Simple Summary
Approximately 50% of high-grade gliomas (HGG) in children are diffuse midline gliomas (DMGs), which carry the worst prognosis of all HGG, with a 2-year survival of less than 10%. DMGs are characterized by H3K27M mutation, rampant genomic instability, infiltrative growth, and radio-resistance. Recent large-scale profiling studies have identified some of the key molecular drivers underpinning DMG biology and therapeutic resistance. Here, we provide a comprehensive overview of studies that focus on DMG in the context of radio-resistance. We speculate that the aberrant activation of DNA damage response pathway (DDR) represents a druggable vulnerability, which could be leveraged to radio-sensitize DMGs.
Abstract
Malignant gliomas (MG) are among the most prevalent and lethal primary intrinsic brain tumors. Although radiotherapy (RT) is the most effective nonsurgical therapy, recurrence is universal. Dysregulated DNA damage response pathway (DDR) signaling, rampant genomic instability, and radio-resistance are among the hallmarks of MGs, with current therapies only offering palliation. A subgroup of pediatric high-grade gliomas (pHGG) is characterized by H3K27M mutation, which drives global loss of di- and trimethylation of histone H3K27. Here, we review the most recent literature and discuss the key studies dissecting the molecular biology of H3K27M-mutated gliomas in children. We speculate that the aberrant activation and/or deactivation of some of the key components of DDR may be synthetically lethal to H3K27M mutation and thus can open novel avenues for effective therapeutic interventions for patients suffering from this deadly disease.
1. Diagnosis and Treatment
Despite the fact that primary pediatric central nervous system (CNS) tumors are rare (approximately 3.2 cases per 100,000 children per year [1]), they represent the most common solid tumor in childhood and account for the highest number of cancer-related deaths in children [2,3]. Among these malignancies, diffuse intrinsic pontine gliomas (DIPG) are the most aggressive subset with an incidence of less than one case per 100,000 children per year [1]. The anatomic location and infiltrative nature of DIPGs precludes successful resection and leads to a variety of progressive neurological deficits during the short life span, ultimately resulting in death [4]. The median age at diagnosis is seven. The median progression-free survival is seven months and once progression is diagnosed, patients die within 2–4 months [5,6].
Recent technological advancement in diagnostics, especially the use of whole genome sequencing (WGS) at diagnosis or post-mortem, has mapped the complexity of pediatric high-grade gliomas (pHGG) at the genomic and epigenomic level, showing that recurring somatic mutations in histone H3 and other key signaling pathways underpin the growth, proliferation, survival, and therapeutic resistance of these tumors [6,7]. Three molecular subtypes—MYCN, Silent, and H3 K27M—have been defined on the basis of the integrative analysis of DNA methylation profiling, WGS, and histopathological and clinical data (Figure 1).
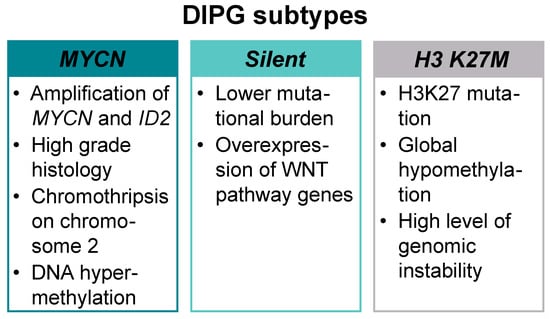
Figure 1.
Molecular subtypes of diffuse intrinsic pontine glioma (DIPG). DIPGs can be divided into three distinct molecular subtypes: MYCN, Silent, and H3 K27M.
A high-grade histology, chromothripsis (high frequency of chromosomal rearrangements acquired in one or more regions) on chromosome 2, DNA hypermethylation, and high-level amplification of MYCN proto-oncogene bHLH transcription factor (MYCN) and inhibitor of DNA binding 2 (ID2) are the hallmarks of MYCN subtype. The H3 K27M subtype is the most prevalent in DIPG and presents with H3K27 mutation, global hypomethylation, and multiple concurrent mutations (Table 1) [8,9].

Table 1.
Concurrent mutations in the H3 K27M subtype [8,9].
The Silent subtype carries lower mutational burden than the other two and shows overexpression of wingless-related integration site (WNT) pathway genes. Patients in the silent subgroup are significantly younger compared to the other two subgroups (5 years for silent, 6.3 years for MYCN, and 7 years for H3 K27M) and present with a low-grade astrocytoma histology [8,10]. In a more recent study, Mackay et al. defined the following molecular subgroups: H3G34R/V, H3K27M, high grade glioma wildtype (HGG WT), isocitrate dehydrogenase 1 (IDH1), low-grade glioma (LGG)-like, pleomorphic xanthoastrocytoma (PXA)-like, and “other”. As reported previously, these subgroups differed in anatomical location, age at diagnosis, and overall survival, with the LGG-like group representing a younger cohort and excellent prognosis [11].
In 2016, the World Health Organization (WHO) reclassified CNS tumors to take account of molecular pathology, with this including definition of a new entity—histone 3 K27M-mutated diffuse midline gliomas (DMGs)—which encompass about 80% of DIPGs [12,13]. Clinical presentation, history, physical neurological examination, and magnetic resonance imaging (MRI) are generally used in diagnosis of DMGs. Typical radiographic characteristics of DIPG on MRI include a T1 hypointense, T2 hyperintense tumor centered in and involving > 50% of the pons, with absence of or irregular gadolinium enhancement (Figure 2) [9,14]. Given the infiltrative nature of these tumors, surgical intervention is substantially limited and therapy has been traditionally instituted on the basis of imaging data, usually without histopathological verification. A series of conflicting studies cited morbidity rates between 0% and 10% and mortality rates between 0% to 3% to be associated with stereotactic biopsy procedure. However, when biopsy data on pediatric patients were extracted, the diagnostic yield ranged from 96% to 100%, with no mortality and morbidity less than 5% for the largest series [15]. Most importantly, molecular analysis from biopsy material has greatly increased our chances for a better understanding of the unique biological behavior of DMGs [16,17].
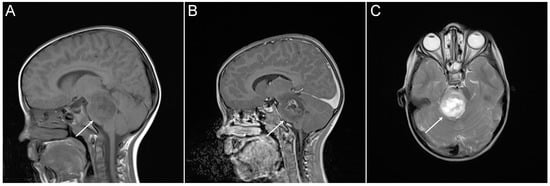
Figure 2.
Magnetic resonance imaging (MRI) of DIPG tumor from an individual patient (white arrows). (A) Sagittal T1-weighted. (B) Sagittal T1-weighted. (C) Axial T2-weighted.
The current standard of care consists of fractionated radiation (delivered most commonly using photon radiation therapy (RT)), typically with a total dose of 54–60 Gy delivered with a daily fraction of 1.8–2 Gy over 6 weeks. Steroids are often used during the course of RT to relieve symptoms of peri-tumoral oedema [16]. Radiation therapy allows transient control of the disease or even symptomatic relief, but recurrence is inevitable. Of note, there is some evidence suggesting a certain degree of radio-sensitivity remains at this stage, since some DMG patients were found to benefit from re-irradiation [18]. Re-irradiation with approximately 25 Gy over 10 fractions has been reported to be safe and suggests a modest survival benefit, as well as stabilization and/or improvement in symptoms [13].
Chemotherapy (such as alkylating agent temozolomide (TMZ), carboplatin, cisplatin, or irinotecan) alone, or as an adjuvant to RT, has not been associated with any additional survival benefit [19,20]. Numerous regimens of either a mono- or combinational chemotherapy have been trialed with overall poor outcome. Agents targeting other molecular pathways, such as erlotinib (tyrosine kinase EGFR inhibitor) and bevacizumab (humanized mouse antibody targeting vascular endothelial growth factor A (VEGFA)) have also been proven ineffective, whether administered alone or in combination with RT [8]. Similarly, radio-sensitizers (e.g., carbogen, motefaxin gadolinium) did not improve the survival of DMG patients [21,22]. Currently in clinical trials for DMGs are inhibitors of histone deacetylases (HDAC), which emerged from functional screen data as well as the observation of consistently elevated histone acetylation in a number of studies [23].
The blood–brain barrier (BBB) is thought to be one of the key reasons why DMG patients respond poorly to chemotherapy and other targeted therapies. In DMGs, the BBB is frequently intact, as evidenced by the lack of gadolinium enhancement. Interestingly, some studies suggest that the brainstem region may have even more robust BBB compared to other areas of the brain [24]. Numerous methods have been developed in an attempt to bypass this barrier—among these, convention-enhanced delivery (CED) or the use of nanoparticles are being extensively tested in pre-clinical as well as the clinical setting [25,26]. The CED approach utilizes a catheter to deliver drugs directly to the target region [26]. There are active clinical trials for DMGs that are exploring the delivery of the conventional chemotherapeutic irinotecan (NCT03086616) and a radioactive iodine-labeled monoclonal antibody omburtamab (NCT01502917) via CED [27,28]. The National Clinical Trial NCT03566199 explores the CED approach in combination with HDAC inhibitor panobinostat in a nanoparticle formulation (MTX110) [29].
Despite intense scientific effort and recent developments in the clinical trial setting, the fact that treatment options for DMG patients are limited stresses the urgent need to develop novel therapeutics and effective delivery mechanisms for this patient group.
2. Mutations and Key Signaling Genes in DMG
Somatic mutations in histone genes in pHGG were first reported by two groups [6,7]. Wu et al. performed WGS on seven DIPGs and targeted sequencing on an additional cohort of 43 DIPGs and 36 non-brainstem pediatric glioblastomas (pGBMs). They found that 78% of DIPGs and 22% of non-brainstem pGBMs contained heterozygous mutations in H3 histone family member 3A (H3F3A) and histone cluster 1 H3B (HIST1H3B), respectively [6]. The study by Schwartzentruber et al. included 48 pGBMs and found that somatic mutations of histone genes, including H3.3K27M and H3.3G34R/V, occurred in 31% of patients [7]. These mutations result in a substitution of histone 3 lysine to methionine at position 27 (H3K27M) or glycine to arginine/valine at position 34 (H3G34R/V). While H3.3K27M-mutated tumors are localized in midline structures (e.g., brainstem, thalamus, cerebellum, and spine), the H3.3G34R/V-mutated tumors locate exclusively in cerebral hemispheres. The histone variant H3.1 (HIST1H3B gene) was found mutated and solely associated with DMG brainstem tumors (H3K27M), occurring in 18% of cases [6,30]. The H3K27M mutation in H3.3 histone occurs with alterations in the p53 pathway, mostly tumor suppressor protein p53 (TP53)- and protein phosphatase magnesium-dependent 1 delta (PPM1D)-inactivating mutations, while H3.1 histone mutation associates with activating mutations in the activin A receptor type 1 (ACVR1A) gene as well as whole chromosome 2 gain and chromosome 16q loss [7,31,32]. DNA topoisomerase III alpha (TOP3A) amplification is TOP3A alterations that were mutually exclusive with ATRX deletion/mutations found in H3.3K27M DIPGs [11,33].
A study by Buczkowicz et al. (2014) revealed that DIPG outcome correlated more accurately with H3 mutation status than with WHO grade, with the overall survival rate for WHO grade II H3.3-mutated patients being comparable with that of WHO grade IV GBM tumors [10]. The oncogenesis of DMGs is driven by the lysine-to-methionine substitution at position 27 of the regulatory tail of histone variants H3F3A, HIST1H3B/C, and HIST2H3A. This mutation is dominant negative and heterozygous, whereas each histone H3 variant mutated at K27 drives a specific oncogenic program [34]. H3K27M mutation drives the global loss of di- and trimethylation of histone H3K27 (H3K27me2/H3K27me3) and affects the enzymatic activity of enhancer of zeste homologue 2 (EZH2) [35,36]. EZH2 is the main component of the polycomb repressive complex 2 (PRC2), which under normal conditions mediates the di- and trimethylation on H3K27 (Figure 3) [36]. A recent study by Harutyunyan et al. showed that the removal of H3K27M mutation restores the levels of H3K27me2/H3K27me3 and impairs the tumorigenic capacity of DMG cells in vivo in a mouse model [37]. Interestingly, the authors found that the loss of H3K27me2 and H3K27me3 induces limited transcriptomic changes, while genes that are actually dysregulated are primarily involved in stemness and neurogenesis [37].
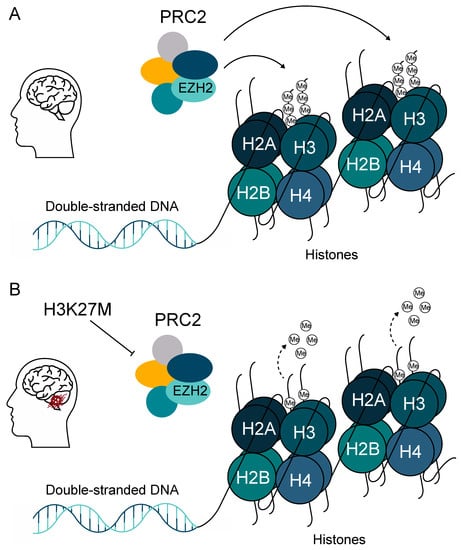
Figure 3.
H3K27M is the major oncogenic driver in pediatric high-grade gliomas (pHGG). (A) The polycomb repressive complex 2 (PRC2) complex mediates the di- and trimethylation of H3K27 in healthy brains. (B) The H3K27M mutation in diffuse midline gliomas (DMG) leads to global loss of di- and trimethylation of histone H3K27 (H3K27me2 and H3K27me3) by inactivating the PRC2 complex.
Similar to other cancer types, DMGs are characterized by frequent aberrations involving a number of signaling pathways downstream of cell surface receptors. Focal amplifications and mutations to the PDGFRA as well as concurrent focal amplifications in other receptor tyrosine kinases (RTKs) such as KIT proto-oncogene receptor tyrosine kinase (KIT)/vascular endothelial growth factor receptor 2 (VEGFR2)/MET/epidermal growth factor receptor (EGFR2) are some of the most common genomic events found in DMGs [4]. Although no single event is prevalent in these tumors, studies have identified mutations in both phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) and phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1), HRas proto-oncogene GTPase (HRAS) amplification, and phosphatase and tensin homolog (PTEN) loss, jointly contributing to an aberrant activation of the phosphatidyl-inosital-3 kinase (PI3K) and/or mitogen-activated protein kinase (MAPK) signaling in these tumors [38].
The abrogation of cell cycle checkpoints leading to dysregulated cell cycle kinetics is among the hallmarks of cancer [39]. The H3K27M mutation reportedly results in epigenetic silencing of cyclin-dependent kinase inhibitor 2A (CDKN2A), a key regulator of G1/S checkpoint. In addition, essential players in the G1/S checkpoint activation, CDK4/6, and associated cyclins have been reported to be amplified in DMGs [40]. Besides the aberrant expression of G1/S checkpoint regulators, DMGs were found to overexpress several key players in the G2/M checkpoint progression such as Wee1 G2 checkpoint kinase (Wee1), maternal embryonic leucine zipper kinase (MELK), polo-like kinase 1 (PLK1), and aurora kinase B (AURKB) [41,42,43].
3. DNA Repair Response Pathway
Error-free and timely regulation of cell cycle progression is crucial for genome integrity maintenance. Each of the ≈1013 cells in the human body receives tens of thousands of DNA lesions per day [44]. These DNA lesions can result from either endogenous (e.g., replication errors, reactive oxygen species (ROS)) or exogenous sources (e.g., ultraviolet light (UV), ionizing radiation (IR), chemical agents) [45]. Exposure to IR inflicts single-stranded DNA breaks (SSBs), double-stranded DNA breaks (DSBs), base damage, and DNA-protein cross-links in the genomic DNA. Among them, DSBs are the most deleterious lesions that, if not repaired correctly, can lead to genomic instability and cell death [46].
A sophisticated network of DNA damage response (DDR) systems has evolved to deal with the fundamental problem of maintaining genomic stability (Figure 4). These include a set of DNA repair mechanisms, damage tolerance processes, and cell-cycle checkpoint pathways. Depending on the type of DNA damage and the outcome of DNA repair, DDR activation can lead to cell survival, apoptosis, cell cycle arrest, or cellular senescence (e.g., permanent cell cycle arrest) [44,47].
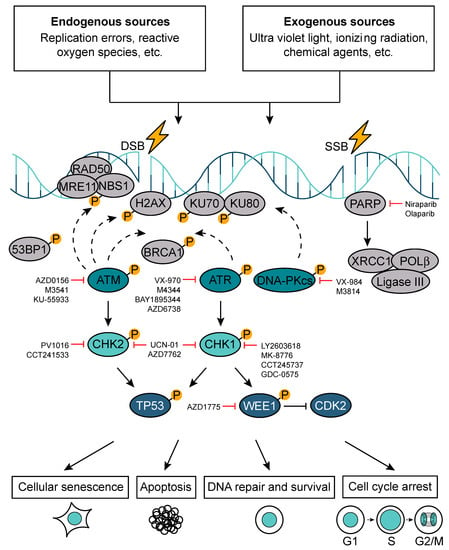
Figure 4.
DNA damage response pathway (DDR). In response to DNA damage, which can be induced by the exposure to either endogenous or exogenous factors, cells activate DDR. Depending on the extent and type of DNA damage (single-strand DNA break (SSB) versus double-strand DNA break (DSB)), the DDR can trigger cell cycle arrest, apoptosis, cellular senescence, or DNA repair. A number of DDR components have been successfully targeted in pre-clinical settings.
The protein kinases ataxia telangiectasia mutated (ATM), ataxia telangiectasia and rad3 related (ATR), and DNA-dependent protein kinase catalytic subunit (DNA-PKcs) are the key regulators of DDR. ATM and ATR activation triggers an activatory phosphorylation cascade, which results in the phosphorylation of DDR mediators (e.g., p53-binding protein 1 (53BP1), H2A histone family member X at Serine 139 (H2AXSer139 or γH2AX), breast cancer type 1 susceptibility protein (BRCA1)) and downstream kinases checkpoint kinases 1/2 (Chk1/Chk2). Chk1 and Chk2 then in turn phosphorylate downstream effectors such as TP53. DNA-PKcs activation leads to the formation of a complex with the Ku heterodimer (Ku70/Ku80), playing an important role in non-homologous end-joining (NHEJ) [48]. There are two major pathways devoted to DSBs repair: NHEJ and homologous recombination DNA repair (HR), which operate in a cell-cycle phase-dependent manner. NHEJ can occur in all phases of the cell cycle (dominates in G1) and is error-prone because it directly ligates broken DNA ends. The error-free repair by HR is restricted to S and G2 phase and it uses sister chromatids as a template for the repair [49]. The G1/S checkpoint is regulated by ATM, Chk2, and p53 and aims at repairing DNA lesions, which may present as obstacles for DNA replication machinery. Replication is a tightly regulated process and, if compromised, it results in replication stress, which is a central cause of genome instability [50,51]. The intra-S phase checkpoint is regulated by ATR, Chk1, DNA-PK, and Wee1. When triggered, this checkpoint delays the replication process to allow the cells to repair and to cope with DNA damage during S phase. The G2/M checkpoint is regulated by Chk1, myelin transcription factor 1 (Myt1), and Wee1, safeguarding genomic stability by preventing cells to exit G2/M cell cycle phase and divide prior repairing existent DNA lesions. The failure to repair DNA damage at G2/M phase followed by an attempt to divide may result in mitotic catastrophe and cell death [51,52].
SSBs are the most common DNA lesions and they are repaired through a process called base excision repair (BER). During BER, SSBs are recognized by poly(ADP-ribose) polymerase (PARP), which induces extensive poly-ADP-ribosylation (PARylation) and thereby initiates their repair [53]. In particular, PARP recruits the scaffold protein X-ray cross-complementing protein 1 (XRCC1) followed by additional enzymes such as DNA polymerase β (POLβ) and DNA ligase 3 (Ligase III) [54,55]. POLβ is responsible for filling in the single-nucleotide gap and preparing the strand for ligation by Ligase III [56]. SSBs can indirectly lead to DSB formation if two of them arise in close proximity of each other. Moreover, if left unrepaired, SSBs can be converted into DSBs, ultimately resulting in replication fork stalling and collapse [57]. Other major pathways involved in the maintenance of genomic stability include nucleotide excision repair (NER) and mismatch repair (MMR). The NER pathway removes helix-distorting lesions and primarily copes with UV-induced damage [58]. The MMR pathway corrects mismatched nucleotides, insertions, and deletions acquired during DNA replication [59].
In cancer, the critical balance between the genomic maintenance in malignant cells and the number and partial overlap of DDR pathways (Figure 4) provide an exciting therapeutic opportunity. Because different DNA repair pathways may functionally overlap, and as one pathway can sometimes “back-up” for defects in another, inhibition of pathways presented in a cancer cell should in some cases have a greater impact on the cancer than on normal tissues. This deficiency in the cancer cells can be exploited using an approach termed synthetic lethality. A paradigm for this is provided by drugs targeting the enzyme PARP1, which binds SSBs and BER intermediates to facilitate DNA repair processes. Notably, PARP inhibitors are relatively nontoxic to normal cells but are strikingly cytotoxic towards HR-defective cells, particularly those with impaired BRCA1 or BRCA2 function [60,61]. In BRCA-deficient cells, PARP inhibition drives the collapse of replication forks and the formation of replication-associated DSBs that can only be repaired by HR [45,60,61,62,63].
4. DNA Repair Gene Expression in DMGs
The rampant inter- and intra-patient heterogeneity with treatment-resistant cancer stem-like cells (CSCs) at the apex is believed to drive the resistance to existent chemo- and radiotherapy [64,65,66,67,68]. A study performed by Alhajala et al. showed that genes involved in stem cell differentiation, angiogenesis, cell proliferation, and cell growth are upregulated in response to irradiation in pGBM with wild-type H3 [69]. Harutyunyan et al. have reported that the loss of H3K27me2 and H3K27me3 dysregulates the expression of genes involved in stemness and neurogenesis, thereby affecting cell differentiation [37]. Larson et al. have shown that H3.3 K27M mutation alone enhances neural stem cell self-renewal in a genetically engineered mouse model [70]. CSCs were found to constitituvely activate DDR signaling, which is believed to protect them from the insult of DNA damaging agents [67]. In H3K27M-mutated tumors, the dysregulation of stem cell maintanence and aberrant DDR activation represent a vulnerability that may be exploited for their sensitization to DNA damaging therapies such as chemo-irradiation.
The identification of histone mutations in a large proportion of HGG from pediatric and adolescent patients has increased the enthusiasm for targeted combinational approaches to tackle these tumors [6,7]. The first major breakthrough came in 2012, when studies using whole-genome and whole-exome sequencing reported that 80% of DIPG and 35% midline glioblastoma (mGBM) possess mutations in histone H3.3/3.1 and that these mutations, together with the abrogation of cell cycle checkpoints, confer radio-resistance and dismal prognosis [6,7,10,71]. Perturbations in DNA damage repair gene expression in H3K27M mutant tumors were first identified in 2010 by Zarghooni et al., who reported a loss of heterozygosity (LOH) of a number of DNA repair genes [7]. In 2017, Mackay et al. performed an integrated genomic analysis, which likewise found that 60.7% of the pHGG harbored genetic alterations in the DNA repair pathway, highlighting DNA repair as a potential therapeutic target [11]. Moreover, Mackay et al. found a diverse set of heterozygous mutations in a number of genes involved in error-free HR repair and numerous Fanconi anemia genes [11]. Their analysis identified, among others, amplifications of cyclin D2 (CCND2) and TOP3A in H3.3K27M DIPG [11]. CCND2 promotes cell cycle progression by phosphorylation of retinoblastoma (Rb), whereas TOP3A is involved in HR [72,73]. It remains unknown as to whether these alterations are involved in the radio-resistance seen in DMGs. A study by Zhang et al. has shown that the loss of H3K27me2 and H3K27me3 reduces the number of 53BP1 foci as well as decreasing the efficiency of NHEJ in human dermal fibroblasts [74]. ATM is necessary for the initiation of DDR and DSB repair and was found mutated in 10.7% of H3.3K27M brainstem DMGs [11], but the consequences of such combination on genomic stability and therapeutic resistance remain elusive [75]. TP53 regulates cell cycle arrest, senescence, apoptosis, autophagy, metabolism, and aging. About 42–77% of DMGs carry the TP53 mutation, a phenotype that is associated with increased radio-resistance, early relapse, and worsened prognosis compared to TP53 wild-type DMGs [76,77]. Moreover, mutations in PPM1D have been identified in up to 25% of DIPGs [31,78]. PPM1D encodes the wild-type p53-induced phosphatase 1 (Wip1), which has been shown to be a negative regulator of the DDR by dephosphorylating ATM [79], TP53 [80], Chk2 [81], Chk1 [80], and γH2AX [82], which together lead to a reduced activation of TP53. This could potentially contribute to the radio-resistance in the DMGs harboring the PPM1D mutation, as seen for the DMGs harboring the TP53 mutation [77].
5. DNA Repair as a Therapeutic Target to Radio-Sensitize DMGs
Whereas RT has proven to be an effective palliative and curative tool in cancer management, it is not without risk of toxicity to normal tissues [83]. A wide variety of tumor types demonstrate resistance to RT, which limits its utility as a stand-alone treatment modality and stresses the unmet need for the identification and successful validation of radio-sensitizing agents. There have been attempts to improve the outcome of RT with the integration of systemic agents before, during, or after IR in a number of solid malignancies. In DMGs, the number of studies interrogating radio-sensitizing agents is limited. Those studies that have been reported thus far did not conclude a clear clinical benefit compared to the standard of care. Taking the vast inter- and intra-tumoral heterogeneity into consideration, better stratification and identification of patients who might benefit from being given a drug–IR combination is necessary, which can be warranted only through international multi-center collaborative efforts. The fact that H3K27M mutation confers radio-resistance warrants further investigation into using radio- and chemo-sensitizing agents in the therapeutic intervention of H3K27mM-mutated MGs. Using synthetic lethal approaches to H3K27M mutation may provide a rational way to develop novel and highly selective radio-sensitizing strategies (Figure 5).
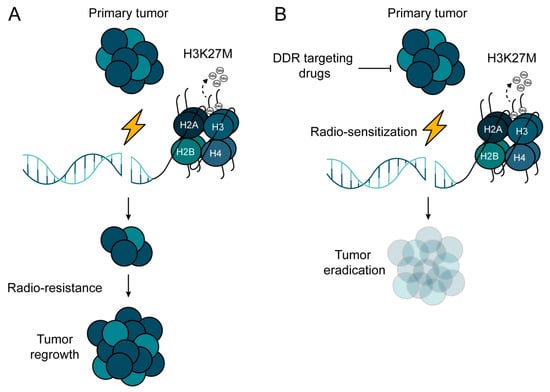
Figure 5.
Potential treatment strategy to overcome radio-resistance in H3K27M-mutated pHGG. (A) H3K27M mutation confers radio-resistance in pHGG due to the de-regulation of DNA repair machineries. (B) Combinational approach, where radiotherapy (RT) is combined with DNA repair inhibitors, may pave the way for overcoming the radio-resistance of H3K27M-mutated pHGG.
The overall limited impact of RT in the management of cancer fueled intensive research efforts, leading to pre-clinical and clinical testing of various agents and/or tool compounds. DNA damage-inducing agents such as TMZ, cyclophosphamide, etoposide, and lomustine have been interrogated in regards to their ability to improve the efficacy of RT, with no success. O6-Methyl-guanine-DNA methyltransferase (MGMT) promoter methylation is associated with TMZ resistance. In a study of 46 DMGs with confirmed H3F3A mutation, none had MGMT promoter methylation. Through a series of in vitro experiments, Abe et al. confirmed that epigenetically driven high expression of MGMT is the main reason for TMZ resistance [84,85].
We and others have shown that the DDR is constitutively active in adult malignant gliomas (MG) due to ongoing oxidative and replication stress [86,87] and that the targeting of DDR components may be of high clinical relevance, as evidenced by extended survival of tumor-bearing animals in in vivo studies [88,89]. A number of reports highlighted the aberrant expression of DNA repair factors in DMGs, indicating impaired DNA repair [11,90]; however, the extent of DDR activation and the functionality of DNA repair mechanisms in DMGs remains elusive. A subset of DMGs has been found to overexpress PARP1, and its inhibition has been studied as a potential therapeutic intervention in DMGs, resulting in radio-sensitization and anti-tumor efficacy both in vitro and in vivo. Unfortunately, the clinical relevance of these findings is tainted by the model choice for some of the key experiments shown in this particular study. The authors used pediatric high-grade astrocytoma cells instead of DMG cells and injected these into the cortex instead of the pons in their in vivo proof-of-concept study [38,91].
A study by Akamandisa et al. (2019) showed that targeting PPM1D effectively eradicates PPM1D mutant DMGs in vitro and in vivo, especially when combined with irradiation [92]. In the presence of DNA damage, DDR activates cell cycle checkpoints to prevent cells from entering mitosis. PLK1 is one of the DNA damage checkpoints [93], while also controlling DNA repair [94]. A study by Amani et al. concluded that targeting PLK1 with small molecule inhibitors, in combination with IR, warrants further investigation on the basis of their in vitro evidence of reduced proliferative index and increased apoptosis in DMGs treated with PLK1 inhibitor BI 6727 in combination with IR [95]. In DMGs, p53 function is often compromised by loss-of-function mutations, genomic deletions, or indirectly as a result of PPM1D mutations [10,38]. Nikolaev et al. (2020) showed that a combination of eprenetapopt (also known as APR-246 or PRIMA-1MET, a small organic molecular that induces oxidative stress and reactivates mutant p53) and histone lysine demethylase inhibitor VIII (GSK-J4, a cell permeable prodrug of GSK-J1, which is a selective inhibitor of the H3K27 histone demethylase of Jumonji histone demethylases) markedly sensitizes DMGs to IR [96]. The mammalian target of rapamycin (mTOR) signaling pathway is one of the key oncogenic drivers in DMGs. A growing body of evidence suggests a close link with DDR and many factors important for the DDR were found to be involved in the mTOR pathway [97]. One study found that a dual mTOR kinase inhibitor sapanisertib (TAK228) inhibited tumorigenicity and enhanced the effect of RT in DMG, supporting this connection [98]. Despite the evidence from pre-clinical studies supporting radio-sensitizing strategies, the number of currently active clinical trials exploring the impact of novel agents in combination with IR is limited. Among the reasons that preclude successful translation is the lack of in vivo validation using patient-derived xenograft models, which are more representative of original tumor architecture than commercially available stable cell lines [99].
An example of a putative radio-sensitizer assessed both in a pre-clinical study and a clinical trial is the selective Wee1 inhibitor adavosertib (MK-1775). Wee1 is a critical component of the G2/M cell cycle checkpoint machinery, which prevents mitotic entry in the presence of DNA damage [100]. In 2013, Mueller et al. reported Wee1 to be overexpressed in DIPGs and proposed the use of the Wee1 inhibitor in combination with IR for the treatment of DIPG [41]. The NCT01922076 study, which is currently running, is a phase I trial assessing the safety, toxicity, and maximum tolerated dose (MTD) for adavosertib administered concurrently with radiation therapy in children with newly diagnosed DIPGs.
B lymphoma Moloney murine leukemia virus insertion region 1 (BMI-1), a core member of polycomb repressive complex 1 (PRC1), has been intensely investigated in the field of cancer epigenetics and DNA repair for decades. The recruitment of BMI-1 to sites of DNA DSBs is critical for efficient HR repair [101]. Kumar et al. have shown that BMI-1 is highly expressed in DIPGs compared to normal tissue and that downregulation in vitro leads to inhibition of neurosphere formation, migration, self-renewal, and cell-cycle signaling. Importantly, this study reported that the targeted inhibition of BMI-1 sensitizes DIPG cells to radiomimetic drug-induced DNA damage [102]. The safety of PTC596 (an orally active inhibitor of BMI-1) taken during radiation in children with newly diagnosed DMGs and HGG is currently being investigated in a phase Ib clinical trial (NCT03605550).
A study by Werbrouck et al. has shown promising data on the efficacy of the Chk1 inhibitor LY2606368 in combination with IR in TP53-mutated DMGs [77]. LY2606368 is also being investigated within a phase I clinical in refractory pediatric tumors including DMGs as a monotherapy, with the aim being to analyze putative biomarkers and estimate the MTD as a primary objective (NCT02808650) [77].
A recently initiated clinical trial in newly diagnosed DIPGs was designed to explore the impact of the chemotherapeutic agent gemcitabine as a monotherapy (NCT02992015) [103]. Gemcitabine (difluorodeoxycytidine; dFdCyd) inhibits DNA replication and was found to radio-sensitize a wide range of cancer cell models in vitro [104,105]. Although the mechanism of sensitization is not yet clear, recent evidence suggests that gemcitabine lowers the threshold for radiation-induced apoptosis [106]. An open-label, single-arm phase I/II trial in newly diagnosed DMGs explored the potential and safety of gemcitabine administered concomitantly with RT. This study demonstrated that weekly gemcitabine concomitant with RT is safe. Unfortunately, due to a limited number of patients included, this study failed to draw any conclusions in terms of the efficacy of this combination [107].
Improved mechanistic understanding on the role of EGFR signaling in the onset of DMGs, prompted the use of nimotuzumab, humanized the immunoglobulin G1 (IgG1) anti-EGFR monoclonal antibody in DMGs. Assuming a potential synergy with both RT and vinorelbine (a semi-synthetic vinca alkaloid), Massimino et al. (2014) launched a non-randomized, open label phase II pilot study with the aim of assessing the safety of this combination. The combination was well tolerated [108]. In 2019, a multinational phase III trial evaluated the safety and efficacy of nimotuzumab in combination with RT in newly diagnosed DMGs [109]. This study concluded that concomitant treatment with RT and nimotuzumab was feasible in an outpatient setting, but the progression-free survival and overall survival were not superior compared to the results achieved with RT and intensive chemotherapy in a hospitalized setting [109].
6. Conclusions
With little improvement in the outcome of H3K27M-mutated glioma (DMGs) over the past four decades, median survival rates remain at approximately 9 to 11 months. There is an unmet need for novel treatment strategies in this patient group. We believe that the H3K27M mutation-driven dysregulation of DDR pathways intrinsic to DMGs represents a unique vulnerability that can be leveraged for their sensitization to DNA damaging therapies such as IR. Unfortunately, our understanding of the functional interplay between DNA repair and radio-resistance in DMGs remains poor. A better comprehension of molecular mechanisms underpinning DNA repair, radio-resistance, and the efficacy, as well as safety of radio-sensitizing strategies, represents a critical step preceding a successful therapeutic intervention and improved survival of DMG patients.
Author Contributions
Conceptualization, writing, editing: H.P. and P.H. Writing and editing: K.S. All authors have read and accepted the published version of the manuscript.
Funding
This research was supported by Børne Hjernecancer Fonden.
Acknowledgments
The authors would like to thank Kirstine Juul Elbæk, Alex Frias Hernandez, and Lina Vardouli for proofreading of the manuscript. We would also like to thank René Mathiasen for providing us with MRI pictures that were acquired with valid informed consent.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Johnson, K.J.; Cullen, J.; Barnholtz-Sloan, J.S.; Ostrom, Q.T.; Langer, C.E.; Turner, M.C.; McKean-Cowdin, R.; Fisher, J.L.; Lupo, P.J.; Partap, S.; et al. Childhood brain tumor epidemiology: A brain tumor epidemiology consortium review. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2716–2736. [Google Scholar] [CrossRef]
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A. International incidence of childhood cancer, 2001-10: A population-based registry study. Lancet Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; de Blank, P.M.; Kruchko, C.; Petersen, C.M.; Liao, P.; Finlay, J.L.; Stearns, D.S.; Wolff, J.E.; Wolinsky, Y.; Letterio, J.J.; et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro Oncol. 2015, 16 (Suppl. S10), x1–x36. [Google Scholar] [CrossRef]
- Duchatel, R.J.; Jackson, E.R.; Alvaro, F.; Nixon, B.; Hondermarck, H.; Dun, M.D. Signal Transduction in Diffuse Intrinsic Pontine Glioma. Proteomics 2019, 19, 1800479. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.H.; Veldhuijzen van Zanten, S.E.; Sanchez Aliaga, E.; Heymans, M.W.; Warmuth-Metz, M.; Hargrave, D.; van der Hoeven, E.J.; Gidding, C.E.; de Bont, E.S.; Eshghi, O.S.; et al. Survival prediction model of children with diffuse intrinsic pontine glioma based on clinical and radiological criteria. Neuro Oncol. 2015, 17, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226. Available online: https://www.nature.com/articles/nature10833#supplementary-information (accessed on 12 March 2020). [CrossRef]
- Mathew, R.K.; Rutka, J.T. Diffuse Intrinsic Pontine Glioma: Clinical Features, Molecular Genetics, and Novel Targeted Therapeutics. J. Korean Neurosurg. Soc. 2018, 61, 343–351. [Google Scholar] [CrossRef]
- Panditharatna, E.; Yaeger, K.; Kilburn, L.B.; Packer, R.J.; Nazarian, J. Clinicopathology of diffuse intrinsic pontine glioma and its redefined genomic and epigenomic landscape. Cancer Genet. 2015, 208, 367–373. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e525. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Vitanza, N.A.; Monje, M. Diffuse Intrinsic Pontine Glioma: From Diagnosis to Next-Generation Clinical Trials. Curr. Treat. Options Neurol. 2019, 21, 37. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, T.C.; Campagna, E.J.; Foreman, N.K.; Handler, M.H. Interpretation of magnetic resonance images in diffuse intrinsic pontine glioma: A survey of pediatric neurosurgeons. J. Neurosurg. Pediatrics 2011, 8, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Puget, S.; Blauwblomme, T.; Grill, J. Is Biopsy Safe in Children with Newly Diagnosed Diffuse Intrinsic Pontine Glioma? Am. Soc. Clin. Oncol. Educ. Book 2012, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Sager, O.; Dincoglan, F.; Demiral, S.; Uysal, B.; Gamsiz, H.; Ozcan, F.; Gundem, E.; Elcim, Y.; Ekmen, A.; Onal, E.; et al. Radiation Therapy (RT) for Diffuse Intrinsic Pontine Glioma (DIPG) in Children. Arch. Can. Res. 2018, 6, 14. [Google Scholar] [CrossRef]
- Puget, S.; Beccaria, K.; Blauwblomme, T.; Roujeau, T.; James, S.; Grill, J.; Zerah, M.; Varlet, P.; Sainte-Rose, C. Biopsy in a series of 130 pediatric diffuse intrinsic Pontine gliomas. Childs Nerv. Syst. 2015, 31, 1773–1780. [Google Scholar] [CrossRef]
- Janssens, G.O.; Gandola, L.; Bolle, S.; Mandeville, H.; Ramos-Albiac, M.; van Beek, K.; Benghiat, H.; Hoeben, B.; Morales La Madrid, A.; Kortmann, R.D.; et al. Survival benefit for patients with diffuse intrinsic pontine glioma (DIPG) undergoing re-irradiation at first progression: A matched-cohort analysis on behalf of the SIOP-E-HGG/DIPG working group. Eur. J. Cancer 2017, 73, 38–47. [Google Scholar] [CrossRef]
- Bailey, S.; Howman, A.; Wheatley, K.; Wherton, D.; Boota, N.; Pizer, B.; Fisher, D.; Kearns, P.; Picton, S.; Saran, F.; et al. Diffuse intrinsic pontine glioma treated with prolonged temozolomide and radiotherapy--results of a United Kingdom phase II trial (CNS 2007 04). Eur. J. Cancer 2013, 49, 3856–3862. [Google Scholar] [CrossRef]
- Chassot, A.; Canale, S.; Varlet, P.; Puget, S.; Roujeau, T.; Negretti, L.; Dhermain, F.; Rialland, X.; Raquin, M.A.; Grill, J.; et al. Radiotherapy with concurrent and adjuvant temozolomide in children with newly diagnosed diffuse intrinsic pontine glioma. J. Neuro-Oncol. 2012, 106, 399–407. [Google Scholar] [CrossRef]
- Aquino-Parsons, C.; Hukin, J.; Green, A. Concurrent carbogen and radiation therapy in children with high-risk brainstem gliomas. Pediatric Blood Cancer 2008, 50, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.A.; Zhou, T.; McNall-Knapp, R.Y.; Jakacki, R.I.; Levy, A.S.; Vezina, G.; Pollack, I.F. Motexafin-gadolinium and involved field radiation therapy for intrinsic pontine glioma of childhood: A children’s oncology group phase 2 study. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, e55–e60. [Google Scholar] [CrossRef] [PubMed]
- Gallitto, M.; Lazarev, S.; Wasserman, I.; Stafford, J.M.; Wolden, S.L.; Terezakis, S.A.; Bindra, R.S.; Bakst, R.L. Role of Radiation Therapy in the Management of Diffuse Intrinsic Pontine Glioma: A Systematic Review. Adv. Radiat. Oncol. 2019, 4, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Subashi, E.; Cordero, F.J.; Halvorson, K.G.; Qi, Y.; Nouls, J.C.; Becher, O.J.; Johnson, G.A. Tumor location, but not H3.3K27M, significantly influences the blood-brain-barrier permeability in a genetic mouse model of pediatric high-grade glioma. J. Neuro-Oncol. 2016, 126, 243–251. [Google Scholar] [CrossRef]
- Bredlau, A.L.; Dixit, S.; Chen, C.; Broome, A.-M. Nanotechnology Applications for Diffuse Intrinsic Pontine Glioma. Curr. Neuropharmacol. 2017, 15, 104–115. [Google Scholar] [CrossRef]
- Himes, B.T.; Zhang, L.; Daniels, D.J. Treatment Strategies in Diffuse Midline Gliomas With the H3K27M Mutation: The Role of Convection-Enhanced Delivery in Overcoming Anatomic Challenges. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Souweidane, M.M.; Kramer, K.; Pandit-Taskar, N.; Zhou, Z.; Zanzonico, P.; Donzelli, M.; Lyashchenko, S.K.; Haque, S.; Thakur, S.B.; Cheung, N.-K.V.; et al. A phase I study of convection-enhanced delivery of 124I-8H9 radio-labeled monoclonal antibody in children with diffuse intrinsic pontine glioma: An update with dose-response assessment. J. Clin. Oncol. 2019, 37, 2008. [Google Scholar] [CrossRef]
- Louis, N.; Liu, S.; He, X.; Drummond, D.C.; Noble, C.O.; Goldman, S.; Mueller, S.; Bankiewicz, K.; Gupta, N.; Hashizume, R. New therapeutic approaches for brainstem tumors: A comparison of delivery routes using nanoliposomal irinotecan in an animal model. J. Neurooncol. 2018, 136, 475–484. [Google Scholar] [CrossRef]
- Singleton, W.G.B.; Bienemann, A.S.; Woolley, M.; Johnson, D.; Lewis, O.; Wyatt, M.J.; Damment, S.J.P.; Boulter, L.J.; Killick-Cole, C.L.; Asby, D.J.; et al. The distribution, clearance, and brainstem toxicity of panobinostat administered by convection-enhanced delivery. J. Neurosurg. Pediatr. 2018, 22, 288–296. [Google Scholar] [CrossRef]
- Jones, C.; Karajannis, M.A.; Jones, D.T.W.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.-J.; Gupta, N.; Hawkins, C.; et al. Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro Oncol. 2017, 19, 153–161. [Google Scholar] [CrossRef]
- Nikbakht, H.; Panditharatna, E.; Mikael, L.G.; Li, R.; Gayden, T.; Osmond, M.; Ho, C.-Y.; Kambhampati, M.; Hwang, E.I.; Faury, D.; et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun. 2016, 7, 11185. [Google Scholar] [CrossRef] [PubMed]
- Fontebasso, A.M.; Gayden, T.; Nikbakht, H.; Neirinck, M.; Papillon-Cavanagh, S.; Majewski, J.; Jabado, N. Epigenetic dysregulation: A novel pathway of oncogenesis in pediatric brain tumors. Acta Neuropathol. 2014, 128, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Grill, J.; Debily, M.-A.; Varlet, P.; Plessier, A.; Le Dret, L.; Beccaria, K.; Puget, S.; Castel, D. The dark matter of diffuse intrinsic pontine gliomas: An update. Expert Opin. Orphan Drugs 2019, 7, 11–20. [Google Scholar] [CrossRef]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.W.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef]
- Meel, M.H.; Kaspers, G.J.L.; Hulleman, E. Preclinical therapeutic targets in diffuse midline glioma. Drug Resist. Updates 2019, 44, 15–25. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Paugh, B.S.; Broniscer, A.; Qu, C.; Miller, C.P.; Zhang, J.; Tatevossian, R.G.; Olson, J.M.; Geyer, J.R.; Chi, S.N.; da Silva, N.S.; et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J. Clin. Oncol. 2011, 29, 3999–4006. [Google Scholar] [CrossRef]
- Mueller, S.; Hashizume, R.; Yang, X.; Kolkowitz, I.; Olow, A.K.; Phillips, J.; Smirnov, I.; Tom, M.W.; Prados, M.D.; James, C.D.; et al. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro Oncol. 2014, 16, 352–360. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Zarghooni, M.; Bartels, U.; Morrison, A.; Misuraca, K.L.; Chan, T.; Bouffet, E.; Huang, A.; Becher, O.; Hawkins, C. Aurora Kinase B Is a Potential Therapeutic Target in Pediatric Diffuse Intrinsic Pontine Glioma. Brain Pathol. 2013, 23, 244–253. [Google Scholar] [CrossRef]
- Meel, M.H.; de Gooijer, M.C.; Guillén Navarro, M.; Waranecki, P.; Breur, M.; Buil, L.C.M.; Wedekind, L.E.; Twisk, J.W.R.; Koster, J.; Hashizume, R.; et al. MELK Inhibition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2018, 24, 5645–5657. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-radiation induced DNA double-strand breaks: A direct and indirect lighting up. Radiother. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef]
- Sulli, G.; Di Micco, R.; d’Adda di Fagagna, F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 2012, 12, 709–720. [Google Scholar] [CrossRef]
- Yoo, S.; Dynan, W.S. Geometry of a complex formed by double strand break repair proteins at a single DNA end: Recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res. 1999, 27, 4679–4686. [Google Scholar] [CrossRef]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509. Available online: https://www.nature.com/articles/nature06337#supplementary-information (accessed on 12 March 2020). [CrossRef]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Castedo, M.; Perfettini, J.-L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef]
- Oei, S.L.; Keil, C.; Ziegler, M. Poly(ADP-ribosylation) and genomic stability. Biochem. Cell Biol. 2005, 83, 263–269. [Google Scholar] [CrossRef]
- Caldecott, K.W. XRCC1 and DNA strand break repair. DNA Repair 2003, 2, 955–969. [Google Scholar] [CrossRef]
- Okano, S.; Kanno, S.-i.; Nakajima, S.; Yasui, A. Cellular Responses and Repair of Single-strand Breaks Introduced by UV Damage Endonuclease in Mammalian Cells. J. Biol. Chem. 2000, 275, 32635–32641. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian single-strand break repair: Mechanisms and links with chromatin. DNA Repair 2007, 6, 443–453. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Löbrich, M. DNA double-strand breaks: Their cellular and clinical impact? Oncogene 2007, 26, 7717–7719. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell. Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef]
- Minchom, A.; Aversa, C.; Lopez, J. Dancing with the DNA damage response: Next-generation anti-cancer therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10, 1758835918786658. [Google Scholar] [CrossRef]
- Faria, J.; Romão, L.; Martins, S.; Alves, T.; Mendes, F.A.; de Faria, G.P.; Hollanda, R.; Takiya, C.; Chimelli, L.; Morandi, V.; et al. Interactive properties of human glioblastoma cells with brain neurons in culture and neuronal modulation of glial laminin organization. Differentiation 2006, 74, 562–572. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef]
- Alhajala, H.S.; Nguyen, H.S.; Shabani, S.; Best, B.; Kaushal, M.; Al-Gizawiy, M.M.; Erin Ahn, E.-Y.; Knipstein, J.A.; Mirza, S.; Schmainda, K.M.; et al. Irradiation of pediatric glioblastoma cells promotes radioresistance and enhances glioma malignancy via genome-wide transcriptome changes. Oncotarget 2018, 9, 34122–34131. [Google Scholar] [CrossRef]
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C.; et al. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 2019, 35, 140–155.e147. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Hawkins, C. Pathology, Molecular Genetics, and Epigenetics of Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2015, 5, 147. [Google Scholar] [CrossRef]
- Zarghooni, M.; Bartels, U.; Lee, E.; Buczkowicz, P.; Morrison, A.; Huang, A.; Bouffet, E.; Hawkins, C. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J. Clin. Oncol. 2010, 28, 1337–1344. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Martin, C.-A.; Sarlós, K.; Logan, C.V.; Thakur, R.S.; Parry, D.A.; Bizard, A.H.; Leitch, A.; Cleal, L.; Ali, N.S.; Al-Owain, M.A.; et al. Mutations in TOP3A Cause a Bloom Syndrome-like Disorder. Am. J. Hum. Genet. 2018, 103, 221–231. [Google Scholar] [CrossRef]
- Zhang, Y.; Chang, J.-F.; Sun, J.; Chen, L.; Yang, X.-M.; Tang, H.-Y.; Jing, Y.-Y.; Kang, X.; He, Z.-M.; Wu, J.-Y.; et al. Histone H3K27 methylation modulates the dynamics of FANCD2 on chromatin to facilitate NHEJ and genome stability. J. Cell Sci. 2018, 131, jcs215525. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Khuong-Quang, D.-A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.-Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef]
- Werbrouck, C.; Evangelista, C.C.S.; Lobón-Iglesias, M.-J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, L.H.; Wan, H.; Yang, R.; Wang, Z.; Feng, J.; Yang, S.; Jones, S.; Wang, S.; Zhou, W.; et al. Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat. Genet. 2014, 46, 726–730. [Google Scholar] [CrossRef]
- Shreeram, S.; Demidov, O.N.; Hee, W.K.; Yamaguchi, H.; Onishi, N.; Kek, C.; Timofeev, O.N.; Dudgeon, C.; Fornace, A.J.; Anderson, C.W.; et al. Wip1 Phosphatase Modulates ATM-Dependent Signaling Pathways. Mol. Cell 2006, 23, 757–764. [Google Scholar] [CrossRef]
- Lu, X.; Nannenga, B.; Donehower, L.A. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005, 19, 1162–1174. [Google Scholar] [CrossRef]
- Fujimoto, H.; Onishi, N.; Kato, N.; Takekawa, M.; Xu, X.Z.; Kosugi, A.; Kondo, T.; Imamura, M.; Oishi, I.; Yoda, A.; et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006, 13, 1170–1180. [Google Scholar] [CrossRef]
- Macurek, L.; Lindqvist, A.; Voets, O.; Kool, J.; Vos, H.R.; Medema, R.H. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene 2010, 29, 2281–2291. [Google Scholar] [CrossRef]
- Santivasi, W.L.; Xia, F. Ionizing Radiation-Induced DNA Damage, Response, and Repair. Antioxid. Redox Signal. 2013, 21, 251–259. [Google Scholar] [CrossRef]
- Abe, H.; Natsumeda, M.; Okada, M.; Watanabe, J.; Tsukamoto, Y.; Kanemaru, Y.; Yoshimura, J.; Oishi, M.; Hashizume, R.; Kakita, A.; et al. MGMT Expression Contributes to Temozolomide Resistance in H3K27M-Mutant Diffuse Midline Gliomas. Front. Oncol. 2020, 9, 1568. [Google Scholar] [CrossRef]
- Banan, R.; Christians, A.; Bartels, S.; Lehmann, U.; Hartmann, C. Absence of MGMT promoter methylation in diffuse midline glioma, H3 K27M-mutant. Acta Neuropathol. Commun. 2017, 5, 98. [Google Scholar] [CrossRef]
- Venere, M.; Hamerlik, P.; Wu, Q.; Rasmussen, R.D.; Song, L.A.; Vasanji, A.; Tenley, N.; Flavahan, W.A.; Hjelmeland, A.B.; Bartek, J.; et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014, 21, 258–269. [Google Scholar] [CrossRef]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef]
- Rasmussen, R.D.; Gajjar, M.K.; Tuckova, L.; Jensen, K.E.; Maya-Mendoza, A.; Holst, C.B.; Møllgaard, K.; Rasmussen, J.S.; Brennum, J.; Bartek, J., Jr.; et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat. Commun. 2016, 7, 13398. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, R.D.; Gajjar, M.K.; Jensen, K.E.; Hamerlik, P. Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol. Oncol. 2016, 10, 751–763. [Google Scholar] [CrossRef]
- Chornenkyy, Y.; Agnihotri, S.; Yu, M.; Buczkowicz, P.; Rakopoulos, P.; Golbourn, B.; Garzia, L.; Siddaway, R.; Leung, S.; Rutka, J.T.; et al. Poly-ADP-Ribose Polymerase as a Therapeutic Target in Pediatric Diffuse Intrinsic Pontine Glioma and Pediatric High-Grade Astrocytoma. Mol. Cancer Ther. 2015, 14, 2560. [Google Scholar] [CrossRef] [PubMed]
- Akamandisa, M.P.; Nie, K.; Nahta, R.; Hambardzumyan, D.; Castellino, R.C. Inhibition of mutant PPM1D enhances DNA damage response and growth suppressive effects of ionizing radiation in diffuse intrinsic pontine glioma. Neuro Oncol. 2019, 21, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Smits, V.A.; Klompmaker, R.; Arnaud, L.; Rijksen, G.; Nigg, E.A.; Medema, R.H. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2000, 2, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Van Vugt, M.A.; Bras, A.; Medema, R.H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell. 2004, 15, 799–811. [Google Scholar] [CrossRef]
- Amani, V.; Prince, E.W.; Alimova, I.; Balakrishnan, I.; Birks, D.; Donson, A.M.; Harris, P.; Levy, J.M.M.; Handler, M.; Foreman, N.K.; et al. Polo-like Kinase 1 as a potential therapeutic target in Diffuse Intrinsic Pontine Glioma. BMC Cancer 2016, 16, 647. [Google Scholar] [CrossRef]
- Nikolaev, A.; Fiveash, J.B.; Yang, E.S. Combined Targeting of Mutant p53 and Jumonji Family Histone Demethylase Augments Therapeutic Efficacy of Radiation in H3K27M DIPG. Int. J. Mol. Sci. 2020, 21, 490. [Google Scholar] [CrossRef]
- Ma, Y.; Vassetzky, Y.; Dokudovskaya, S. mTORC1 pathway in DNA damage response. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1293–1311. [Google Scholar] [CrossRef]
- Miyahara, H.; Yadavilli, S.; Natsumeda, M.; Rubens, J.A.; Rodgers, L.; Kambhampati, M.; Taylor, I.C.; Kaur, H.; Asnaghi, L.; Eberhart, C.G.; et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett. 2017, 400, 110–116. [Google Scholar] [CrossRef]
- Yoshida, G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Do, K.; Doroshow, J.H.; Kummar, S. Wee1 kinase as a target for cancer therapy. Cell Cycle 2013, 12, 3348–3353. [Google Scholar] [CrossRef]
- Ginjala, V.; Nacerddine, K.; Kulkarni, A.; Oza, J.; Hill, S.J.; Yao, M.; Citterio, E.; van Lohuizen, M.; Ganesan, S. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol. Cell. Biol. 2011, 31, 1972–1982. [Google Scholar] [CrossRef]
- Kumar, S.S.; Sengupta, S.; Lee, K.; Hura, N.; Fuller, C.; DeWire, M.; Stevenson, C.B.; Fouladi, M.; Drissi, R. BMI-1 is a potential therapeutic target in diffuse intrinsic pontine glioma. Oncotarget 2017, 8, 62962–62975. [Google Scholar] [CrossRef] [PubMed]
- Green, A.L.; Flannery, P.; Hankinson, T.C.; O’Neill, B.; Amani, V.; DeSisto, J.; Knox, A.; Chatwin, H.; Lemma, R.; Hoffman, L.M.; et al. Preclinical and clinical investigation of intratumoral chemotherapy pharmacokinetics in DIPG using gemcitabine. Neuro-Oncol. Adv. 2020, 2. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Kotsantis, P.; Stewart, G.S.; Groth, P.; Petermann, E. BRCA2 and RAD51 promote double-strand break formation and cell death in response to gemcitabine. Mol. Cancer Ther. 2014, 13, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.S.; Eisbruch, A.; McGinn, C.J.; Fields, M.T.; Shewach, D.S. Radiosensitization by gemcitabine. Oncology 1999, 13, 55–60. [Google Scholar] [PubMed]
- Im, M.M.; Flanagan, S.A.; Ackroyd, J.J.; Shewach, D.S. Drug metabolism and homologous recombination repair in radiosensitization with gemcitabine. Radiat. Res. 2015, 183, 114–123. [Google Scholar] [CrossRef]
- Veldhuijzen van Zanten, S.E.M.; El-Khouly, F.E.; Jansen, M.H.A.; Bakker, D.P.; Sanchez Aliaga, E.; Haasbeek, C.J.A.; Wolf, N.I.; Zwaan, C.M.; Vandertop, W.P.; van Vuurden, D.G.; et al. A phase I/II study of gemcitabine during radiotherapy in children with newly diagnosed diffuse intrinsic pontine glioma. J. Neurooncol. 2017, 135, 307–315. [Google Scholar] [CrossRef]
- Massimino, M.; Biassoni, V.; Miceli, R.; Schiavello, E.; Warmuth-Metz, M.; Modena, P.; Casanova, M.; Pecori, E.; Giangaspero, F.; Antonelli, M.; et al. Results of nimotuzumab and vinorelbine, radiation and re-irradiation for diffuse pontine glioma in childhood. J. Neurooncol. 2014, 118, 305–312. [Google Scholar] [CrossRef]
- Fleischhack, G.; Massimino, M.; Warmuth-Metz, M.; Khuhlaeva, E.; Janssen, G.; Graf, N.; Rutkowski, S.; Beilken, A.; Schmid, I.; Biassoni, V.; et al. Nimotuzumab and radiotherapy for treatment of newly diagnosed diffuse intrinsic pontine glioma (DIPG): A phase III clinical study. J. Neuro-Oncol. 2019, 143, 107–113. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).