PARylation During Transcription: Insights into the Fine-Tuning Mechanism and Regulation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
The Mechanism of Poly-ADP-Ribosylation and the PARP Superfamily
2. PARylation in Transcription Regulation
2.1. The Major Regulatory Steps of Transcription Activation
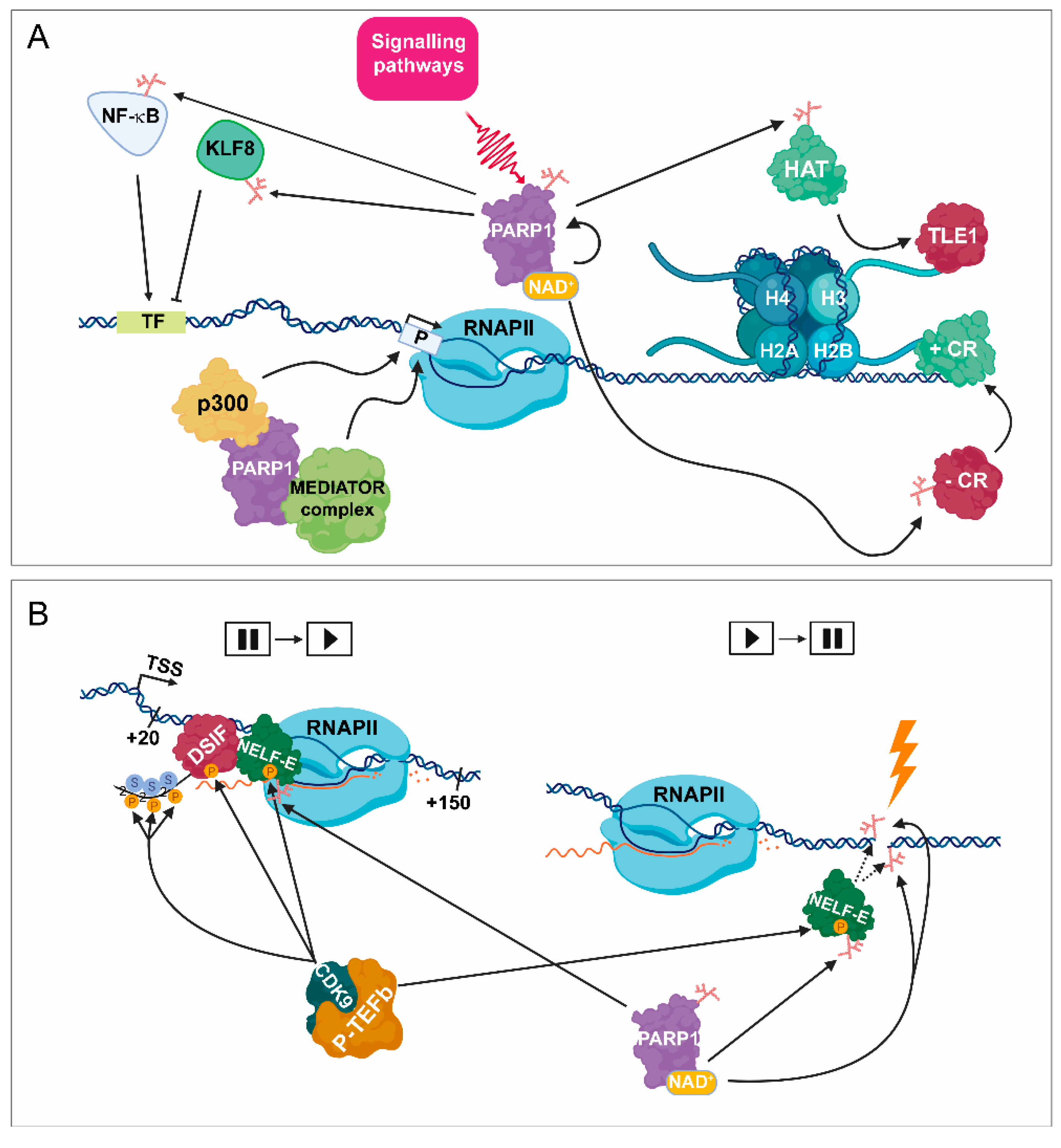
2.2. PARP1 Plays a Key Role in the Fine-Tune Regulation of Transcription Initiation
2.3. PARP1 Mediates Promoter-Proximal Pausing and Transcription Elongation
2.4. PARylation Regulates Transcription Responses during DNA Damage
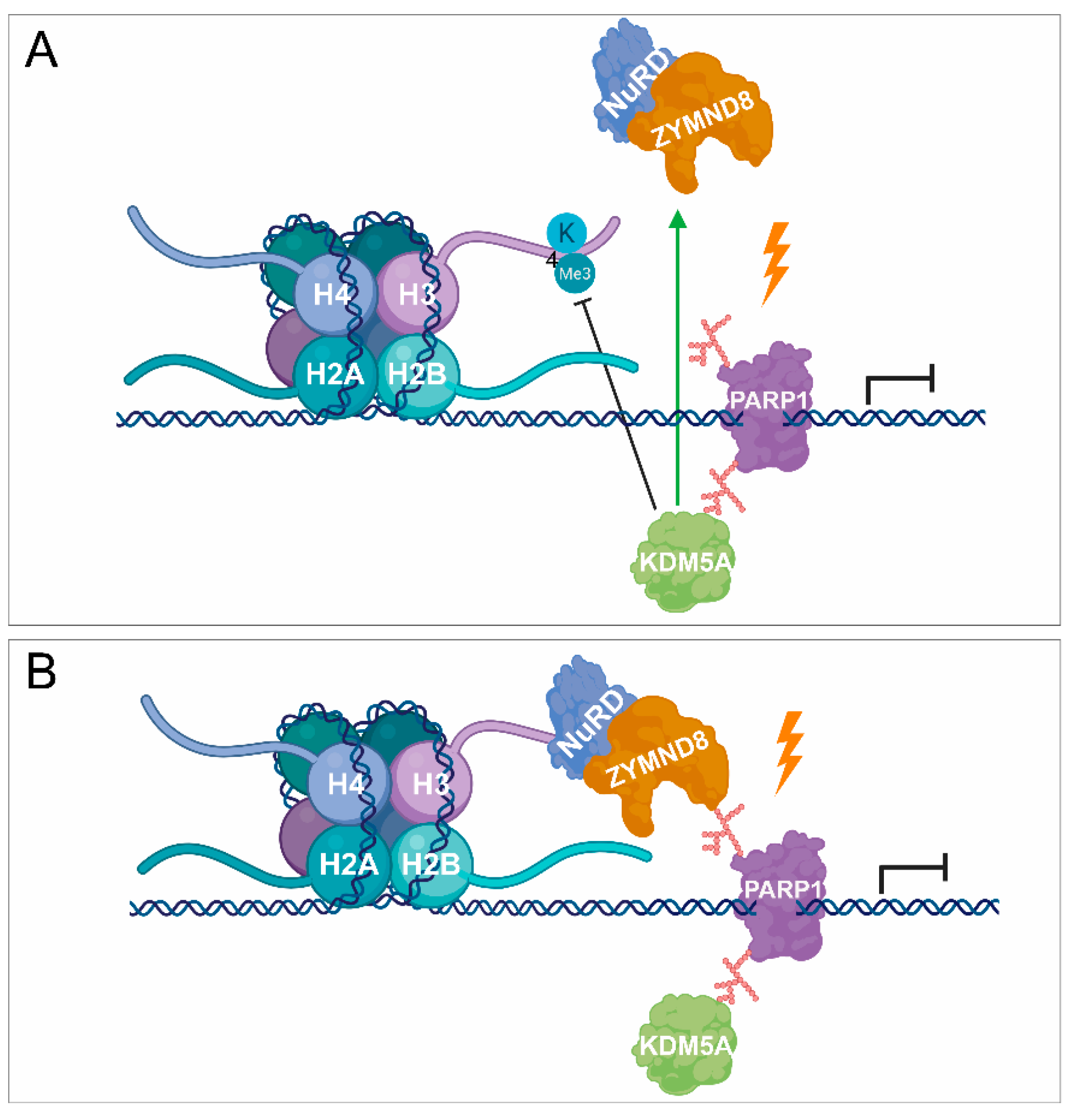
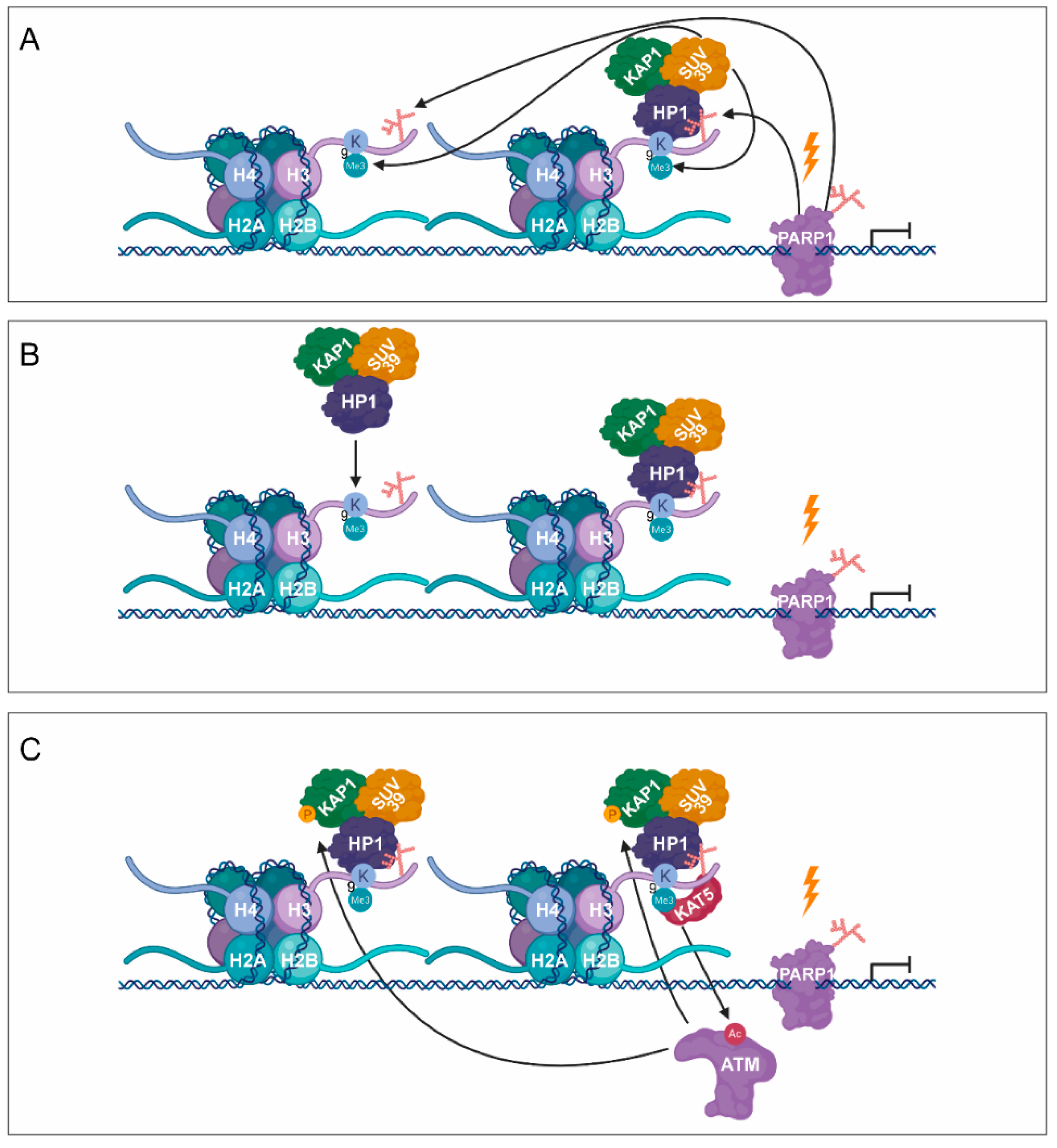
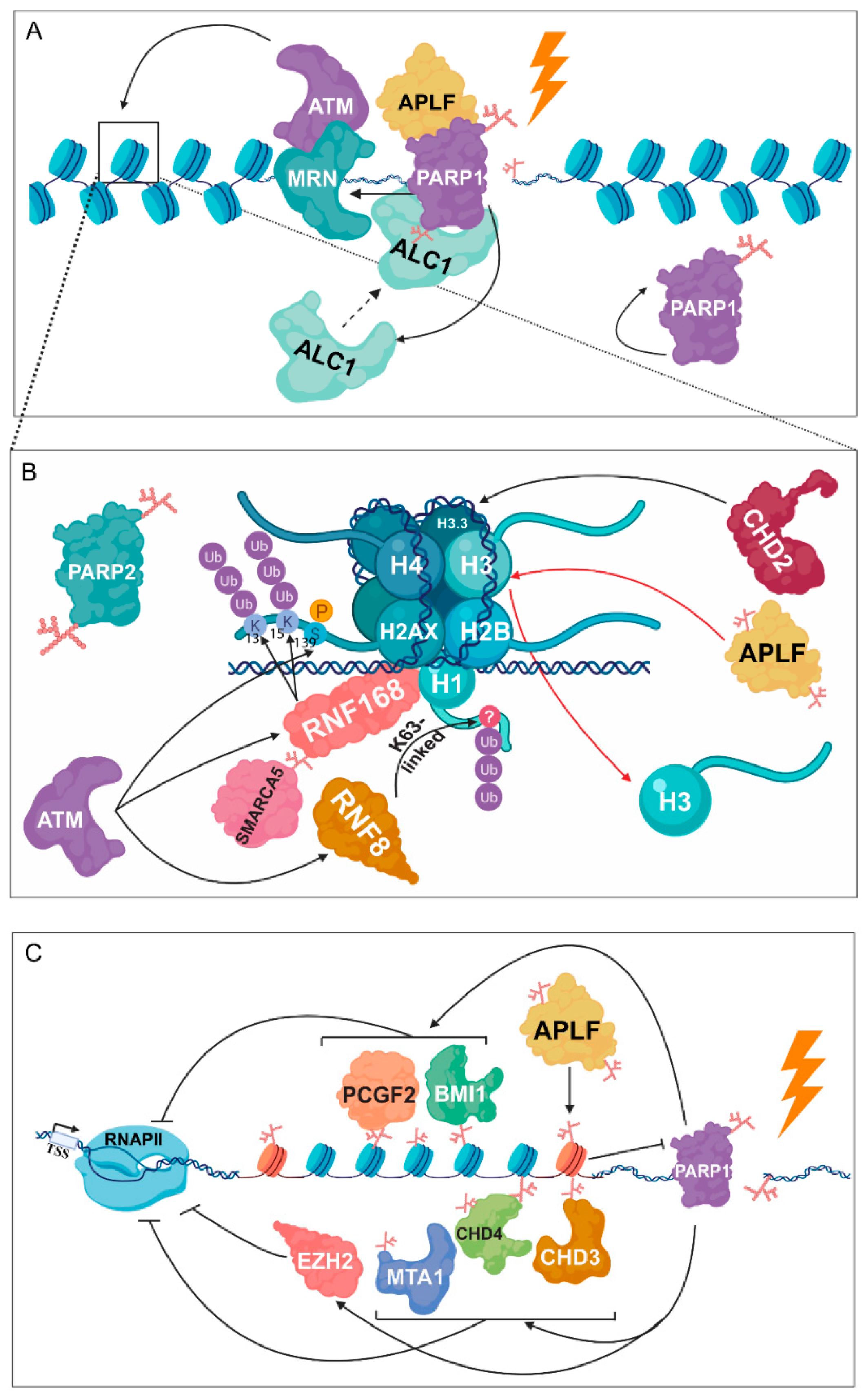
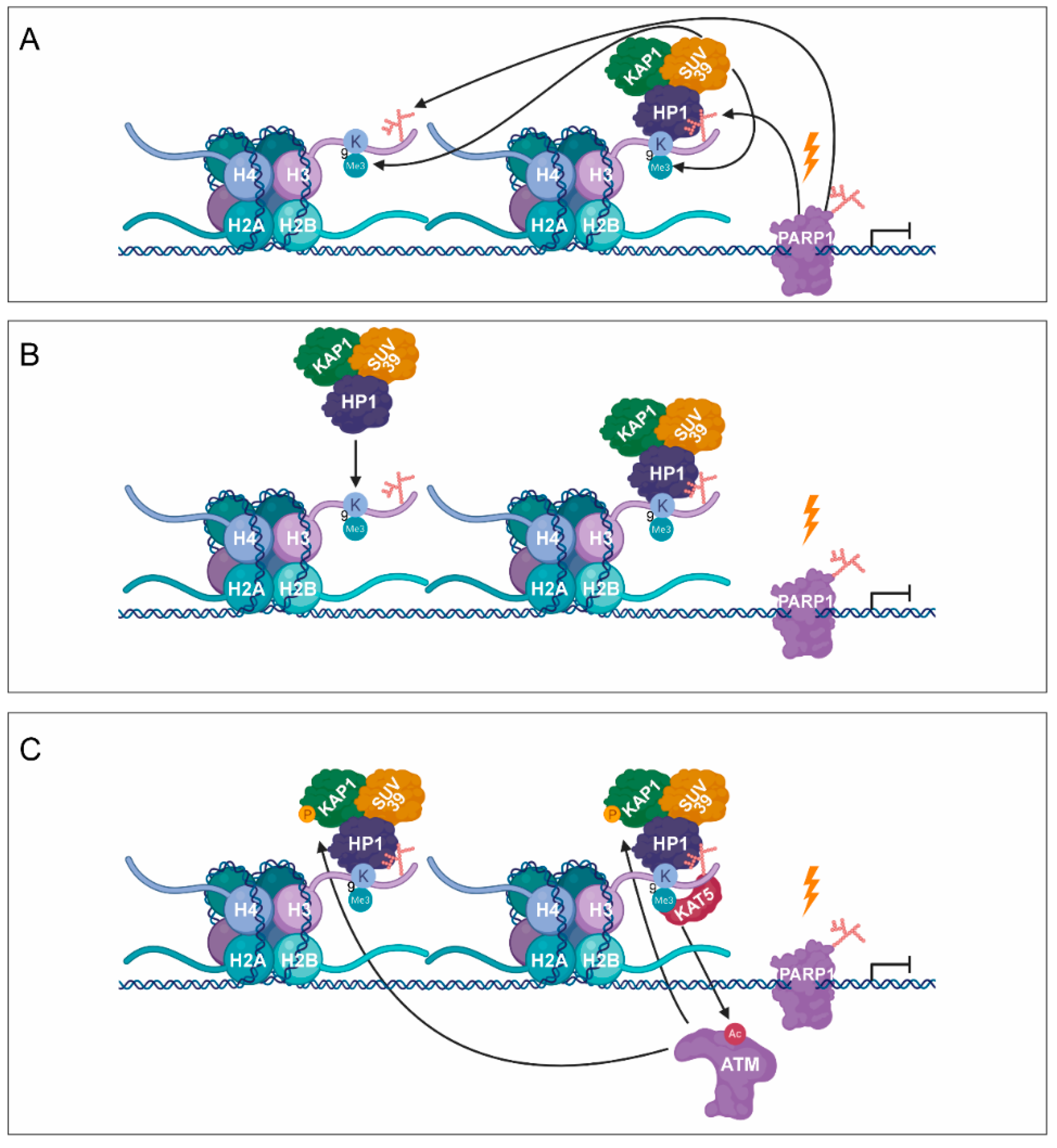
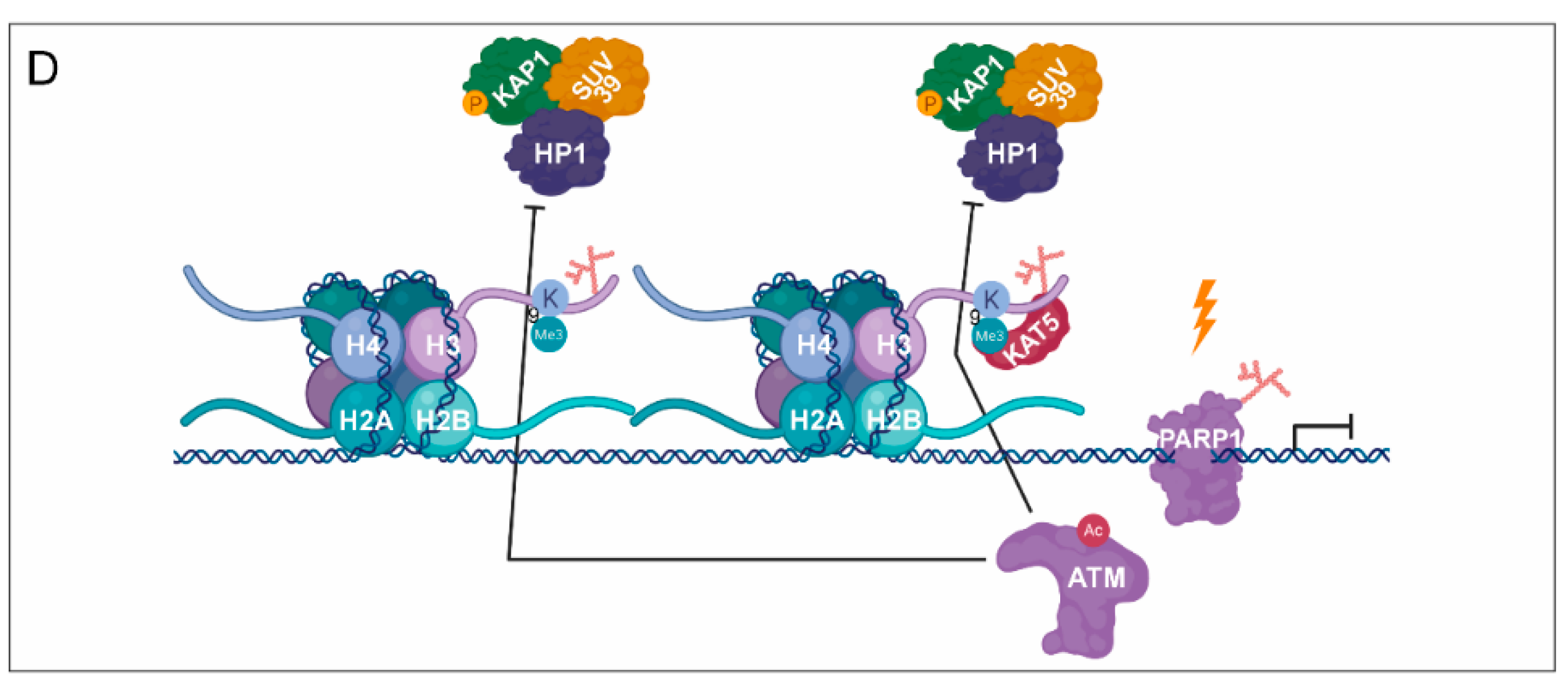
3. PARylation in the Regulation of DNA Damage-Induced Chromatin Structural Changes
4. Role of PARP1 in RNA Metabolism
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef]
- de Murcia, G.; de Murcia, J.M. Poly(ADP-ribose) polymerase: A molecular nick-sensor. Trends Biochem. Sci. 1994, 19, 172–176. [Google Scholar] [CrossRef]
- Kameshita, I.; Matsuda, Z.; Taniguchi, T.; Shizuta, Y. Poly(ADP-ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J. Biol. Chem. 1984, 259, 4770–4776. [Google Scholar] [PubMed]
- Lin, W.; Amé, J.C.; Aboul-Ela, N.; Jacobson, E.L.; Jacobson, M.K. Isolation and characterization of the cDNA encoding bovine poly(ADP- ribose) glycohydrolase. J. Biol. Chem. 1997, 272, 11895–11901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slade, D.; Dunstan, M.S.; Barkauskaite, E.; Weston, R.; Lafite, P.; Dixon, N.; Ahel, M.; Leys, D.; Ahel, I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 2011, 477, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Barkauskaite, E.; Brassington, A.; Tan, E.S.; Warwicker, J.; Dunstan, M.S.; Banos, B.; Lafite, P.; Ahel, M.; Mitchison, T.J.; Ahel, I.; et al. Visualization of poly(ADP-ribose) bound to PARG reveals inherent balance between exo- and endo-glycohydrolase activities. Nat. Commun. 2013, 4, 2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, P.; Bonfiglio, J.J.; Palazzo, L.; Bartlett, E.; Matic, I.; Ahel, I. Serine ADP-ribosylation reversal by the hydrolase ARH3. eLife 2017, 6, e28533. [Google Scholar] [CrossRef]
- Emanuelli, M.; Carnevali, F.; Saccucci, F.; Pierella, F.; Amici, A.; Raffaelli, N.; Magni, G. Molecular cloning, chromosomal localization, tissue mRNA levels, bacterial expression, and enzymatic properties of human NMN adenylyltransferase. J. Biol. Chem. 2001, 276, 406–412. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T.; Ahel, I.; Chang, P. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun. 2014, 5, 4426. [Google Scholar] [CrossRef] [Green Version]
- Cook, B.D.; Dynek, J.N.; Chang, W.; Shostak, G.; Smith, S. Role for the Related Poly(ADP-Ribose) Polymerases Tankyrase 1 and 2 at Human Telomeres. Mol. Cell. Biol. 2002, 22, 332–342. [Google Scholar] [CrossRef] [Green Version]
- Mashimo, M.; Kato, J.; Moss, J. ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 18964–18969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Fang, J.; Taatjes, D.J.; Nogales, E. Structural visualization of key steps in human transcription initiation. Nature 2013, 495, 481–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernecky, C.; Grob, P.; Ebmeier, C.C.; Nogales, E.; Taatjes, D.J. Molecular architecture of the human Mediator-RNA polymerase II-TFIIF assembly. PLoS Biol. 2011, 9, e1000603. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, E.; Lis, J.T. Transcription Factor TFIIH Is Required for Promoter Melting In Vivo. Mol. Cell. Biol. 1999, 19, 5652–5658. [Google Scholar] [CrossRef] [Green Version]
- Lis, J. Promoter-associated pausing in promoter architecture and postinitiation transcriptional regulation. Cold Spring Harb. Symp. on Quant. Biol. 1998, 63, 347–356. [Google Scholar] [CrossRef]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Ferdous, A.; Imai, T.; Hirose, S.; Sugimoto, S.; Yano, K.; Hartzog, G.A.; Winston, F.; et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998, 12, 343–356. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Marshall, N.F.; Price, D.H. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Watanabe, D.; Handa, H. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J. 1998, 17, 7395–7403. [Google Scholar] [CrossRef] [Green Version]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of Human Immunodeficiency Virus Transcription: P-TEFb Phosphorylates RD and Dissociates Negative Effectors from the Transactivation Response Element. Mol. Cell. Biol. 2004, 24, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Ping, Y.H.; Rana, T.M. DSIF and NELF Interact with RNA Polymerase II Elongation Complex and HIV-1 Tat Stimulates P-TEFb-mediated Phosphorylation of RNA Polymerase II and DSIF during Transcription Elongation. J. Biol. Chem. 2001, 276, 12951–12958. [Google Scholar] [CrossRef] [Green Version]
- Hassa, P.O.; Hottiger, M.O. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-κB in inflammatory disorders. Cell. Mol. Life Sci. 2002, 59, 1534–1553. [Google Scholar] [CrossRef]
- Tulin, A.; Spradling, A. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science 2003, 299, 560–562. [Google Scholar] [CrossRef]
- Cervellera, M.N.; Sala, A. Poly(ADP)-ribose) polymerase is a B-MYB coactivator. J. Biol. Chem. 2000, 275, 10692–10696. [Google Scholar] [CrossRef] [Green Version]
- Nie, J.; Sakamoto, S.; Song, D.; Qu, Z.; Ota, K.; Taniguchi, T. Interaction of Oct-1 and automodification domain of poly(ADP-ribose) synthetase. FEBS Lett. 1998, 424, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.S.; Chang, C.W.; Pawlik, K.M.; Zhou, D.; Renfrow, M.B.; Townes, T.M. SRY (sex determining region Y)-box2 (Sox2)/poly ADP-ribose polymerase 1 (Parp1) complexes regulate pluripotency. Proc. Natl. Acad. Sci. USA 2012, 109, 3772–3777. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Wang, X.; Li, T.; Urvalek, A.M.; Yu, L.; Li, J.; Zhu, J.; Lin, Q.; Peng, X.; Zhao, J. Identification of poly (ADP-ribose) polymerase-1 (PARP-1) as a novel krüppel-like factor 8-interactingand-regulating protein. J. Biol. Chem. 2011, 286, 20335–20344. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wang, Y.; Wang, L.; Luo, X.; Huang, K.; Wang, C.; Du, M.; Liu, F.; Luo, T.; Huang, D.; et al. Poly(ADP-ribose) polymerase 1 is a key regulator of estrogen receptor α-dependent gene transcription. J. Biol. Chem. 2013, 288, 11348–11357. [Google Scholar] [CrossRef] [Green Version]
- Le May, N.; Iltis, I.; Amé, J.C.; Zhovmer, A.; Biard, D.; Egly, J.M.; Schreiber, V.; Coin, F. Poly (ADP-Ribose) Glycohydrolase Regulates Retinoic Acid Receptor-Mediated Gene Expression. Mol. Cell 2012, 48, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Roper, S.J.; Chrysanthou, S.; Senner, C.E.; Sienerth, A.; Gnan, S.; Murray, A.; Masutani, M.; Latos, P.; Hemberger, M. ADP-ribosyltransferases Parp1 and Parp7 safeguard pluripotency of ES cells. Nucleic Acids Res. 2014, 42, 8914–8927. [Google Scholar] [CrossRef] [Green Version]
- Slattery, E.; Dignam, J.D.; Matsui, T.; Roeder, R.G. Purification and analysis of a factor which suppresses nick-induced transcription by RNA polymerase II and its identity with poly(ADP-ribose) polymerase. J. Biol. Chem. 1983, 258, 5955–5959. [Google Scholar]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPs, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef] [Green Version]
- Meisterernst, M.; Stelzer, G.; Roeder, R.G. Poly(ADP-ribose) polymerase enhances activator-dependent transcription in vitro. Proc. Natl. Acad. Sci. USA 1997, 94, 2261–2265. [Google Scholar] [CrossRef] [Green Version]
- Rawling, J.M.; Alvarez-Gonzalez, R. TFIIF, a basal eukaryotic transcription factor, is a substrate for poly(ADP-ribosyl)ation. Biochem. J. 1997, 324, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Ju, B.G.; Solum, D.; Song, E.J.; Lee, K.J.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. Activating the PARP-1 sensor component of the groucho/TLE1 corepressor complex mediates a CaMKinase IIδ-dependent neurogenic gene activation pathway. Cell 2004, 119, 815–829. [Google Scholar] [CrossRef] [Green Version]
- Krishnakumar, R.; Kraus, W.L. PARP-1 Regulates Chromatin Structure and Transcription through a KDM5B-Dependent Pathway. Mol. Cell 2010, 39, 736–749. [Google Scholar] [CrossRef] [Green Version]
- Muse, G.W.; Gilchrist, D.A.; Nechaev, S.; Shah, R.; Parker, J.S.; Grissom, S.F.; Zeitlinger, J.; Adelman, K. RNA polymerase is poised for activation across the genome. Nat. Genet. 2007, 39, 1507–1511. [Google Scholar] [CrossRef] [Green Version]
- Fujita, T.; Piuz, I.; Schlegel, W. The transcription elongation factors NELF, DSIF and P-TEFb control constitutive transcription in a gene-specific manner. FEBS Lett. 2009, 583, 2893–2898. [Google Scholar] [CrossRef]
- Awwad, S.W.; Abu-Zhayia, E.R.; Guttmann-Raviv, N.; Ayoub, N. NELF -E is recruited to DNA double-strand break sites to promote transcriptional repression and repair. EMBO Rep. 2017, 18, 745–764. [Google Scholar] [CrossRef] [Green Version]
- Petesch, S.J.; Lis, J.T. Rapid, Transcription-Independent Loss of Nucleosomes over a Large Chromatin Domain at Hsp70 Loci. Cell 2008, 134, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Gibson, B.A.; Zhang, Y.; Jiang, H.; Hussey, K.M.; Shrimp, J.H.; Lin, H.; Schwede, F.; Yu, Y.; Kraus, W.L. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 2016, 353, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Mortusewicz, O.; Ma, H.T.; Herr, P.; Poon, R.R.Y.; Helleday, T.; Qian, C. Timeless Interacts with PARP-1 to Promote Homologous Recombination Repair. Mol. Cell 2015, 60, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiácovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef] [Green Version]
- Spruijt, C.G.; Luijsterburg, M.S.; Menafra, R.; Lindeboom, R.G.H.; Jansen, P.W.T.C.; Edupuganti, R.R.; Baltissen, M.P.; Wiegant, W.W.; Voelker-Albert, M.C.; Matarese, F.; et al. ZMYND8 Co-localizes with NuRD on Target Genes and Regulates Poly(ADP-Ribose)-Dependent Recruitment of GATAD2A/NuRD to Sites of DNA Damage. Cell Rep. 2016, 17, 783–798. [Google Scholar] [CrossRef] [Green Version]
- Gong, F.; Clouaire, T.; Aguirrebengoa, M.; Legube, G.; Miller, K.M. Histone demethylase KDM5A regulates the ZMY ND8-NuRD chromatin remodeler to promote DNA repair. J. Cell Biol. 2017, 216, 1959–1974. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.; Ismail, I.H.; Young, L.C.; Poirier, G.G.; Hendzel, M.J. Polycomb repressive complex 2 contributes to DNA double-strand break repair. Cell Cycle 2013, 12, 2675–2683. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Gui, B.; Xie, G.; Zhang, D.; Shang, Y.; Liang, J. Corepressor protein CDYL functions as a molecular bridge between polycomb repressor complex 2 and repressive chromatin mark trimethylated histone lysine 27. J. Biol. Chem. 2011, 286, 42414–42425. [Google Scholar] [CrossRef] [Green Version]
- Abu-Zhayia, E.R.; Awwad, S.W.; Ben-Oz, B.M.; Khoury-Haddad, H.; Ayoub, N. CDYL1 fosters double-strand break-induced transcription silencing and promotes homology-directed repair. J. Mol. Cell Biol. 2018, 10, 341–357. [Google Scholar] [CrossRef]
- Ayrapetov, M.K.; Gursoy-Yuzugullu, O.; Xu, C.; Xu, Y.; Price, B.D. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. USA 2014, 111, 9169–9174. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M.K.; Moreau, L.A.; Whetstine, J.R.; Price, B.D. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 2009, 11, 1376–1382. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [Green Version]
- Spagnolo, L.; Barbeau, J.; Curtin, N.J.; Morris, E.P.; Pearl, L.H. Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res. 2012, 40, 4168–4177. [Google Scholar] [CrossRef] [Green Version]
- Caron, P.; Pankotai, T.; Wiegant, W.W.; Tollenaere, M.A.X.; Furst, A.; Bonhomme, C.; Helfricht, A.; de Groot, A.; Pastink, A.; Vertegaal, A.C.O.; et al. WWP2 ubiquitylates RNA polymerase II for DNA-PK-dependent transcription arrest and repair at DNA breaks. Genes Dev. 2019, 33, 684–704. [Google Scholar] [CrossRef]
- Izhar, L.; Adamson, B.; Ciccia, A.; Lewis, J.; Pontano-Vaites, L.; Leng, Y.; Liang, A.C.; Westbrook, T.F.; Harper, J.W.; Elledge, S.J. A Systematic Analysis of Factors Localized to Damaged Chromatin Reveals PARP-Dependent Recruitment of Transcription Factors. Cell Rep. 2015, 11, 1486–1500. [Google Scholar] [CrossRef] [Green Version]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Jungmichel, S.; Rosenthal, F.; Altmeyer, M.; Lukas, J.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 2013, 52, 272–285. [Google Scholar] [CrossRef] [Green Version]
- Francia, S.; Michelini, F.; Saxena, A.; Tang, D.; De Hoon, M.; Anelli, V.; Mione, M.; Carninci, P.; D’adda Di Fagagna, F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012, 488, 231–235. [Google Scholar] [CrossRef]
- Michelini, F.; Pitchiaya, S.; Vitelli, V.; Sharma, S.; Gioia, U.; Pessina, F.; Cabrini, M.; Wang, Y.; Capozzo, I.; Iannelli, F.; et al. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol. 2017, 19, 1400–1411. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Goldstein, M. Small RNAs Recruit Chromatin-Modifying Enzymes MMSET and Tip60 to Reconfigure Damaged DNA upon Double-Strand Break and Facilitate Repair. Cancer Res. 2016, 76, 1904–1915. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.T.; Hawley, B.R.; Skalka, G.L.; Baldock, R.A.; Smith, E.M.; Bader, A.S.; Malewicz, M.; Watts, F.Z.; Wilczynska, A.; Bushell, M. Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair. Nat. Commun. 2018, 9, 532. [Google Scholar] [CrossRef] [Green Version]
- Lobrich, M.; Jeggo, P. A Process of Resection-Dependent Nonhomologous End Joining Involving the Goddess Artemis. Trends Biochem. Sci. 2017, 42, 690–701. [Google Scholar] [CrossRef] [Green Version]
- El Hage, A.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Anderson, S.F.; Chao, D.M.; Wei, W.J.; Ye, L.Y.; Young, R.A.; Livingston, D.M.; Parvin, J.D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 5605–5610. [Google Scholar] [CrossRef] [Green Version]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 Recruitment to Transcriptional Pause Sites Is Required for R-Loop-Driven DNA Damage Repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [Green Version]
- Caron, M.C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.P.; Langelier, M.F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef] [Green Version]
- Farmer, H.; McCabe, H.; Lord, C.J.; Tutt, A.H.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- AlHilli, M.M.; Becker, M.A.; Weroha, S.J.; Flatten, K.S.; Hurley, R.M.; Harrell, M.I.; Oberg, A.L.; Maurer, M.J.; Hawthorne, K.M.; Hou, X.; et al. In vivo anti-tumor activity of the PARP inhibitor niraparib in homologous recombination deficient and proficient ovarian carcinoma. Gynecol. Oncol. 2016, 143, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef] [Green Version]
- von Kobbe, C.; Harrigan, J.A.; Schreiber, V.; Stiegler, P.; Piotrowski, J.; Dawut, L.; Bohr, V.A. Poly(ADP-ribose) polymerase 1 regulates both the exonuclease and helicase activities of the Werner syndrome protein. Nucleic Acids Res. 2004, 32, 4003–4014. [Google Scholar] [CrossRef] [Green Version]
- Bunch, H.; Lawney, B.P.; Lin, Y.F.; Asaithamby, A.; Murshid, A.; Wang, Y.Y.E.; Chen, B.P.C.; Calderwood, S.K. Transcriptional elongation requires DNA break-induced signalling. Nat. Commun. 2015, 6, 10191. [Google Scholar] [CrossRef] [Green Version]
- Giaever, G.N.; Wang, J.C. Supercoiling of intracellular DNA can occur in eukaryotic cells. Cell 1988, 55, 849–856. [Google Scholar] [CrossRef]
- Liu, L.F.; Miller, K.G. Eukaryotic DNA topoisomerases: Two forms of type I DNA topoisomerases from HeLa cell nuclei. Proc. Natl. Acad. Sci. USA 1981, 78, 3487–3491. [Google Scholar] [CrossRef] [Green Version]
- Teves, S.S.; Henikoff, S. Transcription-generated torsional stress destabilizes nucleosomes. Nat. Struct. Mol. Biol. 2014, 21, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.Y.; Shyy, S.; Wang, J.C.; Liu, L.F. Transcription generates positively and negatively supercoiled domains in the template. Cell 1988, 53, 433–440. [Google Scholar] [CrossRef]
- Massé, E.; Drolet, M. Escherichia coli DNA topoisomerase I inhibits R-loop formation by relaxing transcription-induced negative supercoiling. J. Biol. Chem. 1999, 274, 16659–16664. [Google Scholar] [CrossRef] [Green Version]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.; van, I.W.F.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.; et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, A.; Garcia-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [Green Version]
- Houlard, M.; Artus, J.; Leguillier, T.; Vandormael-Pournin, S.; Cohen-Tannoudji, M. DNA-RNA hybrids contribute to the replication dependent genomic instability induced by Omcg1 deficiency. Cell Cycle 2011, 10, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; McBride, K.M.; Hensley, S.; Lu, Y.; Chedin, F.; Bedford, M.T. Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol. Cell 2014, 53, 484–497. [Google Scholar] [CrossRef] [Green Version]
- Wahba, L.; Amon, J.D.; Koshland, D.; Vuica-Ross, M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell 2011, 44, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Redinbo, M.R.; Stewart, L.; Kuhn, P.; Champoux, J.J.; Hol, W.G.J. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 1998, 279, 1504–1513. [Google Scholar] [CrossRef]
- Marinello, J.; Chillemi, G.; Bueno, S.; Manzo, S.G.; Capranico, G. Antisense transcripts enhanced by camptothecin at divergent CpG-island promoters associated with bursts of topoisomerase I-DNA cleavage complex and R-loop formation. Nucleic Acids Res. 2013, 41, 10110–10123. [Google Scholar] [CrossRef]
- Marinello, J.; Bertoncini, S.; Aloisi, I.; Cristini, A.; Malagoli Tagliazucchi, G.; Forcato, M.; Sordet, O.; Capranico, G. Dynamic Effects of Topoisomerase I Inhibition on R-Loops and Short Transcripts at Active Promoters. PLoS ONE 2016, 11, e0147053. [Google Scholar] [CrossRef] [Green Version]
- Cristini, A.; Park, J.H.; Capranico, G.; Legube, G.; Favre, G.; Sordet, O. DNA-PK triggers histone ubiquitination and signaling in response to DNA double-strand breaks produced during the repair of transcription-blocking topoisomerase I lesions. Nucleic Acids Res. 2016, 44, 1161–1178. [Google Scholar] [CrossRef] [Green Version]
- Yung, T.M.; Sato, S.; Satoh, M.S. Poly(ADP-ribosyl)ation as a DNA damage-induced post-translational modification regulating poly(ADP-ribose) polymerase-1-topoisomerase I interaction. J. Biol. Chem. 2004, 279, 39686–39696. [Google Scholar] [CrossRef] [Green Version]
- Bauer, P.I.; Chen, H.J.; Kenesi, E.; Kenessey, I.; Buki, K.G.; Kirsten, E.; Hakam, A.; Hwang, J.I.; Kun, E. Molecular interactions between poly(ADP-ribose) polymerase (PARP I) and topoisomerase I (Topo I): Identification of topology of binding. FEBS Lett. 2001, 506, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Malanga, M.; Althaus, F.R. Poly(ADP-ribose) reactivates stalled DNA topoisomerase I and induces DNA strand break resealing. J. Biol. Chem. 2004, 279, 5244–5248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messner, S.; Altmeyer, M.; Zhao, H.; Pozivil, A.; Roschitzki, B.; Gehrig, P.; Rutishauser, D.; Huang, D.; Caflisch, A.; Hottiger, M.O. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010, 38, 6350–6362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Okano, S.; Lan, L.; Yasui, A.; Mori, T.; Caldecott, K.W. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 2003, 23, 3974–3981. [Google Scholar] [CrossRef] [Green Version]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Kassab, M.A.; Dantzer, F.; Yu, X. PARP2 mediates branched poly ADP-ribosylation in response to DNA damage. Nat. Commun. 2018, 9, 3233. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiroli, F.; Vissers, J.H.A.; Van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [Green Version]
- Smeenk, G.; Wiegant, W.W.; Marteijn, J.A.; Luijsterburg, M.S.; Sroczynski, N.; Costelloe, T.; Romeijn, R.J.; Pastink, A.; Mailand, N.; Vermeulen, W.; et al. Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J. Cell Sci. 2013, 126, 889–903. [Google Scholar] [CrossRef] [Green Version]
- Ahel, D.; Hořejší, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W.; et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellou, H.; Lebeaupin, T.; Chapuis, C.; Smith, R.; Hegele, A.; Singh, H.R.; Kozlowski, M.; Bultmann, S.; Ladurner, A.G.; Timinszky, G.; et al. The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage. Mol. Biol. Cell 2016, 27, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.R.; Nardozza, A.P.; Möller, I.R.; Knobloch, G.; Kistemaker, H.A.V.; Hassler, M.; Harrer, N.; Blessing, C.; Eustermann, S.; Kotthoff, C.; et al. A Poly-ADP-Ribose Trigger Releases the Auto-Inhibition of a Chromatin Remodeling Oncogene. Mol. Cell 2017, 68, 860–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottschalk, A.J.; Trivedi, R.D.; Conaway, J.W.; Conaway, R.C. Activation of the SNF2 family ATPase ALC1 by poly(ADP-ribose) in a stable ALC1·PARP1·nucleosome intermediate. J. Biol. Chem. 2012, 287, 43527–43532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahel, I.; Ahel, D.; Matsusaka, T.; Clark, A.J.; Pines, J.; Boulton, S.J.; West, S.C. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 2008, 451, 81–85. [Google Scholar] [CrossRef]
- Mehrotra, P.V.; Ahel, D.; Ryan, D.P.; Weston, R.; Wiechens, N.; Kraehenbuehl, R.; Owen-Hughes, T.; Ahel, I. DNA repair factor APLF is a histone chaperone. Mol. Cell 2011, 41, 46–55. [Google Scholar] [CrossRef] [Green Version]
- Luijsterburg, M.S.; de Krijger, I.; Wiegant, W.W.; Shah, R.G.; Smeenk, G.; de Groot, A.J.L.; Pines, A.; Vertegaal, A.C.O.; Jacobs, J.J.L.; Shah, G.M.; et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell 2016, 61, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Burgess, R.C.; Burman, B.; Kruhlak, M.J.; Misteli, T. Activation of DNA Damage Response Signaling by Condensed Chromatin. Cell Rep. 2014, 9, 1703–1717. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Lv, P.; Yan, G.; Fan, H.; Cheng, L.; Zhang, F.; Dang, Y.; Wu, H.; Wen, B. MacroH2A1 associates with nuclear lamina and maintains chromatin architecture in mouse liver cells. Sci. Rep. 2015, 5, 17186. [Google Scholar] [CrossRef] [Green Version]
- Douet, J.; Corujo, D.; Malinverni, R.; Renauld, J.; Sansoni, V.; Marjanović, M.P.; Cantariño, N.; Valero, V.; Mongelard, F.; Bouvet, P.; et al. MacroH2A histone variants maintain nuclear organization and heterochromatin architecture. J. Cell Sci. 2017, 130, 1570–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sporn, J.C.; Jung, B. Differential regulation and predictive potential of macroH2A1 isoforms in colon cancer. Am. J. Pathol. 2012, 180, 2516–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusinow, D.A.; Hernández-Muñoz, I.; Fazzio, T.G.; Shah, G.M.; Kraus, W.L.; Panning, B. Poly(ADP-ribose) polymerase 1 is inhibited by a histone H2A variant, macroH2A, and contributes to silencing of the inactive X chromosome. J. Biol. Chem. 2007, 282, 12851–12859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timinszky, G.; Till, S.; Hassa, P.O.; Hothorn, M.; Kustatscher, G.; Nijmeijer, B.; Colombelli, J.; Altmeyer, M.; Stelzer, E.H.K.; Scheffzek, K.; et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009, 16, 923–929. [Google Scholar] [CrossRef]
- Kozlowski, M.; Corujo, D.; Hothorn, M.; Guberovic, I.; Mandemaker, I.K.; Blessing, C.; Sporn, J.; Gutierrez-triana, A.; Smith, R.; Portmann, T.; et al. MacroH 2 A histone variants limit chromatin plasticity through two distinct mechanisms. EMBO Rep. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Polo, S.E.; Kaidi, A.; Baskcomb, L.; Galanty, Y.; Jackson, S.P. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010, 29, 3130–3139. [Google Scholar] [CrossRef] [Green Version]
- Caruso, L.B.; Martin, K.A.; Lauretti, E.; Hulse, M.; Siciliano, M.; Lupey-Green, L.N.; Abraham, A.; Skorski, T.; Tempera, I. Poly(ADP-ribose) Polymerase 1, PARP1, modifies EZH2 and inhibits EZH2 histone methyltransferase activity after DNA damage. Oncotarget 2018, 9, 10585–10605. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Du, Y.; Nakai, K.; Ding, M.; Chang, S.-S.; Hsu, J.L.; Yao, J.; Wei, Y.; Nie, L.; Jiao, S.; et al. EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene 2018, 37, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Matveeva, E.; Maiorano, J.; Zhang, Q.; Eteleeb, A.M.; Convertini, P.; Chen, J.; Infantino, V.; Stamm, S.; Wang, J.; Rouchka, E.C.; et al. Involvement of PARP1 in the regulation of alternative splicing. Cell Discov. 2016, 2, 15046. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Tulin, A.V. Poly(ADP-ribosyl)ation of heterogeneous nuclear ribonucleoproteins modulates splicing. Nucleic Acids Res. 2009, 37, 3501–3513. [Google Scholar] [CrossRef] [Green Version]
- Isabelle, M.; Moreel, X.; Gagné, J.P.; Rouleau, M.; Ethier, C.; Gagné, P.; Hendzel, M.J.; Poirier, G.G. Investigation of PARP-1, PARP-2, and PARG interactomes by affinity-purification mass spectrometry. Proteome Sci. 2010, 8, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munnur, D.; Bartlett, E.; Mikolčević, P.; Kirby, I.T.; Matthias Rack, J.G.; Mikoč, A.; Cohen, M.S.; Ahel, I. Reversible ADP-ribosylation of RNA. Nucleic Acids Res. 2019, 47, 5658–5669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotova, E.; Jarnik, M.; Tulin, A.V. Poly (ADP-ribose) polymerase 1 is required for protein localization to Cajal body. PLoS Genet. 2009, 5, e1000387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matveeva, E.A.; Al-Tinawi, Q.M.H.; Rouchka, E.C.; Fondufe-Mittendorf, Y.N. Coupling of PARP1-mediated chromatin structural changes to transcriptional RNA polymerase II elongation and cotranscriptional splicing. Epigenetics Chromatin 2019, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Di Giammartino, D.C.; Shi, Y.; Manley, J.L. PARP1 Represses PAP and Inhibits Polyadenylation during Heat Shock. Mol. Cell 2013, 49, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilal Iqbal, M.; Johns, M.; Cao, J.; Liu, Y.; Yu, S.C.; Hyde, G.D.; Laffan, M.A.; Marchese, F.P.; Cho, S.H.; Clark, A.R.; et al. PARP-14 combines with tristetraprolin in the selective posttranscriptional control of macrophage tissue factor expression. Blood 2014, 124, 3646–3655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, Y.; Han, Y.; Guo, X.; Wen, J.; Wang, K.; Jiang, X.; Tian, X.; Ba, X.; Boldogh, I.; Zeng, X. PARP1 promotes gene expression at the post-transcriptiona level by modulating the RNA-binding protein HuR. Nat. Commun. 2017, 8, 14632. [Google Scholar] [CrossRef] [Green Version]
- Moynahan, M.E.; Cui, T.Y.; Jasin, M. Homology-directed DNA repair, mitomycin-C resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001, 61, 4842–4850. [Google Scholar]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasint, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Tutt, A.; Bertwistle, D.; Valentine, J.; Gabriel, A.; Swift, S.; Ross, G.; Griffin, C.; Thacker, J.; Ashworth, A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001, 20, 4704–4716. [Google Scholar] [CrossRef] [PubMed]
- Hegan, D.C.; Lu, Y.; Stacheleka, G.C.; Crosbya, M.E.; Bindraa, R.S.; Glazer, P.M. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc. Natl. Acad. Sci. USA 2010, 107, 2201–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, R.H.G.; Lioutas, A.; Dily, F.L.; Soronellas, D.; Pohl, A.; Bonet, J.; Nacht, A.S.; Samino, S.; Font-Mateu, J.; Vicent, G.P.; et al. ADP-ribose-derived nuclear ATP synthesis by NUDIX5 is required for chromatin remodeling. Science 2016, 352, 1221–1225. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Páhi, Z.G.; Borsos, B.N.; Pantazi, V.; Ujfaludi, Z.; Pankotai, T. PARylation During Transcription: Insights into the Fine-Tuning Mechanism and Regulation. Cancers 2020, 12, 183. https://doi.org/10.3390/cancers12010183
Páhi ZG, Borsos BN, Pantazi V, Ujfaludi Z, Pankotai T. PARylation During Transcription: Insights into the Fine-Tuning Mechanism and Regulation. Cancers. 2020; 12(1):183. https://doi.org/10.3390/cancers12010183
Chicago/Turabian StylePáhi, Zoltán G., Barbara N. Borsos, Vasiliki Pantazi, Zsuzsanna Ujfaludi, and Tibor Pankotai. 2020. "PARylation During Transcription: Insights into the Fine-Tuning Mechanism and Regulation" Cancers 12, no. 1: 183. https://doi.org/10.3390/cancers12010183
APA StylePáhi, Z. G., Borsos, B. N., Pantazi, V., Ujfaludi, Z., & Pankotai, T. (2020). PARylation During Transcription: Insights into the Fine-Tuning Mechanism and Regulation. Cancers, 12(1), 183. https://doi.org/10.3390/cancers12010183