Oncogenic Orphan Nuclear Receptor NR4A3 Interacts and Cooperates with MYB in Acinic Cell Carcinoma

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. AcCC Tumors Harbor Recurrent Translocations Involving the NR4A3 and NR4A2 Genes
2.2. Confirmation of t(4;9) Translocations by Fluorescence In Situ Hybridization
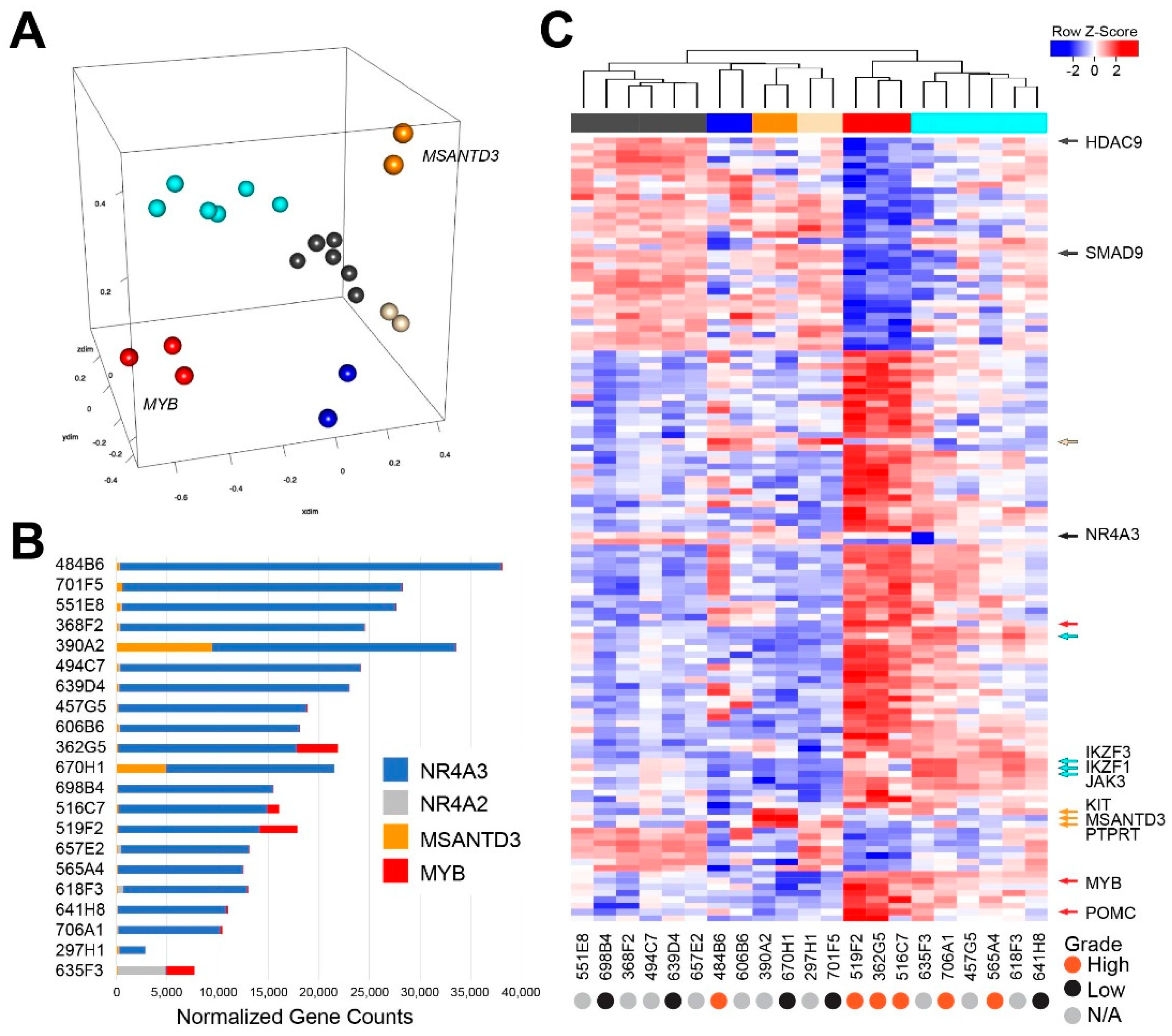
2.3. Transcriptome Analysis Reveals Heterogeneity in Acinic Cell Carcinoma
2.4. Gene Expression Analysis Reveals a Poor Survival Subgroup of AcCC Tumors
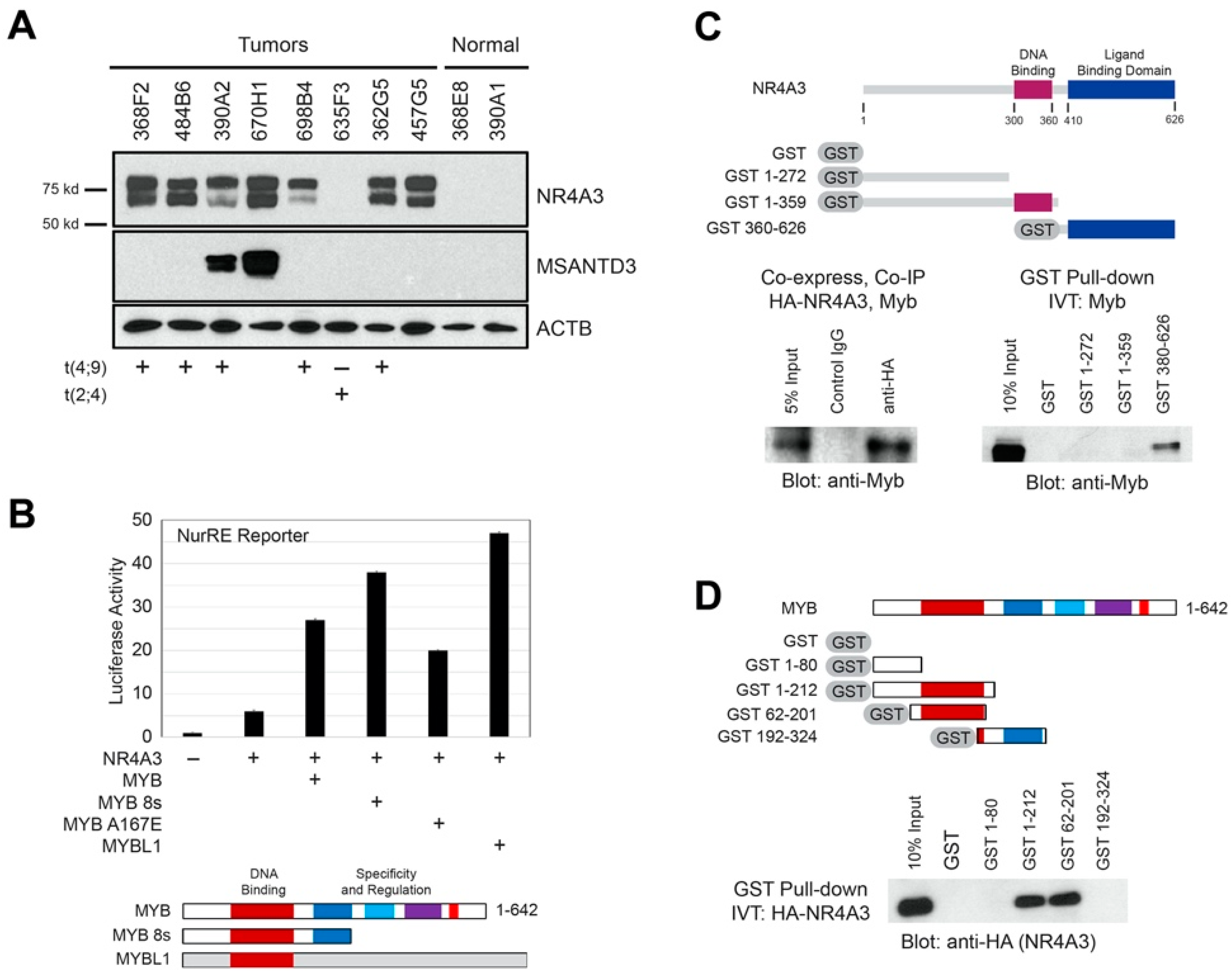
2.5. AcCC Tumors with t(4;9) Overexpress NR4A3 Protein, which Interacts with Myb
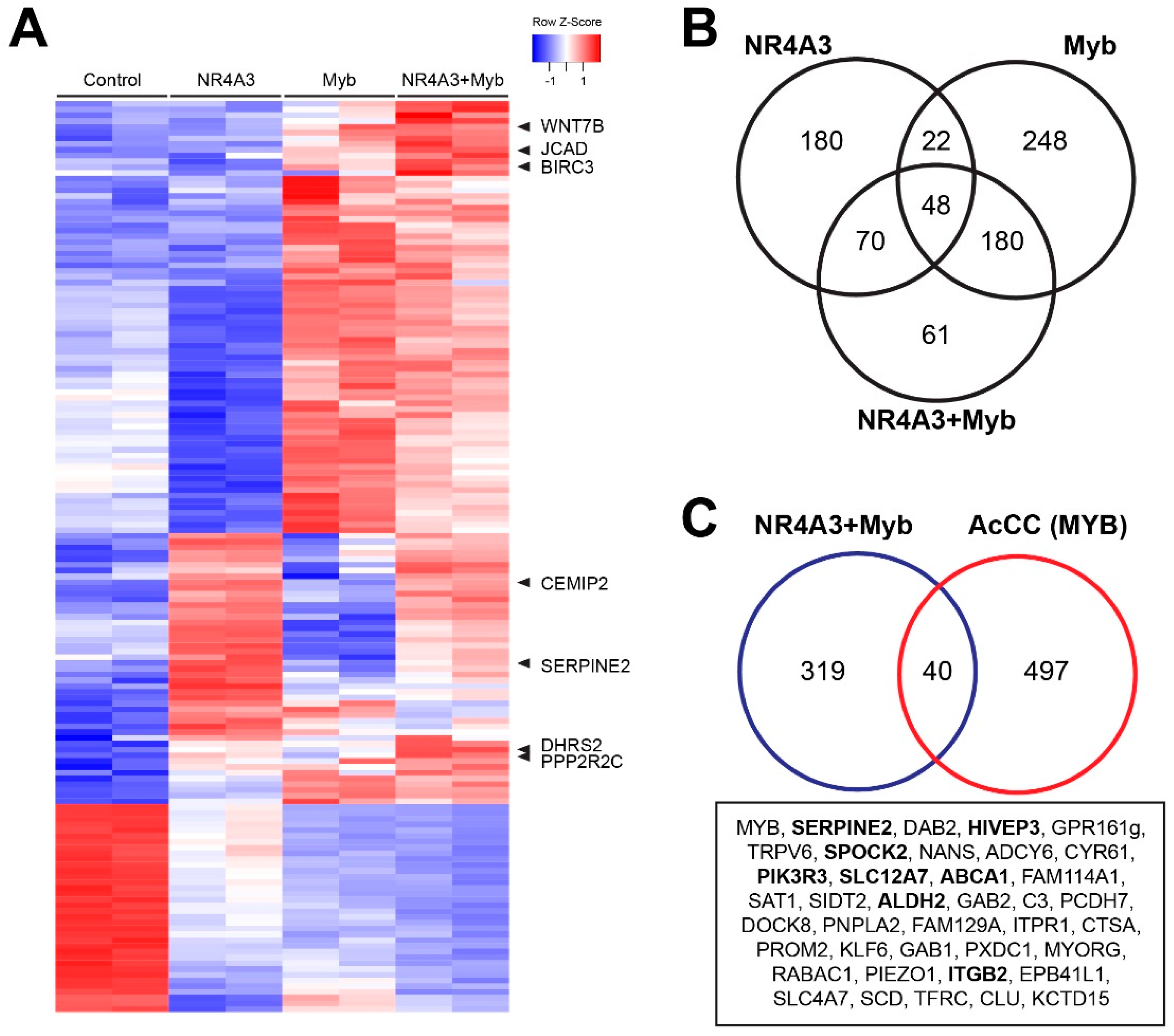
2.6. Transcriptional Cooperativity between NR4A3 and Myb
2.7. Cooperative Gene Activation by NR4A3 and Myb
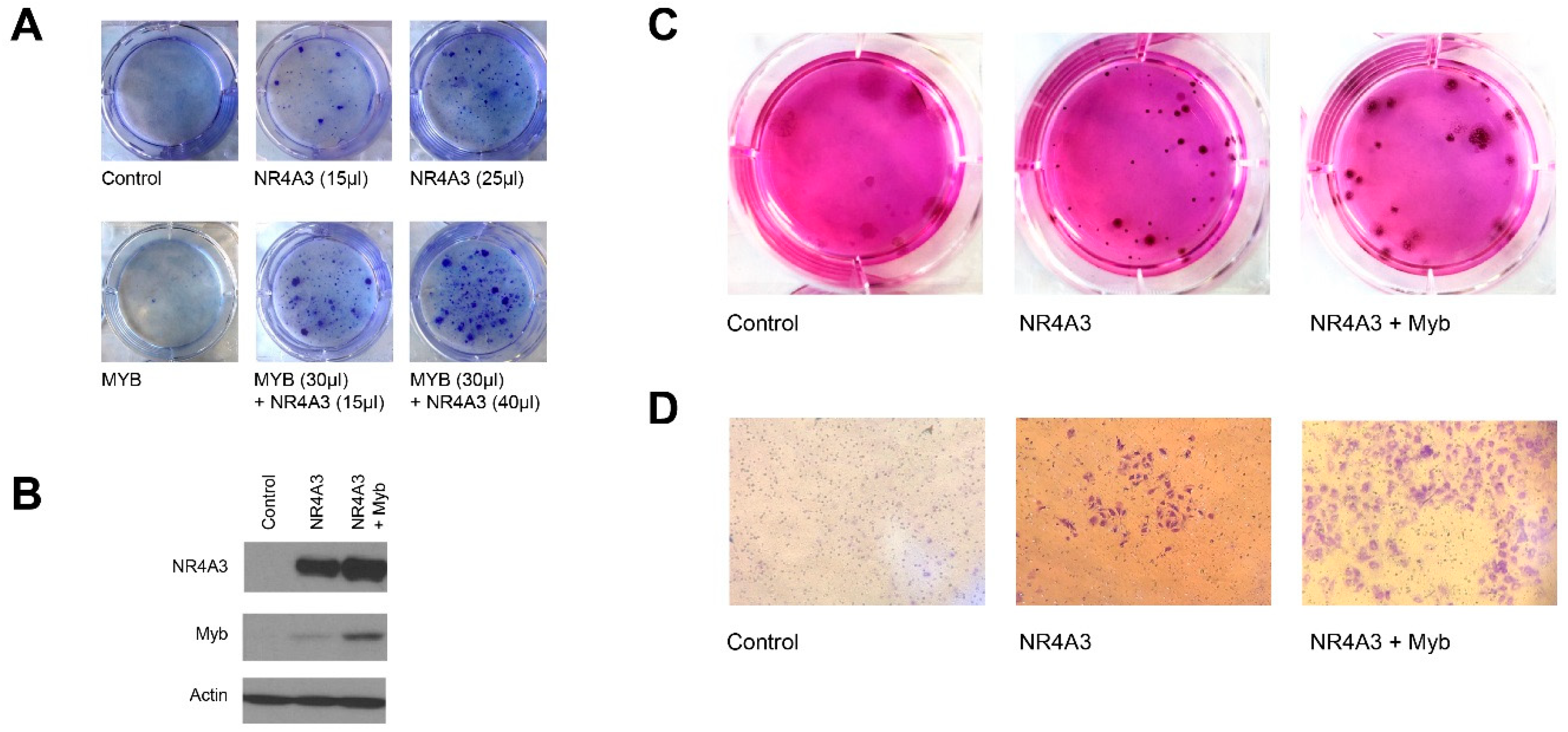
2.8. NR4A3 and Myb Cooperate in Colony Formation Assays
3. Discussion
4. Materials and Methods
4.1. Tissue Collection
4.2. RNA Isolation and Sequencing
4.3. Data Analysis
4.4. Fluorescence In Situ Hybridization (FISH)
4.5. Protein Analysis by Western Blot
4.6. Plasmids
4.7. Cell Culture and Transfections
4.8. Co-immunoprecipitation and GST-Pulldown Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luna, M.A.; Batsakis, J.G.; El-Naggar, A.K. Salivary Gland Tumors in Children. Ann. Otol. Rhinol. Laryngol. 1991, 100, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, N.; Bhattacharyya, N. Clinical Characteristics and Survival for Major Salivary Gland Malignancies in Children. Otolaryngol. Head Neck Surg. 2006, 134, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Batsakis, J.G.; Regezi, J.A.; Luna, M.A.; El-Naggar, A. Histogenesis of salivary gland neoplasms: A postulate with prognostic implications. J. Laryngol. Otol. 1989, 103, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Poorten, V.V.; Triantafyllou, A.; Thompson, L.D.R.; Bishop, J.; Hauben, E.; Hunt, J.; Skalova, A.; Stenman, G.; Takes, R.P.; Gnepp, D.R.; et al. Salivary acinic cell carcinoma: Reappraisal and update. Eur. Arch. Oto Rhino Laryngol. 2015, 273, 3511–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neskey, D.M.; Klein, J.D.; Hicks, S.C.; Garden, A.S.; Bell, D.M.; El-Naggar, A.K.; Kies, M.S.; Weber, R.S.; Kupferman, M.E. Prognostic factors associated with decreased survival in patients with acinic cell carcinoma. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.R.; Sanghvi, S.; Khan, M.N.; Husain, Q.; Baredes, S.; Eloy, J.A. Demographic trends and disease-specific survival in salivary acinic cell carcinoma: An analysis of 1129 cases. Laryngoscope 2014, 124, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Zenga, J.; Parikh, A.S.; Emerick, K.S.; Lin, D.T.; Faquin, W.C.; Deschler, D.G. Close Margins and Adjuvant Radiotherapy in Acinic Cell Carcinoma of the Parotid Gland. JAMA Otolaryngol. Head Neck Surg. 2018, 144, 1011–1016. [Google Scholar] [CrossRef]
- Barasch, N.; Gong, X.; Kwei, K.A.; Varma, S.; Biscocho, J.; Qu, K.; Xiao, N.; Lipsick, J.S.; Pelham, R.J.; West, R.B.; et al. Recurrent rearrangements of the Myb/SANT-like DNA-binding domain containing 3 gene (MSANTD3) in salivary gland acinic cell carcinoma. PLoS ONE 2017, 12, e0171265. [Google Scholar] [CrossRef]
- Haller, F.; Bieg, M.; Will, R.; Körner, C.; Weichenhan, D.; Bott, A.; Ishaque, N.; Lutsik, P.; Moskalev, E.A.; Mueller, S.K.; et al. Enhancer hijacking activates oncogenic transcription factor NR4A3 in acinic cell carcinomas of the salivary glands. Nat. Commun. 2019, 10, 368. [Google Scholar] [CrossRef]
- Haller, F.; Moskalev, E.A.; Kuck, S.; Bieg, M.; Winkelmann, C.; Müller, S.K.; Ihrler, S.; Märkl, B.; Eils, R.; Wiemann, S.; et al. Nuclear NR4A2 (Nurr1) Immunostaining is a Novel Marker for Acinic Cell Carcinoma of the Salivary Glands Lacking the Classic NR4A3 (NOR-1) Upregulation. Am. J. Surg. Pathol. 2020, 44, 1290–1292. [Google Scholar] [CrossRef]
- Haller, F.; Skálová, A.; Ihrler, S.; Märkl, B.; Bieg, M.; Moskalev, E.A.; Erber, R.; Blank, S.; Winkelmann, C.; Hebele, S.; et al. Nuclear NR4A3 Immunostaining Is a Specific and Sensitive Novel Marker for Acinic Cell Carcinoma of the Salivary Glands. Am. J. Surg. Pathol. 2019, 43, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Campos-Melo, D.; Galleguillos, D.; Sánchez, N.; Gysling, K.; Andrés, M.E. Nur transcription factors in stress and addiction. Front. Mol. Neurosci. 2013, 6, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safe, S.; Jin, U.-H.; Hedrick, E.; Reeder, A.; Lee, S.-O. Minireview: Role of orphan nuclear receptors in cancer and potential as drug targets. Mol. Endocrinol. 2014, 28, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Northrop, J.P.; Kuo, M.-H.; Stallcup, M.R. Histone H3 Lysine 9 Methyltransferase G9a Is a Transcriptional Coactivator for Nuclear Receptors. J. Biol. Chem. 2006, 281, 8476–8485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, D.J.; Chauhan, S.; Jimenez-Stinson, D.; Elliott, K.R.; Tsewang, T.D.; Lee, Y.-H.; Marples, B.; Lee, D.Y. Novel CARM1-Interacting Protein, DZIP3, Is a Transcriptional Coactivator of Estrogen Receptor-α. Mol. Endocrinol. 2015, 29, 1708–1719. [Google Scholar] [CrossRef] [Green Version]
- Agaram, N.P.; Zhang, L.; Sung, Y.-S.; Singer, S.; Antonescu, C.R. Extraskeletal myxoid chondrosarcoma with non-EWSR1-NR4A3 variant fusions correlate with rhabdoid phenotype and high-grade morphology. Hum. Pathol. 2014, 45, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Broehm, C.J.; Wu, J.; Gullapalli, R.R.; Bocklage, T. Extraskeletal myxoid chondrosarcoma with a t(9;16)(q22;p11.2) resulting in a NR4A3-FUS fusion. Cancer Genet. 2014, 207, 276–280. [Google Scholar] [CrossRef]
- Davis, E.J.; Wu, Y.-M.; Robinson, D.; Schuetze, S.M.; Baker, L.H.; Athanikar, J.; Cao, X.; Kunju, L.P.; Chinnaiyan, A.M.; Chugh, R. Next generation sequencing of extraskeletal myxoid chondrosarcoma. Oncotarget 2017, 8, 21770–21777. [Google Scholar] [CrossRef] [Green Version]
- Brayer, K.J.; Frerich, C.A.; Kang, H.; Ness, S.A. Recurrent Fusions in MYB and MYBL1 Define a Common, Transcription Factor-Driven Oncogenic Pathway in Salivary Gland Adenoid Cystic Carcinoma. Cancer Discov. 2016, 6, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.B.; Madrid, N.J.; Suzuki, H.; Ness, S.A. Optimized approach for Ion Proton RNA sequencing reveals details of RNA splicing and editing features of the transcriptome. PLoS ONE 2017, 12, e0176675. [Google Scholar] [CrossRef] [Green Version]
- Frerich, C.A.; Brayer, K.J.; Painter, B.M.; Kang, H.; Mitani, Y.; El-Naggar, A.K.; Ness, S.A. Transcriptomes define distinct subgroups of salivary gland adenoid cystic carcinoma with different driver mutations and outcomes. Oncotarget 2018, 9, 7341–7358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labelle, Y.; Zucman-Rossi, J.; Stenman, G.; Kindblom, L.-G.; Knight, J.; Turc-Carel, C.; Dockhorn-Dworniczak, B.; Mandahl, N.; Desmaze, C.; Peter, M.; et al. Oncogenic conversion of a novel orphan nuclear receptor by chromosome translocation. Hum. Mol. Genet. 1995, 4, 2219–2226. [Google Scholar] [CrossRef] [PubMed]
- Drouin, J. 60 YEARS OF POMC: Transcriptional and epigenetic regulation of POMC gene expression. J. Mol. Endocrinol. 2016, 56, T99–T112. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wei, H.; Yan, N.; Dai, S.; Li, J.; Qu, L.; Chen, X.; Guo, M.; Chen, Z.; Chen, Y. Structural basis of NR4A1 bound to the human pituitary proopiomelanocortin gene promoter. Biochem. Biophys. Res. Commun. 2020, 523, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Giraldi, F.P.; Cassarino, F.; Pagliardini, L.; Asnaghi, V.; Cavagnini, F. The human POMC gene promoter: Where do we stand? J. Endocrinol. Investig. 2011, 34, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, M.; Yashiro, T.; Uchida, Y.; Ando, T.; Hara, M.; Arai, H.; Ogawa, H.; Okumura, K.; Kasakura, K.; Nishiyama, C. The Orphan Nuclear Receptor NR4A3 Is Involved in the Function of Dendritic Cells. J. Immunol. 2017, 199, 2958–2967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wansa, K.D.S.A.; Harris, J.M.; Yan, G.; Ordentlich, P.; Muscat, G.E. The AF-1 Domain of the Orphan Nuclear Receptor NOR-1 Mediates Trans-activation, Coactivator Recruitment, and Activation by the Purine Anti-metabolite 6-Mercaptopurine. J. Biol. Chem. 2003, 278, 24776–24790. [Google Scholar] [CrossRef] [Green Version]
- Maira, M.; Martens, C.; Batsché, E.; Gauthier, Y.; Drouin, J. Dimer-Specific Potentiation of NGFI-B (Nur77) Transcriptional Activity by the Protein Kinase A Pathway and AF-1-Dependent Coactivator Recruitment. Mol. Cell. Biol. 2003, 23, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Maira, M.; Martens, C.; Philips, A.; Drouin, J. Heterodimerization between Members of the Nur Subfamily of Orphan Nuclear Receptors as a Novel Mechanism for Gene Activation. Mol. Cell. Biol. 1999, 19, 7549–7557. [Google Scholar] [CrossRef] [Green Version]
- Frampton, J.; Gibson, T.; Ness, S.A.; Döderlein, G.; Graf, T. Proposed structure for the DNA-binding domain of the Myb oncoprotein based on model building and mutational analysis. Protein Eng. Des. Sel. 1991, 4, 891–901. [Google Scholar] [CrossRef]
- Tonon, G.; Modi, S.; Wu, L.; Kubo, A.; Coxon, A.B.; Komiya, T.; O’Neil, K.; Stover, K.; El-Naggar, A.; Griffin, J.D.; et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat. Genet. 2003, 33, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, J.A.; Tenga, A.; Chen, T. The interplay of NR4A receptors and the oncogene–tumor suppressor networks in cancer. Cell. Signal. 2015, 27, 257–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranhotra, H.S. The NR4A orphan nuclear receptors: Mediators in metabolism and diseases. J. Recept. Signal Transduct. 2014, 35, 184–188. [Google Scholar] [CrossRef]
- Murphy, E.P.; Crean, D. Molecular Interactions between NR4A Orphan Nuclear Receptors and NF-κB Are Required for Appropriate Inflammatory Responses and Immune Cell Homeostasis. Biomolecules 2015, 5, 1302–1318. [Google Scholar] [CrossRef] [Green Version]
- Mulholland, D.J.; Dedhar, S.; Coetzee, G.A.; Nelson, C.C. Interaction of Nuclear Receptors with the Wnt/β-Catenin/Tcf Signaling Axis: Wnt You Like to Know? Endocr. Rev. 2005, 26, 898–915. [Google Scholar] [CrossRef]
- Rajalin, A.-M.; Aarnisalo, P. Cross-talk between NR4A orphan nuclear receptors and β-catenin signaling pathway in osteoblasts. Arch. Biochem. Biophys. 2011, 509, 44–51. [Google Scholar] [CrossRef]
- Lin, H.H.; Tu, Z.-J.; Ann, D.K. Involvement of Nuclear Orphan Receptor NGFI-B in Transcriptional Activation of Salivary-specific R15 Gene by cAMP. J. Biol. Chem. 1996, 271, 27637–27644. [Google Scholar] [CrossRef] [Green Version]
- Mohan, H.M.; Aherne, C.M.; Rogers, A.; Baird, A.W.; Winter, D.C.; Murphy, E.P. Molecular Pathways: The Role of NR4A Orphan Nuclear Receptors in Cancer. Clin. Cancer Res. 2012, 18, 3223–3228. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, H.; Mitsuhashi, T.; Seki, K.; Tochigi, N.; Tsuji, M.; Shimoda, T.; Hasegawa, T. Fluorescence in situ hybridization analysis of extraskeletal myxoid chondrosarcomas using EWSR1 and NR4A3 probes. Hum. Pathol. 2010, 41, 336–342. [Google Scholar] [CrossRef]
- Tessem, J.S.; Moss, L.G.; Chao, L.C.; Arlotto, M.; Lu, D.; Jensen, M.V.; Stephens, S.B.; Tontonoz, P.; Hohmeier, H.E.; Newgard, C.B. Nkx6.1 regulates islet β-cell proliferation via Nr4a1 and Nr4a3 nuclear receptors. Proc. Natl. Acad. Sci. USA 2014, 111, 5242–5247. [Google Scholar] [CrossRef] [Green Version]
- Weyrich, P.; Staiger, H.; Stančáková, A.; Schäfer, S.A.; Kirchhoff, K.; Ullrich, S.; Ranta, F.; Gallwitz, B.; Stefan, N.; Machicao, F.; et al. Common polymorphisms within the NR4A3 locus, encoding the orphan nuclear receptor Nor-1, are associated with enhanced β-cell function in non-diabetic subjects. BMC Med. Genet. 2009, 10, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscat, G.E.; Eriksson, N.A.; Byth, K.; Loi, S.; Graham, D.; Jindal, S.; Davis, M.J.; Clyne, C.; Funder, J.W.; Simpson, E.R.; et al. Research Resource: Nuclear Receptors as Transcriptome: Discriminant and Prognostic Value in Breast Cancer. Mol. Endocrinol. 2013, 27, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Wenzl, K.; Troppan, K.; Neumeister, P.; Deutsch, A.J. The Nuclear Orphan Receptor NR4A1 and NR4A3 as Tumor Suppressors in Hematologic Neoplasms. Curr. Drug Targets 2015, 16, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Borup, R.; Rossing, M.; Henao, R.; Yamamoto, Y.; Krogdahl, A.; Godballe, C.; Winther, O.; Kiss, K.; Christensen, L.; Høgdall, E.V.S.; et al. Molecular signatures of thyroid follicular neoplasia. Endocr. Relat. Cancer 2010, 17, 691–708. [Google Scholar] [CrossRef]
- Mullican, S.E.; Zhang, S.; Konopleva, M.; Ruvolo, V.; Andreeff, M.; Milbrandt, J.; Conneely, O.M. Abrogation of nuclear receptors Nr4a3 andNr4a1 leads to development of acute myeloid leukemia. Nat. Med. 2007, 13, 730–735. [Google Scholar] [CrossRef]
- Zhou, L.; Ruvolo, V.R.; McQueen, T.; Chen, W.; Samudio, I.J.; Conneely, O.; Konopleva, M.; Andreeff, M. HDAC inhibition by SNDX-275 (Entinostat) restores expression of silenced leukemia-associated transcription factors Nur77 and Nor1 and of key pro-apoptotic proteins in AML. Leukemia 2012, 27, 1358–1368. [Google Scholar] [CrossRef] [Green Version]
- Kagaya, S.; Ohkura, N.; Tsukada, T.; Miyagawa, M.; Sugita, Y.; Tsujimoto, G.; Matsumoto, K.; Saito, H.; Hashida, R. Prostaglandin A2 Acts as a Transactivator for NOR1 (NR4A3) within the Nuclear Receptor Superfamily. Biol. Pharm. Bull. 2005, 28, 1603–1607. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Dobin, A.; Gingeras, T.R. Optimizing RNA-Seq Mapping with STAR. In Data Mining Techniques for the Life Sciences; Humana Press: New York, NY, USA, 2016; pp. 245–262. [Google Scholar] [CrossRef]
- Homer, N.; Lyons, M.; Shah, M. Torrent Mapping Alignment Program. Available online: https://github.com/iontorrent/TMAP (accessed on 21 February 2019).
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Briefings Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Karolchik, N.; Hinrichs, A.S.; Kent, W.J. The UCSC Genome Browser. Curr. Protoc. Bioinform. 2007, 17, 1.4.1–1.4.24. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Koh, S.S.; Chen, D.; Lee, Y.-H.; Stallcup, M.R. Synergistic Enhancement of Nuclear Receptor Function by p160 Coactivators and Two Coactivators with Protein Methyltransferase Activities. J. Biol. Chem. 2001, 276, 1089–1098. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Number (%) | Fusion | p-Value | |
|---|---|---|---|---|
| (+) | (−) | |||
| Age | ||||
| 35< | 3 (14) | 2 | 1 | 0.39 |
| 35≥ | 18 (86) | 16 | 2 | |
| Sex | ||||
| Male | 4 (19) | 3 | 1 | 1.00 |
| Female | 17 (81) | 15 | 2 | |
| Tumor size | ||||
| 3 cm< | 12 (57) | 11 | 1 | |
| 3 cm≥ | 9 (43) | 7 | 2 | 0.55 |
| Rec/met | ||||
| yes | 5 (24) | 5 | 0 | 1.00 |
| no | 16 (76) | 13 | 3 | |
| 3-year survival | ||||
| alive | 17 (81) | 15 | 2 | 1.00 |
| dead | 4 * (17) | 3 | 1 | |
| Translocation | Case Number (%) | Fusion Type | Number |
|---|---|---|---|
| t(4;9) | 12 (57) | HTN3-NR4A3 | 3 * |
| STATH-NR4A3 | 6 | ||
| CSN2-NR4A3 | 1 | ||
| FDCSP-NR4A3 | 1 | ||
| CSN1S1-NR4A3 HTN3-MSANTD3 | 1 2 ** | ||
| t(9:12) | 4 (19) | PRH1-NR4A3 | 1 |
| PRB3-NR4A3 | 2 * | ||
| PRB1-STX17 | 1 | ||
| t(8;9) | 1 (5) | OXR1-NR4A3 | 1 |
| t(2;4) | 1 (5) | HTN3-NR4A2 | 1 |
| None | 3 (14) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.Y.; Brayer, K.J.; Mitani, Y.; Burns, E.A.; Rao, P.H.; Bell, D.; Williams, M.D.; Ferrarotto, R.; Pytynia, K.B.; El-Naggar, A.K.; et al. Oncogenic Orphan Nuclear Receptor NR4A3 Interacts and Cooperates with MYB in Acinic Cell Carcinoma. Cancers 2020, 12, 2433. https://doi.org/10.3390/cancers12092433
Lee DY, Brayer KJ, Mitani Y, Burns EA, Rao PH, Bell D, Williams MD, Ferrarotto R, Pytynia KB, El-Naggar AK, et al. Oncogenic Orphan Nuclear Receptor NR4A3 Interacts and Cooperates with MYB in Acinic Cell Carcinoma. Cancers. 2020; 12(9):2433. https://doi.org/10.3390/cancers12092433
Chicago/Turabian StyleLee, David Y., Kathryn J. Brayer, Yoshitsugu Mitani, Eric A. Burns, Pulivarthi H. Rao, Diana Bell, Michelle D. Williams, Renata Ferrarotto, Kristen B. Pytynia, Adel K. El-Naggar, and et al. 2020. "Oncogenic Orphan Nuclear Receptor NR4A3 Interacts and Cooperates with MYB in Acinic Cell Carcinoma" Cancers 12, no. 9: 2433. https://doi.org/10.3390/cancers12092433