgenic mouse studies. J. Gastroenterol. 2002, 37 (Suppl. S13), 55–64. [Google Scholar] [CrossRef]
- Chisari, F.V.; Filippi, P.; Buras, J.; McLachlan, A.; Popper, H.; Pinkert, C.A.; Palmiter, R.D.; Brinster, R.L. Structural and pathological effects of synthesis of hepatitis B virus large envelope polypeptide in transgenic mice. Proc. Nat. Acad. Sci. USA 1987, 84, 6909–6913. [Google Scholar] [CrossRef]
- Chisari, F.V.; Klopchin, K.; Moriyama, T.; Pasquinelli, C.; Dunsford, H.A.; Sell, S.; Pinkert, C.A.; Brinster, R.L.; Palmiter, R.D. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 1989, 59, 1145–1156. [Google Scholar] [CrossRef]
- Levrero, M. Viral hepatitis and liver cancer: The case of hepatitis C. Oncogene 2006, 25, 3834–3847. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.B.; Lagging, L.M.; Meyer, K.; Ray, R. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J. Virol. 1996, 70, 4438–4443. [Google Scholar]
- Ray, R.B.; Meyer, K.; Ray, R. Suppression of apoptotic cell death by hepatitis C virus core protein. Virology 1996, 226, 176–182. [Google Scholar] [CrossRef]
- McLauchlan, J. Properties of the hepatitis C virus core protein: A structural protein that modulates cellular processes. J. Viral Hepat. 2000, 7, 2–14. [Google Scholar] [CrossRef]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Aoyama, T. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor alpha in transgenic mice: Implications for HCV-associated hepatocarcinogenesis. Int. J. Cancer 2008, 122, 124–131. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Rice, C.M. Interaction between hepatitis C virus proteins and host cell factors. Curr. Opin. Microbiol. 2002, 5, 419–427. [Google Scholar] [CrossRef]
- Kamegaya, Y.; Hiasa, Y.; Zukerberg, L.; Fowler, N.; Blackard, J.T.; Lin, W.; Choe, W.H.; Schmidt, E.V.; Chung, R.T. Hepatitis C virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis. Hepatology 2005, 41, 660–667. [Google Scholar] [CrossRef]
- Jacob, J.R.; Sterczer, A.; Toshkov, I.A.; Yeager, A.E.; Korba, B.E.; Cote, P.J.; Buendia, M.A.; Gerin, J.L.; Tennant, B.C. Integration of woodchuck hepatitis and N-myc rearrangement determine size and histologic grade of hepatic tumors. Hepatology 2004, 39, 1008–1016. [Google Scholar] [CrossRef]
- Pagano, J.S.; Blaser, M.; Buendia, M.A.; Damania, B.; Khalili, K.; Raab-Traub, N.; Roizman, B. Infectious agents and cancer: Criteria for a causal relation. Semin. Cancer Biol. 2004, 14, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Lerat, H.; Honda, M.; Beard, M.R.; Loesch, K.; Sun, J.; Yang, Y.; Okuda, M.; Gosert, R.; Xiao, S.Y.; Weinman, S.A.; et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 2002, 122, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Morisawa, K.; Nie, J.; Ota, K.; Taniguchi, T.; Saibara, T.; Onishi, S. Transactivation of transforming growth factor alpha gene by hepatitis B virus preS1. Cancer Res. 1998, 58, 1813–1816. [Google Scholar] [PubMed]
- Lou, D.Q.; Molina, T.; Bennoun, M.; Porteu, A.; Briand, P.; Joulin, V.; Vasseur-Cognet, M.; Cavard, C. Conditional hepatocarcinogenesis in mice expressing SV 40 early sequences. Cancer Lett. 2005, 229, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Sandgren, E.P.; Quaife, C.J.; Pinkert, C.A.; Palmiter, R.D.; Brinster, R.L. Oncogene-induced liver neoplasia in transgenic mice. Oncogene 1989, 4, 715–724. [Google Scholar]
- De La Coste, A.; Romagnolo, B.; Billuart, P.; Renard, C.A.; Buendia, M.A.; Soubrane, O.; Fabre, M.; Chelly, J.; Beldjord, C.; Kahn, A.; et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Nat. Acad. Sci. USA 1998, 95, 8847–8851. [Google Scholar] [CrossRef]
- Jhappan, C.; Stahle, C.; Harkins, R.N.; Fausto, N.; Smith, G.H.; Merlino, G.T. TGF alpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell 1990, 61, 1137–1146. [Google Scholar] [CrossRef]
- Murakami, H.; Sanderson, N.D.; Nagy, P.; Marino, P.A.; Merlino, G.; Thorgeirsson, S.S. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: Interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993, 53, 1719–1723. [Google Scholar]
- Calvisi, D.F.; Thorgeirsson, S.S. Molecular mechanisms of hepatocarcinogenesis in transgenic mouse models of liver cancer. Toxicol. Pathol. 2005, 33, 181–184. [Google Scholar] [CrossRef]
- Santoni-Rugiu, E.; Jensen, M.R.; Thorgeirsson, S.S. Disruption of the pRb/E2F pathway and inhibition of apoptosis are major oncogenic events in liver constitutively expressing c-myc and transforming growth factor alpha. Cancer Res. 1998, 58, 123–134. [Google Scholar] [PubMed]
- Calvisi, D.F.; Factor, V.M.; Ladu, S.; Conner, E.A.; Thorgeirsson, S.S. Disruption of beta-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterology 2004, 126, 1374–1386. [Google Scholar] [CrossRef] [PubMed]
- Conner, E.A.; Lemmer, E.R.; Sanchez, A.; Factor, V.M.; Thorgeirsson, S.S. E2F1 blocks and c-Myc accelerates hepatic ploidy in transgenic mouse models. Biochem. Biophys. Res. Commun. 2003, 302, 114–120. [Google Scholar] [CrossRef]
- Tonjes, R.R.; Lohler, J.; O’Sullivan, J.F.; Kay, G.F.; Schmidt, G.H.; Dalemans, W.; Pavirani, A.; Paul, D. Autocrine mitogen IgEGF cooperates with c-myc or with the Hcs locus during hepatocarcinogenesis in transgenic mice. Oncogene 1995, 10, 765–768. [Google Scholar]
- Colnot, S.; Decaens, T.; Niwa-Kawakita, M.; Godard, C.; Hamard, G.; Kahn, A.; Giovannini, M.; Perret, C. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc. Nat. Acad. Sci. USA 2004, 101, 17216–17221. [Google Scholar] [CrossRef]
- Harada, N.; Oshima, H.; Katoh, M.; Tamai, Y.; Oshima, M.; Taketo, M.M. Hepatocarcinogenesis in mice with beta-catenin and Ha-ras gene mutations. Cancer Res. 2004, 64, 48–54. [Google Scholar] [CrossRef]
- Watanabe, S.; Horie, Y.; Kataoka, E.; Sato, W.; Dohmen, T.; Ohshima, S.; Goto, T.; Suzuki, A. Non-alcoholic steatohepatitis and hepatocellular carcinoma: Lessons from hepatocyte-specific phosphatase and tensin homolog (PTEN)-deficient mice. J. Gastroenterol. Hepatol. 2007, 22, S96–s100. [Google Scholar] [CrossRef]
- Jonkers, J.; Berns, A. Conditional mouse models of sporadic cancer. Nat. Rev. Cancer 2002, 2, 251–265. [Google Scholar] [CrossRef]
- Hardouin, S.N.; Nagy, A. Mouse models for human disease. Clin. Genet. 2000, 57, 237–244. [Google Scholar] [CrossRef]
- Tumurbaatar, B.; Sun, Y.; Chan, T.; Sun, J. Cre-estrogen receptor-mediated hepatitis C virus structural protein expression in mice. J. Virol. Methods 2007, 146, 5–13. [Google Scholar] [CrossRef][Green Version]
- Hayashi, S.; McMahon, A.P. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: A tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 2002, 244, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.J.; Liu, R.S.; Hwu, L.; Ke, C.C.; Wang, F.H.; Wang, H.E.; Chen, F.D. Cre/loxP system controlled by specific promoter for radiation-mediated gene therapy of hepatoma. Anticancer Res. 2007, 27, 1571–1579. [Google Scholar] [PubMed]
- Wang, Y.; Krushel, L.A.; Edelman, G.M. Targeted DNA recombination in vivo using an adenovirus carrying the cre recombinase gene. Proc. Nat. Acad. Sci. USA 1996, 93, 3932–3936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Budker, V.; Wolff, J.A. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum. Gene Ther. 1999, 10, 1735–1737. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvak, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef]
- Bell, J.B.; Podetz-Pedersen, K.M.; Aronovich, E.L.; Belur, L.R.; McIvor, R.S.; Hackett, P.B. Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat. Protoc. 2007, 2, 3153–3165. [Google Scholar] [CrossRef]
- Aronovich, E.L.; Bell, J.B.; Khan, S.A.; Belur, L.R.; Gunther, R.; Koniar, B.; Schachern, P.A.; Parker, J.B.; Carlson, C.S.; Whitley, C.B.; et al. Systemic correction of storage disease in MPS I NOD/SCID mice using the sleeping beauty transposon system. Mol. Ther. 2009, 17, 1136–1144. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Investig. 2004, 113, 1774–1783. [Google Scholar] [CrossRef]
- Friemel, J.; Frick, L.; Unger, K.; Egger, M.; Parrotta, R.; Boge, Y.T.; Adili, A.; Karin, M.; Luedde, T.; Heikenwalder, M.; et al. Characterization of HCC Mouse Models: Towards an Etiology-Oriented Subtyping Approach. Mol. Cancer Res. 2019, 17, 1493–1502. [Google Scholar] [CrossRef]
- Thorgeirsson, S.S.; Santoni-Rugiu, E. Transgenic mouse models in carcinogenesis: Interaction of c-myc with transforming growth factor alpha and hepatocyte growth factor in hepatocarcinogenesis. Br. J. Clin. Pharmacol. 1996, 42, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.K.; Fan, L.L.; Chen, X.; Bishop, J.M. Oncogene-specific formation of chemoresistant murine hepatic cancer stem cells. Hepatology 2012, 56, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.I.; Moon, H.; Kim, D.Y.; Cho, K.J.; Ju, H.L.; Kim, D.Y.; Ahn, S.H.; Han, K.H.; Ro, S.W. Development of a transgenic mouse model of hepatocellular carcinoma with a liver fibrosis background. BMC Gastroenterol. 2016, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; De Reynies, A.; Villanueva, A.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Decaens, T.; Franco, D.; Imbeaud, S.; Rousseau, F.; et al. A hepatocellular carcinoma 5-gene score associated with survival of patients after liver resection. Gastroenterology 2013, 145, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Xu, E.; Zhao, Y.; Singh, S.; Li, X.; Couchy, G.; Chen, X.; Zucman-Rossi, J.; Chikina, M.; Monga, S.P. Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant beta-catenin. Hepatology 2016, 64, 1587–1605. [Google Scholar] [CrossRef]
- Abitbol, S.; Dahmani, R.; Coulouarn, C.; Ragazzon, B.; Mlecnik, B.; Senni, N.; Savall, M.; Bossard, P.; Sohier, P.; Drouet, V.; et al. AXIN deficiency in human and mouse hepatocytes induces hepatocellular carcinoma in the absence of beta-catenin activation. J. Hepatol. 2018, 68, 1203–1213. [Google Scholar] [CrossRef]
- Mason, J.M.; Morrison, D.J.; Basson, M.A.; Licht, J.D. Sprouty proteins: Multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006, 16, 45–54. [Google Scholar] [CrossRef]
- Moon, H.; Ju, H.L.; Chung, S.I.; Cho, K.J.; Eun, J.W.; Nam, S.W.; Han, K.H.; Calvisi, D.F.; Ro, S.W. Transforming Growth Factor-beta Promotes Liver Tumorigenesis in Mice via Up-regulation of Snail. Gastroenterology 2017, 153, 1378–1391.e1376. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Peters, M.; Ding, N.; Ribback, S.; Utpatel, K.; Cigliano, A.; Dombrowski, F.; Xu, M.; Chen, X.; et al. Loss of Fbxw7 synergizes with activated Akt signaling to promote c-Myc dependent cholangiocarcinogenesis. J. Hepatol. 2019, 71, 742–752. [Google Scholar] [CrossRef]
- Zhang, S.; Song, X.; Cao, D.; Xu, Z.; Fan, B.; Che, L.; Hu, J.; Chen, B.; Dong, M.; Pilo, M.G.; et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J. Hepatol. 2017, 67, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, J.; Song, X.; Utpatel, K.; Zhang, Y.; Wang, P.; Lu, X.; Zhang, J.; Xu, M.; Su, T.; et al. Combined Treatment with MEK and mTOR Inhibitors is Effective in In Vitro and In Vivo Models of Hepatocellular Carcinoma. Cancers 2019, 11, 930. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wei, W.; Ma, L.; Yang, B.; Gill, R.M.; Chua, M.S.; Butte, A.J.; So, S. Computational Discovery of Niclosamide Ethanolamine, a Repurposed Drug Candidate That Reduces Growth of Hepatocellular Carcinoma Cells In Vitro and in Mice by Inhibiting Cell Division Cycle 37 Signaling. Gastroenterology 2017, 152, 2022–2036. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G.; et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qi, X.; Zeng, Z.; Wang, L.; Wang, J.; Zhang, T.; Xu, Q.; Shen, C.; Zhou, G.; Yang, S.; et al. CRISPR/Cas9-mediated p53 and Pten dual mutation accelerates hepatocarcinogenesis in adult hepatitis B virus transgenic mice. Sci. Rep. 2017, 7, 2796. [Google Scholar] [CrossRef]
- Engelholm, L.H.; Riaz, A.; Serra, D.; Dagnaes-Hansen, F.; Johansen, J.V.; Santoni-Rugiu, E.; Hansen, S.H.; Niola, F.; Frodin, M. CRISPR/Cas9 Engineering of Adult Mouse Liver Demonstrates That the Dnajb1-Prkaca Gene Fusion Is Sufficient to Induce Tumors Resembling Fibrolamellar Hepatocellular Carcinoma. Gastroenterology 2017, 153, 1662–1673.e1610. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).