The Oncoprotein SKI Acts as A Suppressor of NK Cell-Mediated Immunosurveillance in PDAC

 , and
, and 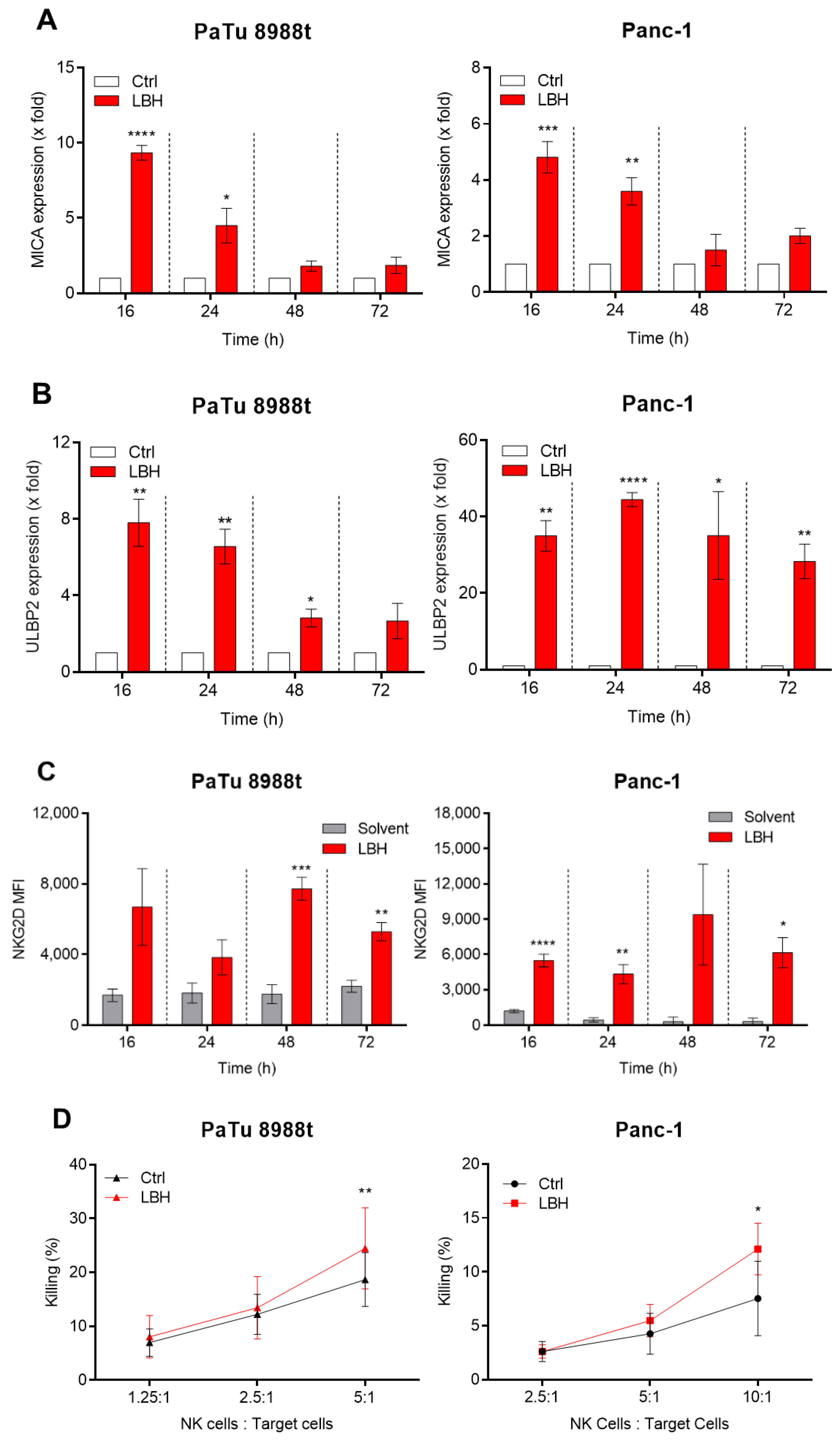{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results and Discussion
3. Material and Methods
3.1. Isolation of Primary NK Cells and Culture of Tumor Cell Lines
3.2. Killing Assay
3.3. Detection of NKG2D-L on Tumor Cells
3.4. Quantitative Real-Time PCR (qRT-PCR) of NKG2D-L
3.5. SKI Knockdown Using siRNA and ChIP-Seq
3.6. Western Blot Analysis
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperb, N.; Tsesmelis, M.; Wirth, T. Crosstalk between Tumor and Stromal Cells in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 5486. [Google Scholar] [CrossRef]
- Schober, M.; Jesenofsky, R.; Faissner, R.; Weidenauer, C.; Hagmann, W.; Michl, P.; Heuchel, R.L.; Haas, S.L.; Löhr, J.-M. Desmoplasia and Chemoresistance in Pancreatic Cancer. Cancers 2014, 6, 2137–2154. [Google Scholar] [CrossRef] [Green Version]
- Griesmann, H.; Drexel, C.; Milosevic, N.; Sipos, B.; Rosendahl, J.; Gress, T.M.; Michl, P. Pharmacological macrophage inhibition decreases metastasis formation in a genetic model of pancreatic cancer. Gut 2016, 66, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Kleeff, J. Immunotherapy of pancreatic cancer. Prog. Mol. Biol. Transl. Sci. 2019, 164, 189–216. [Google Scholar]
- Lim, S.A.; Kim, J.; Jeon, S.; Shin, M.H.; Kwon, J.; Kim, T.-J.; Im, K.; Han, Y.; Kwon, W.; Kim, S.-W.; et al. Defective Localization With Impaired Tumor Cytotoxicity Contributes to the Immune Escape of NK Cells in Pancreatic Cancer Patients. Front. Immunol. 2019, 10, 496. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Huang, M.; Xie, F.; Zhou, H.; Yang, J.; Huang, Q. Gemcitabine inhibits immune escape of pancreatic cancer by down regulating the soluble ULBP2 protein. Oncotarget 2016, 7, 70092–70099. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S. Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of Ligands for the NKG2D Activating Receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [Green Version]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–333. [Google Scholar] [CrossRef]
- Eagle, R.A.; Traherne, J.A.; Hair, J.R.; Jafferji, I.; Trowsdale, J. ULBP6/RAET1L is an additional human NKG2D ligand. Eur. J. Immunol. 2009, 39, 3207–3216. [Google Scholar] [CrossRef] [PubMed]
- Eagle, R.A.; Flack, G.; Warford, A.; Martinez-Borra, J.; Jafferji, I.; Traherne, J.A.; Ohashi, M.; Boyle, L.H.; Barrow, A.D.; Caillat-Zucman, S.; et al. Cellular expression, trafficking, and function of two isoforms of human ULBP5/RAET1G. PLoS ONE 2009, 4, e4503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalupny, N.J.; Sutherland, C.L.; Lawrence, W.A.; Rein-Weston, A.; Cosman, D. ULBP4 is a novel ligand for human NKG2D. Biochem. Biophys. Res. Commun. 2003, 305, 129–135. [Google Scholar] [CrossRef]
- Champsaur, M.; Lanier, L.L. Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 2010, 235, 267–285. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Sauer, M.; Schuldner, M.; Hoffmann, N.; Cetintas, A.; Reiners, K.S.; Shatnyeva, O.; Hallek, M.; Hansen, H.P.; Gasser, S.; Von Strandmann, E.P. CBP/p300 acetyltransferases regulate the expression of NKG2D ligands on tumor cells. Oncogene 2016, 36, 933–941. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Lam, S.S.; Srinath, H.; Schiffer, C.A.; Royer, W.E.; Lin, K. Competition between Ski and CREB-binding Protein for Binding to Smad Proteins in Transforming Growth Factor-beta Signaling. J. Boil. Chem. 2007, 282, 11365–11376. [Google Scholar] [CrossRef] [Green Version]
- Tecalco-Cruz, A.C.; Rios-Lopez, D.G.; Vazquez-Victorio, G.; Rosales-Alvarez, R.E.; Marcias-Silva, M. Transcriptional cofactors Ski and SnoN are major regulators of the TGF-beta/Smad signaling pathway in health and disease. Signal Transduct. Target Ther. 2018, 3, 15. [Google Scholar] [CrossRef] [Green Version]
- Heider, T.R.; Lyman, S.; Schoonhoven, R.; Behrns, K.E. Ski Promotes Tumor Growth Through Abrogation of Transforming Growth Factor-?? Signaling in Pancreatic Cancer. Ann. Surg. 2007, 246, 61–68. [Google Scholar] [CrossRef]
- Wang, P.; Chen, Z.; Meng, Z.; Fan, J.; Luo, J.-M.; Liang, W.; Lin, J.-H.; Zhou, Z.-H.; Chen, H.; Wang, K.; et al. Dual role of Ski in pancreatic cancer cells: Tumor-promoting versus metastasis-suppressive function. Carcinogenesis 2009, 30, 1497–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, P.; Yin, T.; Zhou, F.; Cui, P.; Gou, S.; Wang, C.-Y. Valproic acid sensitizes pancreatic cancer cells to natural killer cell-mediated lysis by upregulating MICA and MICB via the PI3K/Akt signaling pathway. BMC Cancer 2014, 14, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boll, B.; Eltaib, F.; Reiners, K.S.; von Tresckow, B.; Tawadros, S.; Simhadri, V.R.; Burrows, F.; Lundgren, K.; Hansen, H.; Engert, A.; et al. Heat shock protein 90 inhibitor BIIB021 (CNF2024) depletes NF-kappaB and sensitizes Hodgkin’s lymphoma cells for natural killer cell-mediated cytotoxicity. Clin. Cancer Res. 2009, 15, 5108–5116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höring, E.; Podlech, O.; Silkenstedt, B.; Rota, I.A.; Adamopoulou, E.; Naumann, U. The histone deacetylase inhibitor trichostatin a promotes apoptosis and antitumor immunity in glioblastoma cells. Anticancer. Res. 2013, 33, 1351–1360. [Google Scholar] [PubMed]
- Huang, B.; Sikorski, R.; Sampath, P.; Thorne, S.H.; Thorne, S.H. Modulation of NKG2D-ligand Cell Surface Expression Enhances Immune Cell Therapy of Cancer. J. Immunother. 2011, 34, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Cerwenka, A.; Baron, J.L.; Lanier, L.L. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 11521–11526. [Google Scholar] [CrossRef] [Green Version]
- Vyas, M.; Schneider, A.-C.; Shatnyeva, O.; Reiners, K.S.; Tawadros, S.; Kloess, S.; Köhl, U.; Hallek, M.; Hansen, H.P.; Von Strandmann, E.P. Mono- and dual-targeting triplebodies activate natural killer cells and have anti-tumor activity in vitro and in vivo against chronic lymphocytic leukemia. OncoImmunology 2016, 5, e1211220. [Google Scholar] [CrossRef] [Green Version]
- Feld, C.; Sahu, P.; Frech, M.; Finkernagel, F.; Nist, A.; Stiewe, T.; Bauer, U.-M.; Neubauer, A. Combined cistrome and transcriptome analysis of SKI in AML cells identifies SKI as a co-repressor for RUNX1. Nucleic Acids Res. 2018, 46, 3412–3428. [Google Scholar] [CrossRef]
- Masuyama, J.; Murakami, T.; Iwamoto, S.; Fujita, S. Ex vivo expansion of natural killer cells from human peripheral blood mononuclear cells co-stimulated with anti-CD3 and anti-CD52 monoclonal antibodies. Cytotherapy 2016, 18, 80–90. [Google Scholar] [CrossRef]
- Lin, M.; Liang, S.; Wang, X.; Liang, Y.; Zhang, M.; Chen, J.; Niu, L.; Xu, K. Percutaneous irreversible electroporation combined with allogeneic natural killer cell immunotherapy for patients with unresectable (stage III/IV) pancreatic cancer: A promising treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 2607–2618. [Google Scholar] [CrossRef]
- Gürlevik, E.; Fleischmann-Mundt, B.; Brooks, J.; Demir, I.E.; Steiger, K.; Ribback, S.; Yevsa, T.; Woller, N.; Kloos, A.; Ostroumov, D.; et al. Administration of Gemcitabine After Pancreatic Tumor Resection in Mice Induces an Antitumor Immune Response Mediated by Natural Killer Cells. Gastroenterology 2016, 151, 338–350.e7. [Google Scholar]
- Idso, J.M.; Lao, S.; Schloemer, N.J.; Knipstein, J.; Burns, R.; Thakar, M.S.; Malarkannan, S. Entinostat augments NK cell functions via epigenetic upregulation of IFIT1-STING-STAT4 pathway. Oncotarget 2020, 11, 1799–1815. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Johnsen, S.A.; Siveke, J.T.; Ellenrieder, V. Epigenetic treatment of pancreatic cancer: Is there a therapeutic perspective on the horizon? Gut 2016, 66, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Gómez, V.; Mustapha, R.; Ng, K.; Ng, T. Radiation therapy and the innate immune response: Clinical implications for immunotherapy approaches. Br. J. Clin. Pharmacol. 2020, 86, 1726–1735. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponath, V.; Frech, M.; Bittermann, M.; Al Khayer, R.; Neubauer, A.; Brendel, C.; Pogge von Strandmann, E. The Oncoprotein SKI Acts as A Suppressor of NK Cell-Mediated Immunosurveillance in PDAC. Cancers 2020, 12, 2857. https://doi.org/10.3390/cancers12102857
Ponath V, Frech M, Bittermann M, Al Khayer R, Neubauer A, Brendel C, Pogge von Strandmann E. The Oncoprotein SKI Acts as A Suppressor of NK Cell-Mediated Immunosurveillance in PDAC. Cancers. 2020; 12(10):2857. https://doi.org/10.3390/cancers12102857
Chicago/Turabian StylePonath, Viviane, Miriam Frech, Mathis Bittermann, Reem Al Khayer, Andreas Neubauer, Cornelia Brendel, and Elke Pogge von Strandmann. 2020. "The Oncoprotein SKI Acts as A Suppressor of NK Cell-Mediated Immunosurveillance in PDAC" Cancers 12, no. 10: 2857. https://doi.org/10.3390/cancers12102857
APA StylePonath, V., Frech, M., Bittermann, M., Al Khayer, R., Neubauer, A., Brendel, C., & Pogge von Strandmann, E. (2020). The Oncoprotein SKI Acts as A Suppressor of NK Cell-Mediated Immunosurveillance in PDAC. Cancers, 12(10), 2857. https://doi.org/10.3390/cancers12102857