PARP1 Inhibition Augments UVB-Mediated Mitochondrial Changes—Implications for UV-Induced DNA Repair and Photocarcinogenesis

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
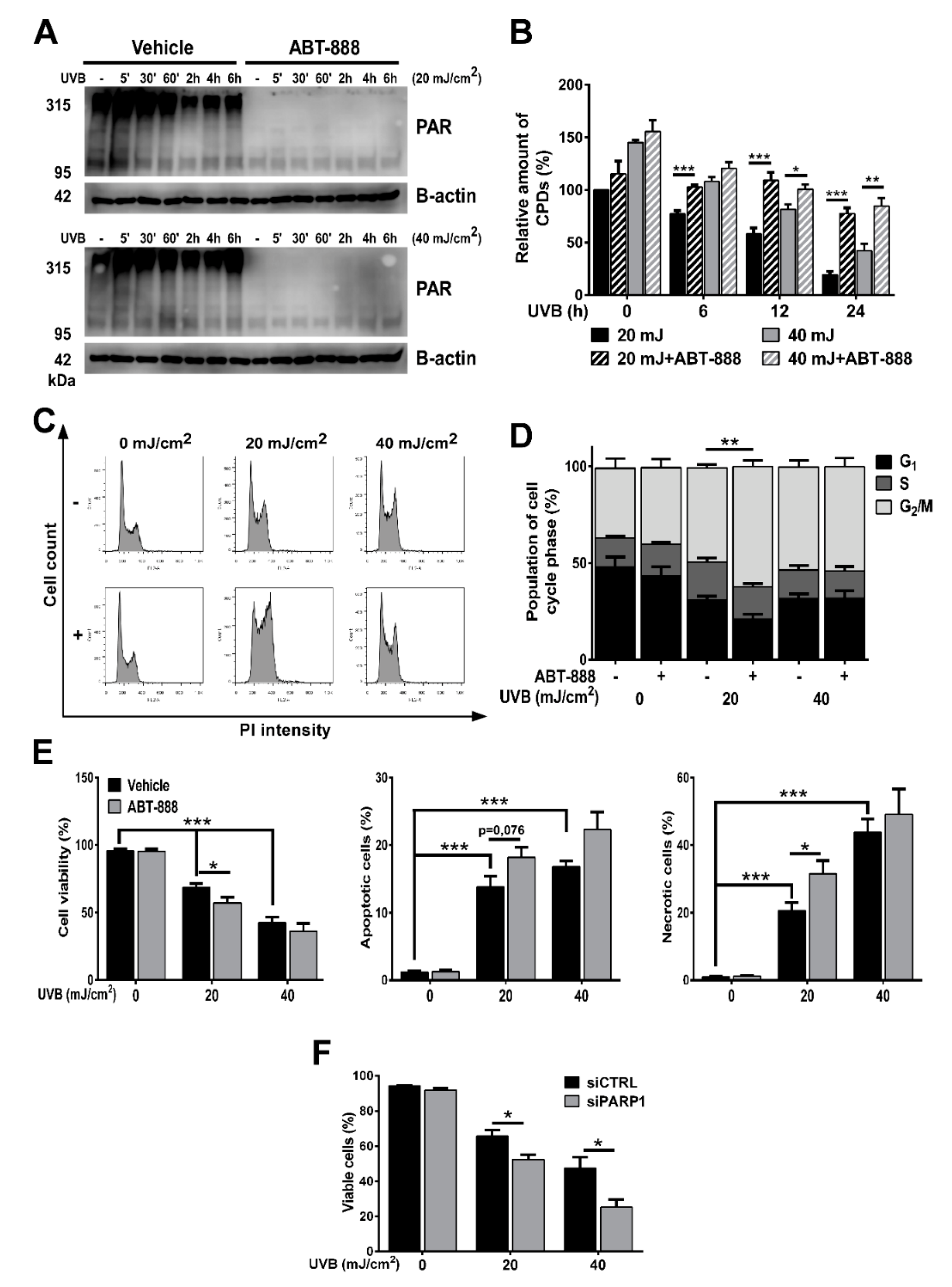
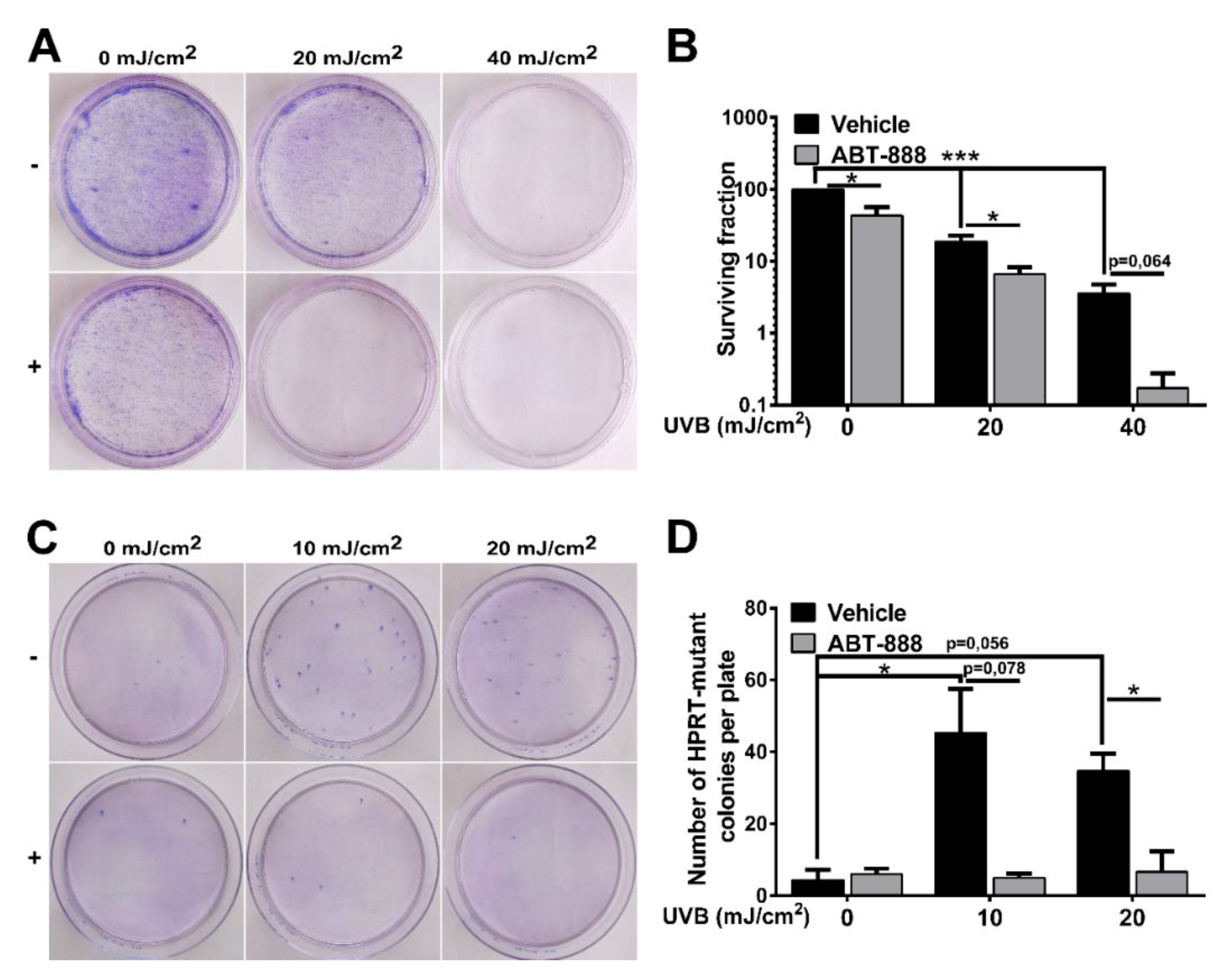
2.1. PARP Inhibition Impairs CPD Repair, Augments UVB-Induced Cell Cycle Block, Apoptosis and Reduces Keratinocyte Proliferation
2.2. PARP Inhibition Enhances UVB-Mediated Mitochondrial Biogenesis
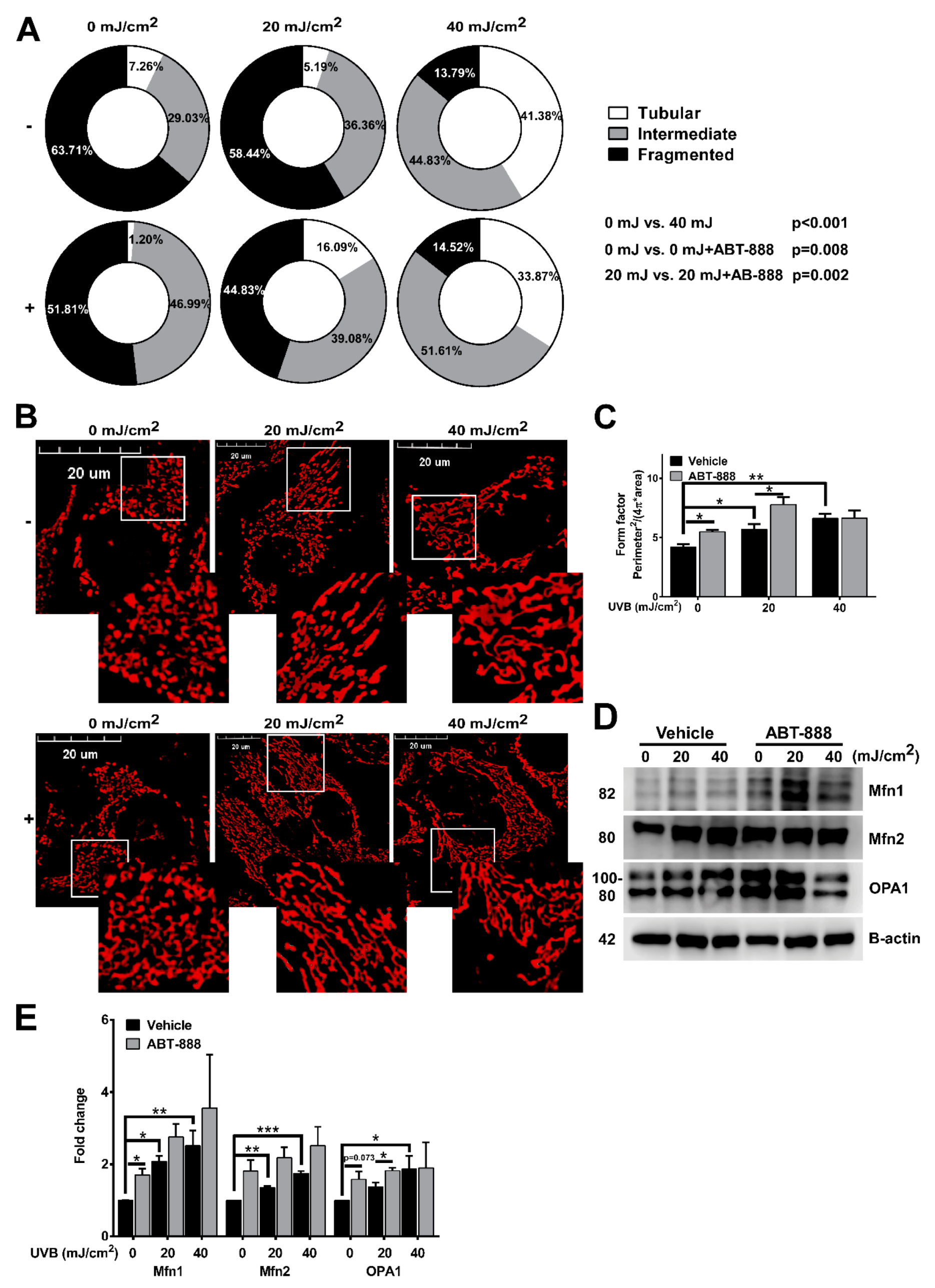
2.3. PARP Inhibition Augments UVB-Mediated Mitochondrial Fusion
2.4. PARP Inhibition and UVB Induces Bulk Autophagy but Not Mitophagy
2.5. PARP Inhibition Boosts UVB-Mediated Mitochondrial Bioenergetic Changes
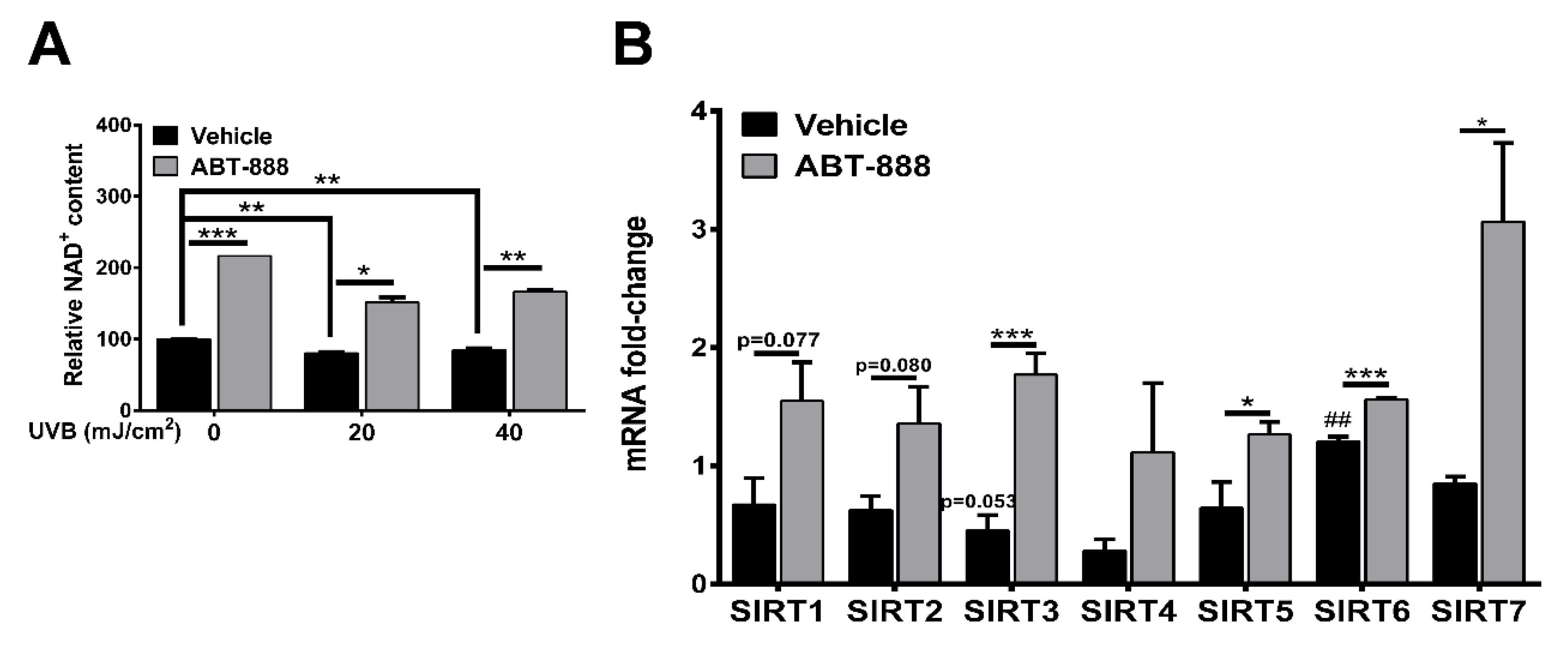
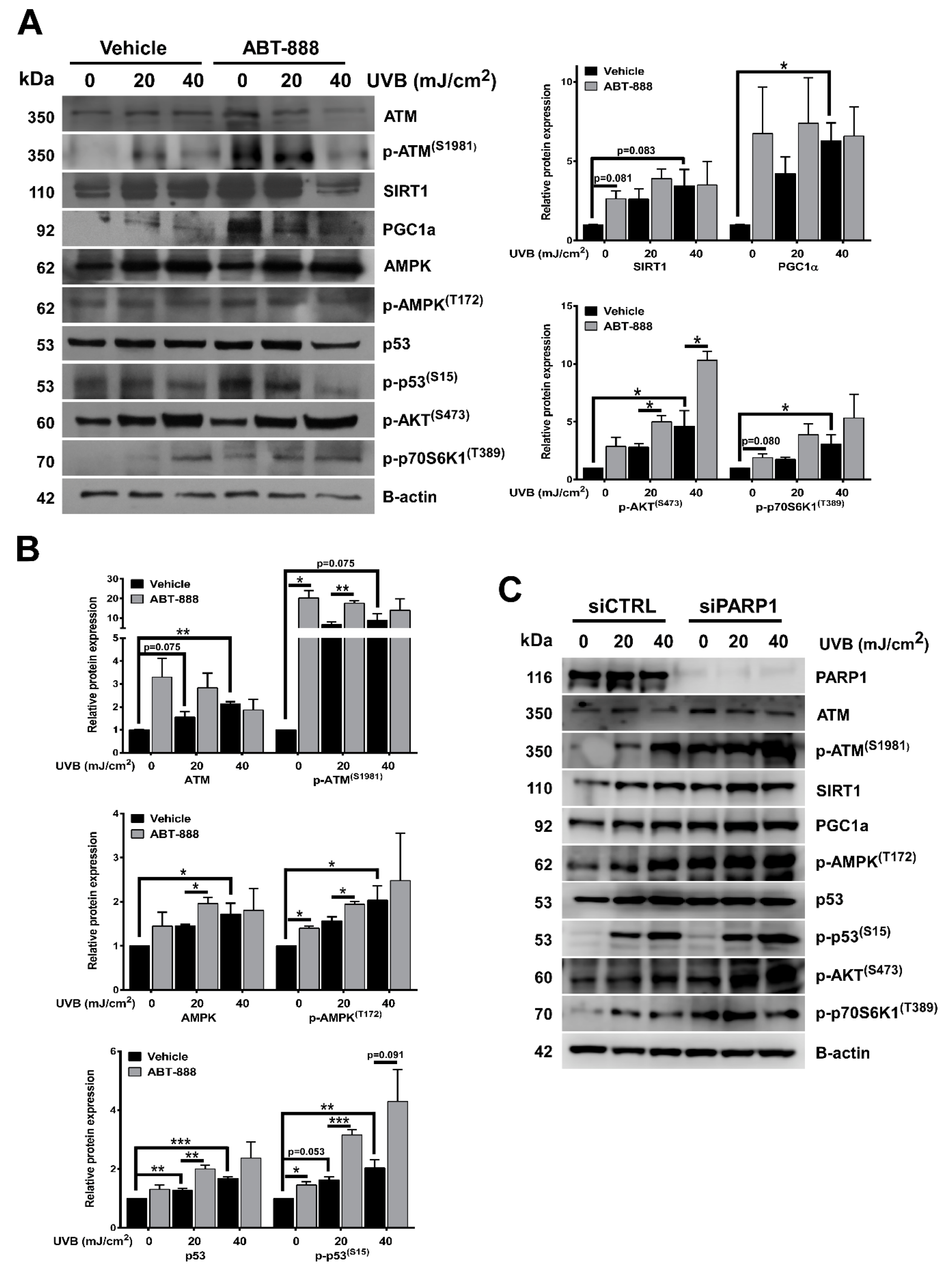
2.6. PARP Inhibition Restores NAD+ Level and SIRTUIN Expression
2.7. PARP Inhibition Enhances UVB-Mediated Upregulation of Metabolic Proteins
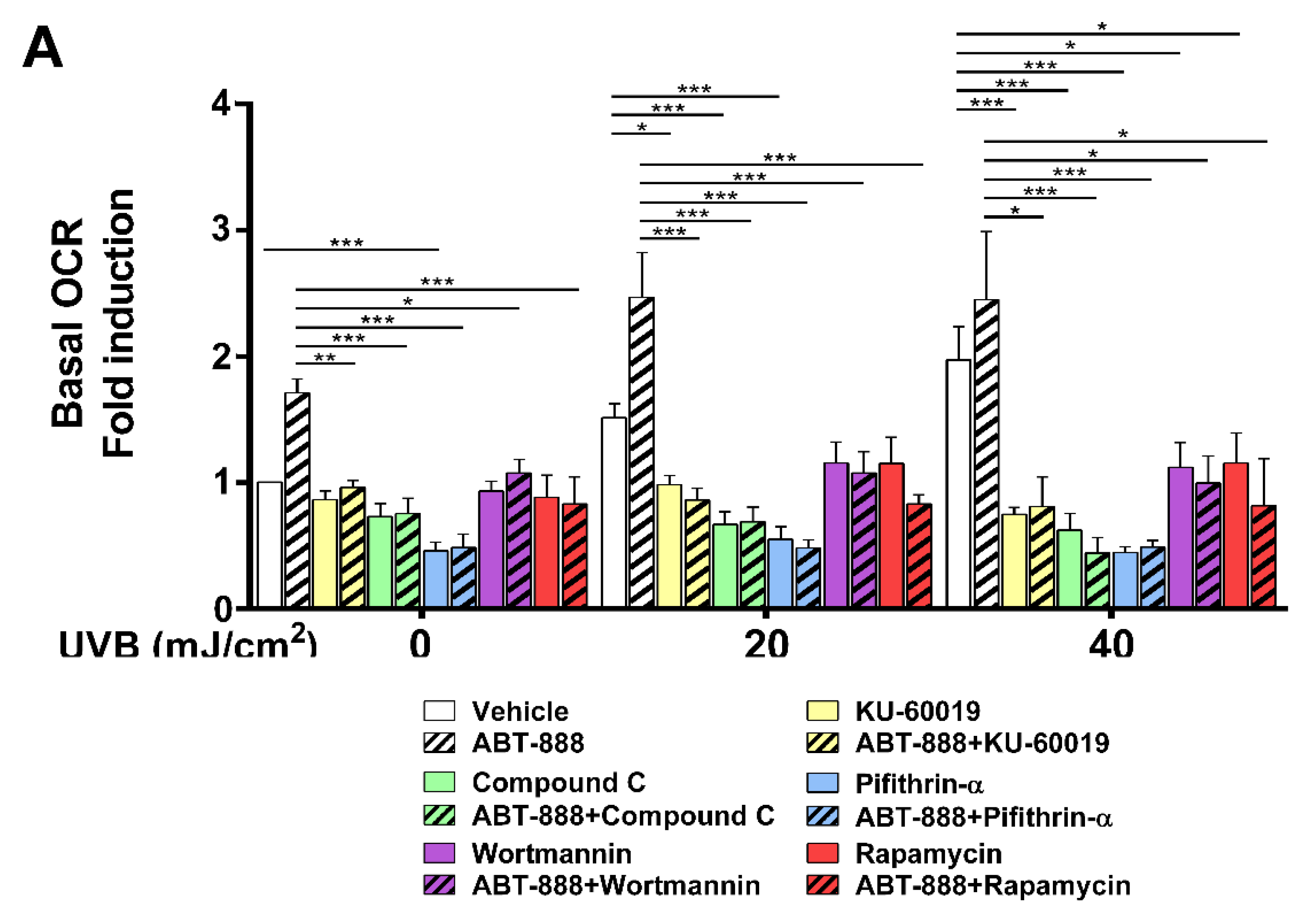
2.8. PARP Inhibition and UVB-Induced Oxidative Phosphorylation and Autophagy Are Dependent on ATM, AMPK, p53, AKT, and mTOR Activation
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Treatment
4.4. Gene Silencing
4.5. Cell Viability and Proliferation
4.6. Cell Cycle Analysis
4.7. HPRT Mutation Assay
4.8. CPD-Specific Enzyme-Linked Immunosorbent Assay (ELISA)
4.9. Real-Time Quantitative RT-PCR
4.10. Western Blot
4.11. Mitochondrial Mass
4.12. Determination of Mitochondrial Membrane Potential
4.13. Determination of Mitochondrial Ultrastructure, Mitochondrial Number, and Area by Transmission Electron Microscopy (TEM)
4.14. Assessment of Mitochondrial Morphology and Autophagy by Confocal Microscopy
4.15. Measurement of Citrate Synthase Activity
4.16. Analysis of Oxygen Consumption and Extracellular Acidification
4.17. Determination of NAD+ Level
4.18. Measurement of ATP Content
4.19. Statistical Analysis
5. Conclusions
Data Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CPD | cyclobutane pyrimidine dimer |
| PAR | poly (ADP-ribose) polymer |
| PARP1 | poly (ADP-ribose) polymerase 1 |
| ATM kinase | ataxia-telangiectasia-mutated kinase |
| AMPK | adenosine monophosphate-activated kinase |
| mTOR | mammalian target of rapamycin |
| p70S6K1 | ribosomal protein S6 kinase beta-1 |
| HPRT | hypoxanthine-guanine phosphoribosyltransferase |
| PGC1A | peroxisome proliferator activated receptor gamma coactivator-1 alpha |
| MTCO1 | mitochondrially encoded cytochrome C oxidase I |
| CS | citrate synthase |
| TCA | tricarboxylic acid |
| SDHA | succinate dehydrogenase complex, subunit A |
| PGK1 | phosphoglycerate kinase 1 |
| NRF2 | nuclear respiratory factor 2 |
| ERRA | estrogen-related receptor alpha |
| Tfam | mitochondrial transcription factor A |
| OCR | oxygen consumption rate |
| ECAR | extracellular acidification rate |
| OXPHOS | oxidative phosphorylation |
| CHO | Chinese hamster ovary |
| Mfn1 | mitofusin 1 |
| Mfn2 | mitofusin 2 |
| OPA1 | optic atrophy 1 |
| LC3A/B | microtubule-associated proteins 1A/1B light chain 3B |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| PI3K | phosphoinositide 3-kinase |
| DiOC6(3) | 3,3′-dihexyloxacarbocyanine iodide |
References
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [PubMed] [Green Version]
- Dominy, J.E.; Puigserver, P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a015008. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial quality control as a therapeutic target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef] [PubMed]
- Mears, J.A. Mitochondrial biogenesis and quality control. In The Structural Basis of Biological Energy Generation; Hohmann-Marriott, M.F., Ed.; Springer: Dordrecht, The Netherlands, 2014; pp. 451–476. [Google Scholar]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; de la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; de Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Sánchez-Alcázar, J.A. Mitochondrial dynamics in mitochondrial diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Feichtinger, R.G.; Sperl, W.; Bauer, J.W.; Kofler, B. Mitochondrial dysfunction: A neglected component of skin diseases. Exp. Dermatol. 2014, 23, 607–614. [Google Scholar] [CrossRef]
- Clayton, D.A.; Doda, J.N.; Friedberg, E.C. The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 1974, 71, 2777–2781. [Google Scholar] [CrossRef] [Green Version]
- LeDoux, S.P.; Wilson, G.L.; Beecham, E.J.; Stevnsner, T.; Wassermann, K.; Bohr, V.A. Repair of mitochondrial DNA after various types of DNA damage in chinese hamster ovary cells. Carcinogenesis 1992, 13, 1967–1973. [Google Scholar] [CrossRef]
- Krutmann, J.; Schroeder, P. Role of mitochondria in photoaging of human skin: The defective powerhouse model. J. Investig. Dermatol. Symp. Proc. 2009, 14, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, K.; Hanna, R.; Birch-Machin, M.A. What is the role of mitochondrial dysfunction in skin photoaging? Exp. Dermatol. 2018, 27, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Fujimoto, A.; Hoon, D.S. Detection of mitochondrial DNA alterations in plasma of malignant melanoma patients. Ann. N. Y. Acad. Sci. 2004, 1022, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Lee, H.C.; Chung, J.G.; Wei, Y.H. Mitochondrial DNA mutations in light-associated skin tumors. Anticancer Res. 2004, 24, 1753–1758. [Google Scholar]
- Durham, S.E.; Krishnan, K.J.; Betts, J.; Birch-Machin, M.A. Mitochondrial DNA damage in non-melanoma skin cancer. Br. J. Cancer 2003, 88, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. Mitochondrial deficiency in cockayne syndrome. Mech. Ageing Dev. 2013, 134, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in xpa via parp-1 hyperactivation and nad(+)/sirt1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [Green Version]
- Croteau, D.L.; Rossi, M.L.; Canugovi, C.; Tian, J.; Sykora, P.; Ramamoorthy, M.; Wang, Z.M.; Singh, D.K.; Akbari, M.; Kasiviswanathan, R.; et al. Recql4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell 2012, 11, 456–466. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Wan, S.; Lyu, Y.L.; Liu, L.F.; Qi, H. Etoposide induces atm-dependent mitochondrial biogenesis through ampk activation. PLoS ONE 2008, 3, e2009. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.J.; Dawson, T.M.; Dawson, V.L. Nuclear and mitochondrial conversations in cell death: Parp-1 and aif signaling. Trends Pharmacol. Sci. 2004, 25, 259–264. [Google Scholar] [CrossRef]
- Bai, P. Biology of poly(adp-ribose) polymerases: The factotums of cell maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Kraus, W.L. On par with parp: Cellular stress signaling through poly(adp-ribose) and parp-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Quesada, R.; Munoz-Gamez, J.A.; Martin-Oliva, D.; Peralta, A.; Valenzuela, M.T.; Matinez-Romero, R.; Quiles-Perez, R.; Menissier-de Murcia, J.; de Murcia, G.; Ruiz de Almodovar, M.; et al. Interaction between atm and parp-1 in response to DNA damage and sensitization of atm deficient cells through parp inhibition. BMC Mol. Biol. 2007, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, M.T.; Guerrero, R.; Núñez, M.I.; Ruiz de Almodóvar, J.M.; Sarker, M.; de Murcia, G.; Oliver, F.J. Parp-1 modifies the effectiveness of p53-mediated DNA damage response. Oncogene 2002, 21, 1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, A.; Kurokawa, S.; Takehashi, M.; Maeda, A.; Fukuda, K.; Kubo, Y.; Nogusa, H.; Takatani-Nakase, T.; Okuda, S.; Ueda, K.; et al. Poly(adp-ribose) polymerase inhibitors activate the p53 signaling pathway in neural stem/progenitor cells. BMC Neurosci. 2017, 18, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, N.A. Poly(adp-ribose) in the cellular response to DNA damage. Radiat. Res. 1985, 101, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ng, S.; Huang, Q.; Wu, Y.T.; Li, Z.; Yao, S.Q.; Shen, H.M. Ampk mediates a pro-survival autophagy downstream of parp-1 activation in response to DNA alkylating agents. FEBS Lett. 2013, 587, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brace, L.E.; Vose, S.C.; Stanya, K.; Gathungu, R.M.; Marur, V.R.; Longchamp, A.; Treviño-Villarreal, H.; Mejia, P.; Vargas, D.; Inouye, K.; et al. Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech. Dis. 2016, 2, 16022. [Google Scholar] [CrossRef]
- Pantovic, A.; Krstic, A.; Janjetovic, K.; Kocic, J.; Harhaji-Trajkovic, L.; Bugarski, D.; Trajkovic, V. Coordinated time-dependent modulation of ampk/akt/mtor signaling and autophagy controls osteogenic differentiation of human mesenchymal stem cells. Bone 2013, 52, 524–531. [Google Scholar] [CrossRef]
- Tao, R.; Gong, J.; Luo, X.; Zang, M.; Guo, W.; Wen, R.; Luo, Z. Ampk exerts dual regulatory effects on the pi3k pathway. J. Mol. Signal. 2010, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Bai, P.; Nagy, L.; Fodor, T.; Liaudet, L.; Pacher, P. Poly(adp-ribose) polymerases as modulators of mitochondrial activity. Trends Endocrinol. Metab. TEM 2015, 26, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, I.; Yoshida, Y.; Suda, M.; Minamino, T. DNA damage response and metabolic disease. Cell Metab. 2014, 20, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. Parp-1 inhibition increases mitochondrial metabolism through sirt1 activation. Cell Metab 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, P.; Canto, C.; Brunyanszki, A.; Huber, A.; Szanto, M.; Cen, Y.; Yamamoto, H.; Houten, S.M.; Kiss, B.; Oudart, H.; et al. Parp-2 regulates sirt1 expression and whole-body energy expenditure. Cell Metab. 2011, 13, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, J.S.; Hajira, A.; Pardo, P.S.; Boriek, A.M. Microrna-149 inhibits parp-2 and promotes mitochondrial biogenesis via sirt-1/pgc-1alpha network in skeletal muscle. Diabetes 2014, 63, 1546–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. Nad(+)-dependent activation of sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [Green Version]
- Pirinen, E.; Canto, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological inhibition of poly(adp-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef] [Green Version]
- Kam, T.-I.; Mao, X.; Park, H.; Chou, S.-C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(adp-ribose) drives pathologic α-synuclein neurodegeneration in parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [Green Version]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in patients with advanced breast cancer and a germline brca mutation. New Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Osborne, C.; Pippen, J.E.; Yoffe, M.; Patt, D.; Rocha, C.; Koo, I.C.; Sherman, B.M.; Bradley, C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. New Engl. J. Med. 2011, 364, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Wagner, L.M. Profile of veliparib and its potential in the treatment of solid tumors. Oncotargets Ther. 2015, 8, 1931–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhute, V.J.; Ma, Y.; Bao, X.; Palecek, S.P. The poly (adp-ribose) polymerase inhibitor veliparib and radiation cause significant cell line dependent metabolic changes in breast cancer cells. Sci. Rep. 2016, 6, 36061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engert, F.; Schneider, C.; Weibeta, L.M.; Probst, M.; Fulda, S. Parp inhibitors sensitize ewing sarcoma cells to temozolomide-induced apoptosis via the mitochondrial pathway. Mol. Cancer Ther. 2015, 14, 2818–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jugé, R.; Breugnot, J.; Da Silva, C.; Bordes, S.; Closs, B.; Aouacheria, A. Quantification and characterization of uvb-induced mitochondrial fragmentation in normal primary human keratinocytes. Sci. Rep. 2016, 6, 35065. [Google Scholar] [CrossRef] [PubMed]
- Zanchetta, L.M.; Garcia, A.; Lyng, F.; Walsh, J.; Murphy, J.E. Mitophagy and mitochondrial morphology in human melanoma-derived cells post exposure to simulated sunlight. Int. J. Radiat. Biol. 2011, 87, 506–517. [Google Scholar] [CrossRef]
- Paz, M.L.; Gonzalez Maglio, D.H.; Weill, F.S.; Bustamante, J.; Leoni, J. Mitochondrial dysfunction and cellular stress progression after ultraviolet b irradiation in human keratinocytes. Photodermatol. Photoimmunol. Photomed. 2008, 24, 115–122. [Google Scholar] [CrossRef]
- Denning, M.F.; Wang, Y.; Tibudan, S.; Alkan, S.; Nickoloff, B.J.; Qin, J.Z. Caspase activation and disruption of mitochondrial membrane potential during uv radiation-induced apoptosis of human keratinocytes requires activation of protein kinase c. Cell Death Differ. 2002, 9, 40–52. [Google Scholar] [CrossRef]
- Jing, L.; He, M.-T.; Chang, Y.; Mehta, S.L.; He, Q.-P.; Zhang, J.-Z.; Li, P.A. Coenzyme q10 protects astrocytes from ros-induced damage through inhibition of mitochondria-mediated cell death pathway. Int. J. Biol. Sci. 2015, 11, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. Slp-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [Green Version]
- Szanto, M.; Rutkai, I.; Hegedus, C.; Czikora, A.; Rozsahegyi, M.; Kiss, B.; Virag, L.; Gergely, P.; Toth, A.; Bai, P. Poly(adp-ribose) polymerase-2 depletion reduces doxorubicin-induced damage through sirt1 induction. Cardiovasc. Res. 2011, 92, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Shieh, W.M.; Ame, J.C.; Wilson, M.V.; Wang, Z.Q.; Koh, D.W.; Jacobson, M.K.; Jacobson, E.L. Poly(adp-ribose) polymerase null mouse cells synthesize adp-ribose polymers. J. Biol. Chem. 1998, 273, 30069–30072. [Google Scholar] [CrossRef] [Green Version]
- Zarkovic, G.; Belousova, E.A.; Talhaoui, I.; Saint-Pierre, C.; Kutuzov, M.M.; Matkarimov, B.T.; Biard, D.; Gasparutto, D.; Lavrik, O.I.; Ishchenko, A.A. Characterization of DNA adp-ribosyltransferase activities of parp2 and parp3: New insights into DNA adp-ribosylation. Nucleic Acids Res. 2018, 46, 2417–2431. [Google Scholar] [CrossRef]
- King, B.S.; Cooper, K.L.; Liu, K.J.; Hudson, L.G. Poly(adp-ribose) contributes to an association between poly(adp-ribose) polymerase-1 and xeroderma pigmentosum complementation group a in nucleotide excision repair. J. Biol. Chem. 2012, 287, 39824–39833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, M.; Kumar, A. Chapter 4—In vitro gene genotoxicity test methods. In In Vitro Toxicology; Dhawan, A., Kwon, S., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 67–89. [Google Scholar]
- Johnson, G. Mammalian cell hprt gene mutation assay: Test methods. In Genetic Toxicology; Springer: New York, NY, USA, 2012; Volume 817, pp. 55–67. [Google Scholar]
- Hsie, A.W.; Couch, D.B.; O’Neill, J.P. Utilization of a Quantitative Mammalian Cell Mutation System, Cho/Hgprt, in Experimental Mutagenesis and Genetic Toxicology. Available online: https://www.osti.gov/biblio/7278027 (accessed on 13 December 2019).
- Hu, T.; Miller, C.M.; Ridder, G.M.; Aardema, M.J. Characterization of p53 in chinese hamster cell lines cho-k1, cho-wbl, and chl: Implications for genotoxicity testing. Mutat. Res. 1999, 426, 51–62. [Google Scholar] [CrossRef]
- Tzang, B.S.; Lai, Y.C.; Hsu, M.; Chang, H.W.; Chang, C.C.; Huang, P.C.; Liu, Y.C. Function and sequence analyses of tumor suppressor gene p53 of cho.K1 cells. DNA Cell Biol. 1999, 18, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Lehman, T.A.; Modali, R.; Boukamp, P.; Stanek, J.; Bennett, W.P.; Welsh, J.A.; Metcalf, R.A.; Stampfer, M.R.; Fusenig, N.; Rogan, E.M.; et al. P53 mutations in human immortalized epithelial cell lines. Carcinogenesis 1993, 14, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Onishi, Y.; Ueha, T.; Kawamoto, T.; Hara, H.; Toda, M.; Harada, R.; Minoda, M.; Kurosaka, M.; Akisue, T. Regulation of mitochondrial proliferation by pgc-1α induces cellular apoptosis in musculoskeletal malignancies. Sci. Rep. 2014, 4, 3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleem, A.; Adhihetty, P.J.; Hood, D.A. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol. Genom. 2009, 37, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dam, A. The Effect of Mitochondrial Biogenesis on Apoptotic Susceptibility in l6 Myoblasts. Master’s Thesis, University of Waterloo, Waterloo, ON, Canada, 2010. [Google Scholar]
- Gong, B.; Chen, Q.; Almasan, A. Ionizing radiation stimulates mitochondrial gene expression and activity. Radiat. Res. 1998, 150, 505–512. [Google Scholar] [CrossRef]
- Yu, J.; Wang, Q.; Chen, N.; Sun, Y.; Wang, X.; Wu, L.; Chen, S.; Yuan, H.; Xu, A.; Wang, J. Mitochondrial transcription factor a regulated ionizing radiation-induced mitochondrial biogenesis in human lung adenocarcinoma a549 cells. J. Radiat. Res. 2013, 54, 998–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA damage response and autophagy: A meaningful partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, L.; Zhao, B.; Shah, P.; Sample, A.; Yang, S.; He, Y.Y. Autophagy positively regulates DNA damage recognition by nucleotide excision repair. Autophagy 2016, 12, 357–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-T.; Lin, J.-H.; Yang, C.-H.; Haung, C.-H.; Weng, C.-W.; Maan-Yuh Lin, A.; Lo, Y.-L.; Chen, W.-S.; Tang, M.-S. Acrolein induces mtdna damages, mitochondrial fission and mitophagy in human lung cells. Oncotarget 2017, 8, 70406–70421. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vargas, J.M.; Rodríguez, M.I.; Majuelos-Melguizo, J.; García-Diaz, Á.; González-Flores, A.; López-Rivas, A.; Virág, L.; Illuzzi, G.; Schreiber, V.; Dantzer, F.; et al. Autophagy requires poly(adp-ribosyl)ation-dependent ampk nuclear export. Cell Death Differ. 2016, 23, 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Vargas, J.M.; Ruiz-Magaña, M.J.; Ruiz-Ruiz, C.; Majuelos-Melguizo, J.; Peralta-Leal, A.; Rodríguez, M.I.; Muñoz-Gámez, J.A.; de Almodóvar, M.R.; Siles, E.; Rivas, A.L.; et al. Ros-induced DNA damage and parp-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 2012, 22, 1181–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Gámez, J.A.; Rodríguez-Vargas, J.M.; Quiles-Pérez, R.; Aguilar-Quesada, R.; Martín-Oliva, D.; de Murcia, G.; de Murcia, J.M.; Almendros, A.; de Almodóvar, M.R.; Oliver, F.J. Parp-1 is involved in autophagy induced by DNA damage. Autophagy 2009, 5, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyrsch, P.; Blenn, C.; Bader, J.; Althaus, F.R. Cell death and autophagy under oxidative stress: Roles of poly(adp-ribose) polymerases and Ca2+. Mol. Cell. Biol. 2012, 32, 3541–3553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajawat, J.; Shukla, N.; Mishra, D.P. Therapeutic targeting of poly(adp-ribose) polymerase-1 (parp1) in cancer: Current developments, therapeutic strategies, and future opportunities. Med. Res. Rev. 2017, 37, 1461–1491. [Google Scholar] [CrossRef]
- Arun, B.; Akar, U.; Gutierrez-Barrera, A.M.; Hortobagyi, G.N.; Ozpolat, B. The parp inhibitor azd2281 (olaparib) induces autophagy/mitophagy in brca1 and brca2 mutant breast cancer cells. Int. J. Oncol. 2015, 47, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biel, T.G.; Rao, V.A. Mitochondrial dysfunction activates lysosomal-dependent mitophagy selectively in cancer cells. Oncotarget 2018, 9, 995–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyschen, J.; Vitoux, B.; Marraud, M.; Cung, M.T.; Branlant, G. Engineered glycolytic glyceraldehyde-3-phosphate dehydrogenase binds the anti conformation of nad+ nicotinamide but does not experience a-specific hydride transfer. Arch. Biochem. Biophys. 1999, 364, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Santidrian, A.F.; Matsuno-Yagi, A.; Ritland, M.; Seo, B.B.; LeBoeuf, S.E.; Gay, L.J.; Yagi, T.; Felding-Habermann, B. Mitochondrial complex i activity and nad+/nadh balance regulate breast cancer progression. J. Clin. Investig. 2013, 123, 1068–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.-I.; Guarente, L. It takes two to tango: Nad+ and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [Green Version]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef]
- Canto, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(adp-ribose) polymerase and sirtuin enzymes. Mol. Asp. Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef] [Green Version]
- Luna, A.; Aladjem, M.I.; Kohn, K.W. Sirt1/parp1 crosstalk: Connecting DNA damage and metabolism. Genome Integr. 2013, 4, 6. [Google Scholar] [CrossRef]
- Ming, M.; Soltani, K.; Shea, C.R.; Li, X.; He, Y.Y. Dual role of sirt1 in uvb-induced skin tumorigenesis. Oncogene 2015, 34, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Lu, S.; Kivlin, R.; Wallin, B.; Card, E.; Bagdasarian, A.; Tamakloe, T.; Wang, W.J.; Song, X.; Chu, W.M.; et al. Sirt1 confers protection against uvb- and h2o2-induced cell death via modulation of p53 and jnk in cultured skin keratinocytes. J. Cell. Mol. Med. 2009, 13, 3632–3643. [Google Scholar] [CrossRef] [Green Version]
- Benavente, C.A.; Schnell, S.A.; Jacobson, E.L. Effects of niacin restriction on sirtuin and parp responses to photodamage in human skin. PLoS ONE 2012, 7, e42276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scher, M.B.; Vaquero, A.; Reinberg, D. Sirt3 is a nuclear nad+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007, 21, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Peterson, L.M.; Wilking-Busch, M.J.; Ndiaye, M.A.; Philippe, C.G.A.; Setaluri, V.; Ahmad, N. Sirtuins in skin and skin cancers. Ski. Pharmacol. Physiol. 2017, 30, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.; Shah, R.G.; Purohit, N.K.; Zhou, P.; Naegeli, H.; Shah, G.M. Poly(adp-ribose) polymerase 1 escorts xpc to uv-induced DNA lesions during nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, E6847–e6856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robu, M.; Shah, R.G.; Petitclerc, N.; Brind’Amour, J.; Kandan-Kulangara, F.; Shah, G.M. Role of poly(adp-ribose) polymerase-1 in the removal of uv-induced DNA lesions by nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2013, 110, 1658–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pines, A.; Vrouwe, M.G.; Marteijn, J.A.; Typas, D.; Luijsterburg, M.S.; Cansoy, M.; Hensbergen, P.; Deelder, A.; de Groot, A.; Matsumoto, S.; et al. Parp1 promotes nucleotide excision repair through ddb2 stabilization and recruitment of alc1. J. Cell Biol. 2012, 199, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccarone, F.; Zampieri, M.; Caiafa, P. Parp1 orchestrates epigenetic events setting up chromatin domains. Semin. Cell Dev. Biol. 2017, 63, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for poly(adp-ribose) polymerase (parp) inhibitors in combination therapy with camptothecins or temozolomide based on parp trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, C.B.; Prasad, S.B.; Yadav, S.S.; Pandey, L.K.; Singh, S.; Pradhan, S.; Narayan, G. Olaparib modulates DNA repair efficiency, sensitizes cervical cancer cells to cisplatin and exhibits anti-metastatic property. Sci. Rep. 2017, 7, 12876. [Google Scholar] [CrossRef]
- Jelinic, P.; Levine, D.A. New insights into parp inhibitors’ effect on cell cycle and homology-directed DNA damage repair. Mol. Cancer Ther. 2014, 13, 1645–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of parp1 and parp2 by clinical parp inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helleday, T. The underlying mechanism for the parp and brca synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.K.; Wilson, S.H. Strategic combination of DNA-damaging agent and parp inhibitor results in enhanced cytotoxicity. Front. Oncol. 2013, 3, 257. [Google Scholar] [CrossRef] [Green Version]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the cytotoxic effects of parp inhibitors with DNA demethylating agents—A potential therapy for cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, R.; Dua, D.; Cresti, N.; Suder, A.; Drew, Y.; Prathapan, V.; Stephens, P.; Thornton, J.K.; de las Heras, B.; Ink, B.; et al. Phase 1 study of the parp inhibitor e7449 as a single agent in patients with advanced solid tumors or b-cell lymphoma. J. Clin. Oncol. 2014, 32, e19531. [Google Scholar] [CrossRef]
- O’Cearbhaill, R.E. Using parp inhibitors in advanced ovarian cancer. Oncology 2018, 32, 339–343. [Google Scholar]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. Nad+ depletion is necessary and sufficient for poly(adp-ribose) polymerase-1-mediated neuronal death. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular atp levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar]
- Zamaraeva, M.V.; Sabirov, R.Z.; Maeno, E.; Ando-Akatsuka, Y.; Bessonova, S.V.; Okada, Y. Cells die with increased cytosolic atp during apoptosis: A bioluminescence study with intracellular luciferase. Cell Death Differ. 2005, 12, 1390–1397. [Google Scholar] [CrossRef]
- Fu, D.; Lippincott-Schwartz, J.; Arias, I.M. Increased mitochondrial fusion and autophagy help isolated hepatocytes repolarize in collagen sandwich cultures. Autophagy 2013, 9, 2154–2155. [Google Scholar] [CrossRef] [Green Version]
- Thomas, H.E.; Zhang, Y.; Stefely, J.A.; Veiga, S.R.; Thomas, G.; Kozma, S.C.; Mercer, C.A. Mitochondrial complex i activity is required for maximal autophagy. Cell Rep. 2018, 24, 2404–2417.e2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montecucco, A.; Zanetta, F.; Biamonti, G. Molecular mechanisms of etoposide. EXCLI J. 2015, 14, 95–108. [Google Scholar]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagenesis 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, A.; Milum, K.; Battu, A.; Wani, G.; Wani, A.A. Ner initiation factors, ddb2 and xpc, regulate uv radiation response by recruiting atr and atm kinases to DNA damage sites. DNA Repair 2013, 12, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Connors, K.E.; Yang, D.Q. Aicar induces phosphorylation of ampk in an atm-dependent, lkb1-independent manner. Mol. Cell. Biochem. 2007, 306, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Kozlov, S. Atm activation and DNA damage response. Cell Cycle 2007, 6, 931–942. [Google Scholar] [CrossRef]
- Viniegra, J.G.; Martinez, N.; Modirassari, P.; Hernandez Losa, J.; Parada Cobo, C.; Sanchez-Arevalo Lobo, V.J.; Aceves Luquero, C.I.; Alvarez-Vallina, L.; Ramon y Cajal, S.; Rojas, J.M.; et al. Full activation of pkb/akt in response to insulin or ionizing radiation is mediated through atm. J. Biol. Chem. 2005, 280, 4029–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, H.; Tummala, H.; Zhelev, N. ATM in focus: A damage sensor and cancer target. Biodiscovery 2012, 5. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. Amp-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Liu, X.; Dagda, R.K.; Zhang, Y. How ampk and pka interplay to regulate mitochondrial function and survival in models of ischemia and diabetes. Oxidative Med. Cell. Longev. 2017, 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.B.; Kinoshita, C.; Kinoshita, Y.; Morrison, R.S. P53 and mitochondrial function in neurons. Biochim. Biophys. Acta 2014, 1842, 1186–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, V.; Verdejo, H.E.; Iglewski, M.; Del Campo, A.; Troncoso, R.; Jones, D.; Zhu, Y.; Kuzmicic, J.; Pennanen, C.; Lopez-Crisosto, C.; et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the akt-mtor-nfkappab-opa-1 signaling pathway. Diabetes 2014, 63, 75–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Prudent, J.; Basu, K.; Goyon, V.; Katsumura, S.; Hulea, L.; Pearl, D.; Siddiqui, N.; Strack, S.; McGuirk, S.; et al. Mtor controls mitochondrial dynamics and cell survival via mtfp1. Mol. Cell 2017, 67, 922–935.e925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Boros, G.; Miko, E.; Muramatsu, H.; Weissman, D.; Emri, E.; Rozsa, D.; Nagy, G.; Juhasz, A.; Juhasz, I.; van der Horst, G.; et al. Transfection of pseudouridine-modified mrna encoding cpd-photolyase leads to repair of DNA damage in human keratinocytes: A new approach with future therapeutic potential. J. Photochem. Photobiol. B Biol. 2013, 129, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Chomczynski, P.; Sacchi, N. Single-step method of rna isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Balogh, A.; Paragh, G., Jr.; Juhasz, A.; Kobling, T.; Torocsik, D.; Miko, E.; Varga, V.; Emri, G.; Horkay, I.; Scholtz, B.; et al. Reference genes for quantitative real time pcr in uvb irradiated keratinocytes. J. Photochem. Photobiol. B Biol. 2008, 93, 133–139. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. Imagej2: Imagej for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Merrill, R.A.; Flippo, K.H.; Strack, S. Measuring mitochondrial shape with imagej. In Techniques to Investigate Mitochondrial Function in Neurons; Strack, S., Usachev, Y.M., Eds.; Springer: New York, NY, USA, 2017; pp. 31–48. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hegedűs, C.; Boros, G.; Fidrus, E.; Kis, G.N.; Antal, M.; Juhász, T.; Janka, E.A.; Jankó, L.; Paragh, G.; Emri, G.; et al. PARP1 Inhibition Augments UVB-Mediated Mitochondrial Changes—Implications for UV-Induced DNA Repair and Photocarcinogenesis. Cancers 2020, 12, 5. https://doi.org/10.3390/cancers12010005
Hegedűs C, Boros G, Fidrus E, Kis GN, Antal M, Juhász T, Janka EA, Jankó L, Paragh G, Emri G, et al. PARP1 Inhibition Augments UVB-Mediated Mitochondrial Changes—Implications for UV-Induced DNA Repair and Photocarcinogenesis. Cancers. 2020; 12(1):5. https://doi.org/10.3390/cancers12010005
Chicago/Turabian StyleHegedűs, Csaba, Gábor Boros, Eszter Fidrus, Gréta Nikoletta Kis, Miklós Antal, Tamás Juhász, Eszter Anna Janka, Laura Jankó, György Paragh, Gabriella Emri, and et al. 2020. "PARP1 Inhibition Augments UVB-Mediated Mitochondrial Changes—Implications for UV-Induced DNA Repair and Photocarcinogenesis" Cancers 12, no. 1: 5. https://doi.org/10.3390/cancers12010005
APA StyleHegedűs, C., Boros, G., Fidrus, E., Kis, G. N., Antal, M., Juhász, T., Janka, E. A., Jankó, L., Paragh, G., Emri, G., Bai, P., & Remenyik, É. (2020). PARP1 Inhibition Augments UVB-Mediated Mitochondrial Changes—Implications for UV-Induced DNA Repair and Photocarcinogenesis. Cancers, 12(1), 5. https://doi.org/10.3390/cancers12010005