1. Introduction
Metastatic disease contributes to 66–90% of cancer deaths [
1,
2]; therefore, understanding the complex interactions of cancer cells and their microenvironment at the primary and metastatic sites will be vital in providing effective treatment. The process of metastasis is multi-step, complex, and significantly driven by the microenvironment. Cells are embedded in a 3D scaffold within tissues known as the extra-cellular matrix (ECM) [
3]. Cell-ECM interactions play a fundamental role in the proper regulation of cell behaviour and homeostasis, especially in response to environmental changes and stresses [
4]. In particular, cancer-cell–ECM interactions are critical for metastasis since cancer cells must interact with ECM at every stage of the metastatic cascade, eventually attaching to the ECM of the secondary organ to initiate growth; the so-called seed (cancer cells) and soil (organ ECM) hypothesis [
5].
The tumour microenvironment is comprised of many different cell types, which together interact and regulate the ECM. Fibroblasts are highly involved in the production and remodelling of the ECM in vivo, and during normal development and physiology, these cells produce mainly connective tissue ECM [
6,
7]. Specifically, ECM at distinct organ niches is different in composition, structure, and biophysical properties, often related to function in normal physiology. The ECM composition of the lung, for example, is rich in fibrillar collagens and elastin, which are important to allow the necessary ‘recoil’ [
8]. At the primary site in breast cancer, the tumour’s ECM is often stiffer [
9] and characterised by an increase of collagen I remodelling and deposition, which facilitates local invasion due to increased linearisation of fibres [
10]. During tumorigenesis, resident fibroblasts become ‘activated’, similar to when they are activated during normal wound healing [
11]. The global composition of the ECM has been catalogued using both in silico and biophysical approaches leading to what is known as the ‘matrisome’; a database containing 1056 genes describing 278 ‘core’ ECM proteins [
12]. Recently it has become possible to produce native ECM from fibroblasts in vitro and use the ECM that they produce in cancer investigations [
13].
Cells interact with the ECM via integrins and glycoproteins, which in turn transduce signals internally, leading to the activation of intracellular kinases [
4]. Kinase activation via protein phosphorylation within cells is a key post-translational modification that is involved in the regulation of intracellular signalling, which leads to the induction of gene expression, metabolic changes, proliferation, differentiation, and membrane transport [
14]. Over 500 kinases are identified in the human genome [
15] and are divided into two categories: tyrosine kinases (PTKs) and serine/threonine kinases (STKs) [
16]. Tyrosine kinases transfer a phosphate molecule from ATP to the tyrosine residue [
17]. Aberrant activation of receptor tyrosine kinases has been described in carcinogenesis, examples of which include the overexpression of human epidermal growth factor receptor (EFGR) and platelet-derived growth factor receptor α/β (PDGFR α/β) [
18,
19]. There are currently (as of September 2020) 52 receptor tyrosine kinase inhibitors currently approved by the US-FDA, 46 of which are used in the treatment of neoplastic diseases [
20], indicating an increased use and focus within cancer treatment.
There has been an increasing number of in vitro approaches to anti-cancer drug testing, often balancing increasing complexity that better represents tumours and the ability to screen at a high throughput level. These approaches range from simple viability assays, where cancer cells on plastic are treated with a prospective compound, and cytotoxicity is measured [
21], to complex patient-derived 3D multicellular cultures [
22]. Issues with cancer cells treated on a two-dimensional monolayer are that they often do not reflect complex three-dimensional tissue architecture or cell–ECM interactions [
23]. However, with increasing complexity and dimensionality, issues with maintenance and control during testing lead to decreased screening capacity. Therefore, it is crucial to continue to investigate new in vitro experimental approaches that consider the importance of the tumour microenvironment and especially the ECM in anti-cancer drug testing.
Many breast cancer types have a poorer clinical outcome if the disease has progressed to stage IV (metastatic disease) [
24,
25]. Breast cancer typically metastasises to the lymph nodes, lungs, liver, bones, and brain, and currently, treatment options for distant metastasis include surgery, radiation, and a small number of chemotherapeutic agents [
26]. However, these treatments often have limited efficacy and impact on patient survival.
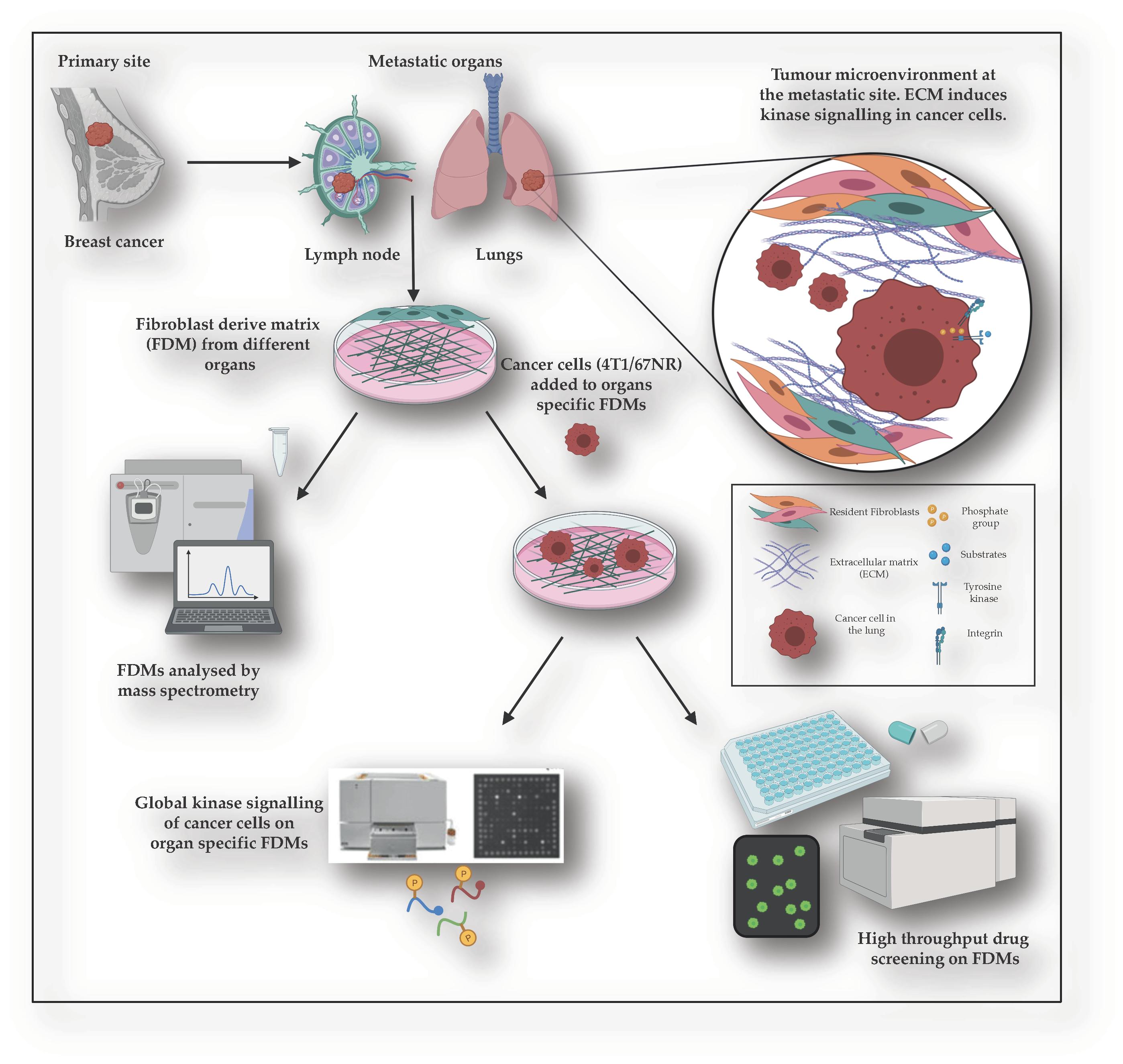
In this study, we have focused on breast cancer and used the highly metastatic 4T1 mouse mammary carcinoma cell line (and 67NR non-metastatic counterpart) [
27], which readily metastasises to lymph nodes, lungs, and liver, and most closely resembles the basal/TNBC (triple-negative breast cancer) breast cancer subtype [
28]. Here, we have used fibroblasts isolated from mouse lymph node, lung, and mammary gland tissue to generate 3D fibroblast-derived matrices (FDMs) that closely resemble in vivo mesenchymal matrices [
13,
29]. We can use these FDMs to expose breast cancer cells to different ECM organ environments (primary and metastatic sites), analyse induced global kinase signalling in the cancer cells, the proliferation of cancer cells, and importantly, conduct a large-scale drug screening.
In this study, we show that despite differences in FDM composition between primary and metastatic sites, global kinase signalling induced in breast cancer cells on FDMs remains relatively unchanged between different organ ECMs. We observe that when exposed to the ECM, 4T1 breast cancer cells induce different global kinase signalling signatures comparative to plastic, which correlates with drug response. Importantly, we reveal that the drug response differs significantly between cancer cells treated on the FDMs and plastic, calling into question the relevance of using plastic for large-scale drug screening in the pre-clinical setting. We show that while the changes in lung/mammary FDM we produce overlap nicely with in vivo protein composition of the lung vs. mammary decellularised organs, we can elucidate similar responses in kinase signalling and drug response when breast cancer cells are exposed to the ‘simpler’ commercially available matrices. This paves the way for simple improvements in drug testing that considers the important role of the ECM.
2. Materials and Methods
2.1. Cell Lines and Cell Culture
4T1 and 67NR cancer cell lines were kindly gifted by Fred Miller (Wayne State University) and cultured in Dulbecco’s modified Eagle medium glutaMAX (DMEM GlutaMAX; Gibco, Thermo Fisher Scientific, cat. no. 10566016, Grand Island, NY, USA) supplemented with 1% penicillin-streptomycin (100 U/mL, Gibco, Thermo Fisher Scientific) and 10% foetal bovine serum (FBS, Gibco, Thermo Fisher Scientific). The 4T1-H2B-GFP+ cells were previously generated by stable transfection of 4T1 cells with a pBOS-H2BGFP vector (BD Pharmingen, San Jose, CA, USA) and were cultured in the same conditions as the 4T1 cells.
BALB/c primary lung (cat. no. BALB-5013), lymph (cat. no. BALB-5070) and mammary (cat. no. BALB-5072) fibroblasts (isolated from healthy mice) were purchased from Cell Biologics (Chicago, IL, USA) and were cultured in Complete Fibroblast Medium with supplement kit (cat. no. M2267, Cell Biologics), which includes 0.5 mL FGF (Fibroblast Growth Factor), 0.5 mL Hydrocortisone, 5 mL L-Glutamine, 5 mL Antibiotic Solution and 50 mL FBS. Primary fibroblasts were cultured on 0.2% gelatin (Sigma-Aldrich, Saint Louis, MO, USA, Gelatin solution, Type B, 2% in H2O, G1393-100 mL) coated cell culture dishes. Mouse cancer-associated mammary fibroblasts, mCAF1, were a kind gift from Erik Sahai (The Francis Crick Institute, London, UK). They were isolated from a mammary fat pad of a tumour bearing FVB/n MMTV-PyMT mouse line and immortalised. mCAF1s were cultured in DMEM high glucose (Gibco, Thermo Fisher Scientific) with 10% FBS, 1% Insulin Transferrin Selenium Solution (ITS-G; Gibco, Thermo Fisher Scientific) and 1% penicillin-streptomycin (100 U/mL, Gibco, Thermo Fisher Scientific). All cell lines were maintained at 37 °C, in a humidified atmosphere with 5% CO2 and were routinely tested for mycoplasma.
2.2. Generating Fibroblast-Derived Matrices
Fibroblast-derived matrices (FDMs) were generated as previously described [
13]. For the primary lung, lymph, and mammary BALB/c fibroblasts, the protocol was adjusted to allow deposition of ECM (extracellular matrix) for 21 days, with media change (+ ascorbic acid) every second day. Additionally, during the process of removing the fibroblasts from the deposited ECM, after the addition of extraction buffer (NH4OH-Triton-X-100), FDMs were washed twice in PBS (gently) and 0.5% sodium deoxycholate (Sigma-Aldrich, D6750-500 mg) was added for 60 min at room temperature. Sodium deoxycholate was then removed, the FDMs were washed in PBS and then DNase I was added as previously described. mCAF1 fibroblasts were cultured for 7 days to allow the deposition of ECM, as previously described, and denuded in the same way as the primary fibroblasts, with the additional lysing step.
2.3. Immunofluorescence Staining and Imaging Analysis
FDMs for the lung, mammary and lymph nodes were generated in Corning® (Kennebunk, ME, USA) high content imaging 96-well imaging plates (cat. no. CLS4580-10). To image the FDMs before the fibroblast lysing, after ECM deposition, cells were fixed in 4% Paraformaldehyde Solution (PFA), 4% in PBS (Affymetrix, cat. no. 19943 Santa Clara, CA, USA) for 10 min at room temperature in a laminar flow hood. PFA was then removed, and samples washed in PBS. Cells were permeabilised in 0.2% Triton-X-100 in PBS (Sigma-Aldrich, cat. no. T8787-50 mL) for 2–5 min. Cells were blocked in 3% BSA (Bovine Serum Albumin, Sigma- Aldrich cat. no. A8806-5G) for 60 min at room temperature. Primary antibodies were prepared in 1% BSA. Blocking BSA was removed, and cells washed in PBS, after which primary antibodies were added and incubated overnight at 4 °C. After 16 h primary antibody was removed, and cells were washed 3 times PBS-T (0.05% TWEEN® 20 (Croda Internation PLC, UK) in PBS) (TWEEN® 20, Merck, cat. no. P9416-100ML, Darmstadt, Germany). Secondary antibodies were made up in 1% BSA and incubated with the samples for 4–6 h at room temperature (kept dark), after which they were washed 3 times in PBS-T. Samples were then placed in PBS at 4 °C before imaging.
Immunofluorescence imaging was performed on the inverted confocal microscope (Leica SP8, Wetzlar, Germany) with a WLL and HC PL APO CS2 20×/0.75 IMM oil objective. Images were acquired at 1024 × 1024 resolution.
Nidogen 1 antibody was used at a concentration of 1:500 (Rabbit Polyclonal-Acris-cat. no. AP02274SU, Origene, Rockville, MD, USA). Phalloidin (Phalloidin AlexaFluor 633, cat. no. A22287, Invitrogen, Thermofisher Fisher Scientific) was used to visualise actin and was used at a concentration of 1:500. The secondary antibody Alexa 488 (cat. no. A11001, Invitrogen, Thermo Fisher Scientific) was used at a concentration of 1:400. DAPI (Thermo Fisher Scientific, cat. no. 62248) was used at a concentration of 1:1000.
2.4. Imaging of Drug Screen
All the following steps were performed on a Microlab Star liquid handling station (Hamilton, Bonaduz, Switzerland). Drug screen plates were fixed on day 3 after drug library added. Plates were fixed in 100 μL/well 10% formalin (Formalin solution 10% neutral buffered, Sigma-Aldrich, cat. no. HT501128-4L) for 10 min. Cells were permeabilised in 0.2% Triton-X-100 in PBS (Sigma-Aldrich, cat. no. T8787-50 mL) for 2–5 min. Next 2× PBS washes are performed, after which DAPI was added at 1 μg/mL for 90 min at room temperature. Plates were then washed ×3 in PBS and placed in 100 μL PBS and stored at 4 °C (dark) until imaging. Imaging was performed on the INCell Analyzer 2200 (GE Healthcare Life Sciences). Images were analysed using the INCell Analyzer Worktstation 1000 software (GE Healthcare Life Sciences). Nuclei were segmented based on the DAPI staining using the Tophat segmentation method. The mean intensity of GFP and DAPI in each nucleus was measured, and the number of GFP positive and DAPI positive cells were counted and compared to controls.
2.5. Mass Spectrometry of Fibroblast-Derived Matrices
FDMs were generated in triplicate from the primary lung, lymph, and mammary fibroblasts, following which the fibroblasts were lysed to leave behind the ECM. FDMs were washed in PBS, scraped, and collected. FDMs were then centrifuged at 8000×
g for 10 min. PBS was then removed, and samples were resuspended in 20 μL GdCl lysis buffer (6M Guanidinium Hydrochloride, 10 mM TCEP, 40 mM CAA, 100 mM Tris, pH 8.5). Samples were then vortexed and boiled at 95 °C for 5 min, then sonicated using the Bioruptor Next Gen (Diagenode SA, Seraing, Belgium) on maximum energy setting (5 × 30 s on/30 s off). Samples were then centrifuged for 1 min at 13,000 rpm and snap-frozen in liquid nitrogen. Sample preparation and mass spectrometry were then performed as previously described [
30]. Briefly, after determining protein concentration with Bradford (Sigma-Aldrich, cat. No. 23236), 5 μg was taken forward for digestion. Samples were diluted 1:3 with 10% Acetonitrile, 50 mM HEPES pH 8.5, LysC (MS grade, cat. no. 125-05061 Wako, Osaka, Japan) added in a 1:50 (enzyme to protein) ratio, and samples were incubated at 37 °C for 4 h. Samples were further diluted to 1:10 with 10% Acetonitrile, 50 mM HEPES pH 8.5; trypsin (MS grade, Promega, Madison, WN USA) was added in a 1:100 (enzyme to protein) ratio, and samples were incubated overnight at 37 °C. Enzyme activity was quenched by adding 2% trifluoroacetic acid (TFA) to a final concentration of 1%. Prior to mass spectrometry analysis, the peptides were desalted on in-house-packed C18 Stagetips [
31]. For each sample, 2 discs of C18 material (3M Empore) were packed in a 200 μL tip, and the C18 material was activated with 40 μL of 100% Methanol (HPLC grade, Sigma-Aldrich), then 40 μL of 80% Acetonitrile, 0.1% formic acid. The tips were subsequently equilibrated 2× with 40 μL of 1% TFA, 3% Acetonitrile, after which the samples were loaded using centrifugation at 4000× rpm. After washing the tips twice with 100 μL of 0.1% formic acid, the peptides were eluted into clean 500 μL Eppendorf tubes using 40% Acetonitrile and 0.1% formic acid. The eluted peptides were concentrated in an Eppendorf Speedvac, and re-constituted in 1% TFA, 2% Acetonitrile for Mass Spectrometry (MS) analysis.
2.5.1. Mass Spectrometry Data Acquisition
For each sample, peptides were loaded onto a 2 cm C18 trap column (Thermo Fisher Scientific, cat. no. 164705), connected in-line to a 50 cm C18 reverse-phase analytical column (Thermo Fisher Scientific, EasySpray cat. no. ES803) using 100% Buffer A (0.1% formic acid in water) at 750 bar, using the Thermo EasyLC 1200 HPLC system, and the column oven operating at 45 °C. Peptides were eluted over a 140 min gradient ranging from 6 to 60% of 80% acetonitrile, 0.1% formic acid at 250 nL/min, and the Orbitrap Fusion instrument (Thermo Fisher Scientific) was run in a DD-MS2 top speed method. Full MS spectra were collected at a resolution of 120,000, with an AGC target of 4 × 105 or maximum injection time of 50 ms and a scan range of 400–1500 m/z. The MS2 spectra were obtained in the ion trap operating at rapid speed, with an AGC target value of 1 × 104 or maximum injection time of 35 ms, a normalised HCD collision energy of 30, and an intensity threshold of 1.7 × 104. Dynamic exclusion was set to 60 s, and ions with a charge state <2, >7 or unknown were excluded. MS2 performance was verified for consistency by running complex cell lysate quality control standards, and chromatography was monitored to check for reproducibility.
2.5.2. Data Processing
Raw MS/MS spectra were processed using MaxQuant version 1.6.1.0 (Max-Planck-Institut, Germany) and searched against the mus musculus reference proteome obtained from Uniprot. Label-free quantitation (LFQ) was enabled, dynamic modifications were set as Oxidation (M) and Acety. on protein N-termini. Cysteine carbamidomethyl was set as a static modification. All results were filtered to a 1% FDR (False Discovery Rate), and common contaminants removed, resulting in a final protein matrix of 3913 proteins identified. For downstream processing, protein identifications with N = 3 in at least one group were retained, and missing values imputed using the built-in imputation algorithm in Perseus (v. 1.5.0.9). The MS proteomics data have been deposited to the ProteomeXchange Consortium (Cambridge, UK) via the PRIDE partner repository with the dataset identifier PXD025786.
2.5.3. ISDoT Mass Spectrometry Acquisition and Data Processing
ISDoT (In Situ Decellularisation of Tissues) and FDM mass spectrometry sample preparation and analysis were performed the same way as described, with the following notable differences [
32]. Organs from 8-week-old Balb/c female mice were used (three per condition).
“Following ISDoT decellularisation, organs were thoroughly washed and dissected free of extraneous tissue before being ground in liquid nitrogen to a fine powder, using a chilled mortar and pestle. Solubilised proteins were reduced in 5 mM DTT (Sigma-Aldrich) and alkylated with 10 mM chloroacetamide (Sigma-Aldrich) before deglycolsylation with PNGase F overnight at 37 °C with shaking (100 U per 10 mg tissue).
The Q-Exactive (Thermo Fisher Scientific) was run using the DD-MS2 top10 method. Full MS spectra were collected at a resolution of 70,000, with an Automatic Gain Control (AGC) target of 3 × 106 or a maximum injection time of 20 ms and a scan range of 300–1750 m/z. The MS2 spectra were obtained at a resolution of 17,500, with an AGC target value of 1 × 106 or a maximum injection time of 60 ms, a normalised collision energy of 25%, and an underfill ratio of 0.1%. Dynamic exclusion was set to 45 s, and ions with a charge state of <2 or unknown were excluded.
Raw MS/MS spectra were processed using MaxQuant version 1.5.1.2 software (Max- Planck Institute, Germany) and searched against the mouse protein database UP000000589_10090 (released 25/02/16). Functional-enrichment analysis was performed using High-Throughput GoMiner software57. One thousand randomisations were performed, and the data were thresholded for a 5% false-discovery rate. Over-represented biological-process and cellular-component terms with ≥5 and ≤500 assigned proteins were reported. The fold change in diseased relative to healthy tissue was mapped onto proteins assigned to each over-represented term”.
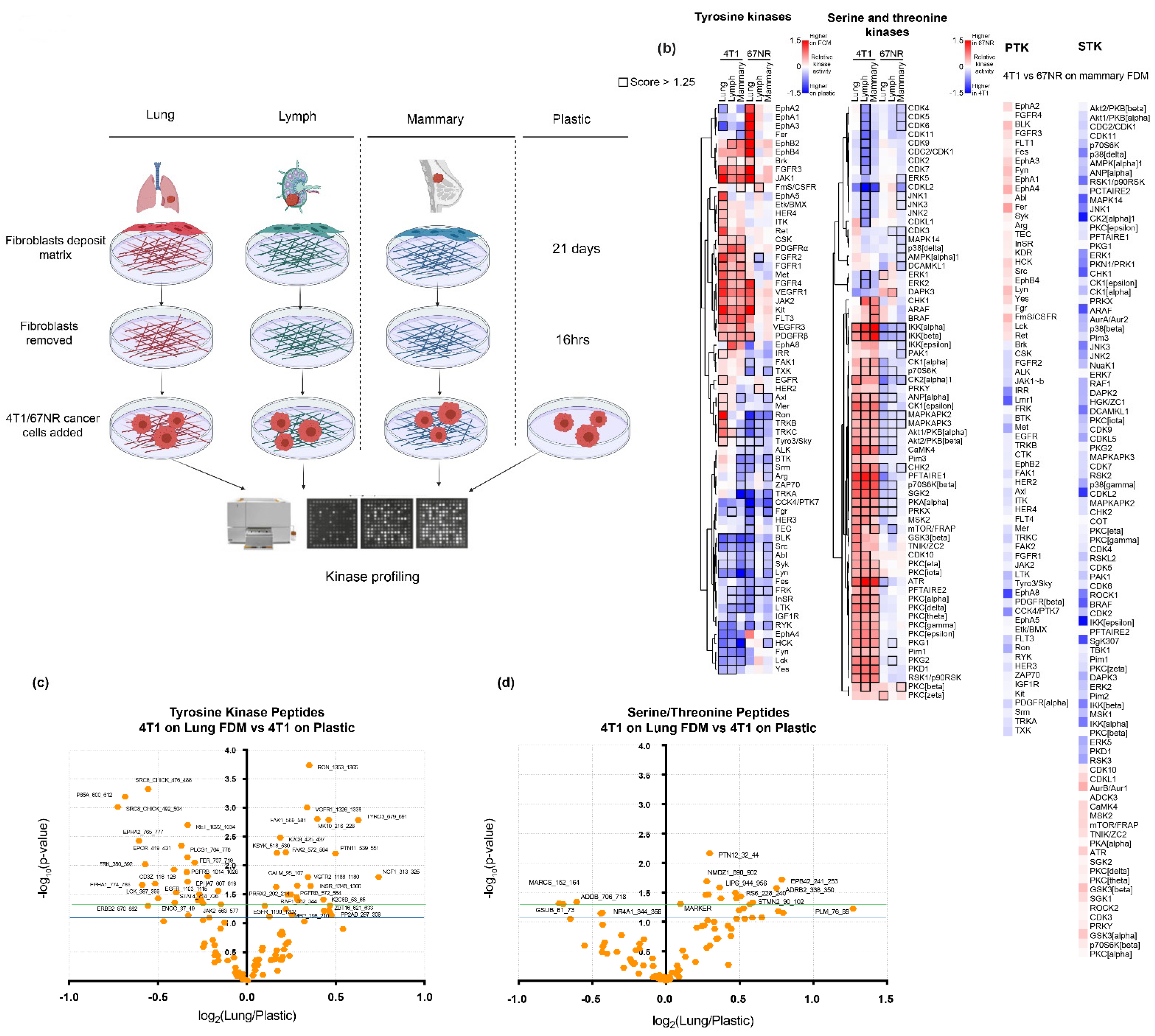
2.6. Kinase Profiling and Analysis
Mammary, lung, lymph node, and mCAF1 FDMs were generated in duplicate and denuded as previously described in 35 mm plastic dishes. Dishes were also coated with Matrigel (Cultrex 3-D culture matrix cat. no. 3445-005-01, lot no. 1519214, R&D Systems, MN, USA), which was diluted in serum-free media (1:1000) to obtain 15 μg/mL overnight at 4 °C. Plates were also coated with 0.2% gelatin (Sigma-Aldrich, Gelatin solution, Type B, 2% in H2O, G1393-100 mL) and 0.2% gelatin followed by Matrigel for 4 h. 4T1 cancer cells were then seeded in serum-free media, the same as used for the 4T1 cells (200,000 cells/35 mm well). After 16 h, 4T1 cells on the matrices were lysed and collected on ice in 100 μL Mammalian Protein Extraction Reagent (of M-PER) (ThermoFisher) with ×1 EDTA-free protease and ×1 phosphatase inhibitor cocktails (Halt, ThermoFisher). Samples were then centrifuged (10 min, at 4 °C, 16,000× g) and supernatant collected. Protein concentration was determined by Bradford Assay.
A unit of 5 μg of protein was mixed with FITC-conjugated anti-phophotyrosine antibody PY20 from each sample and was loaded onto either the PTK (protein tyrosine kinases) or STK (serine/threonine kinases) PamChips (Pamgene, Hertogenbosch, The Netherlands, which contain porous membrane that contains peptides with known phosphorylation sites. When loaded onto the Pamstation12 (Hertogenbosch, The Netherlands), the samples were then pumped through the membrane of the chips, and which allowed peptides on the membrane to be phosphorylated. Fluorescence emitted was then imaged by a built-in CCD camera [
33,
34].
Raw images were then analysed using BioNavigator software (Pamgene, Hertogenbosch, The Netherlands).
T-test of the relative signal intensities of peptides changed was performed (
Table S5, performed in GraphPad Prism v. 9.02, (San Diego, CA, USA). Predicted upstream kinases were analysed using the Upstream Kinase Analysis App (v.2018, BioNavigator, Pamgene), where kinase activity is based on multiple upstream kinase databases as is reported as a mean kinase statistic. Hierarchical clustering of the kinase activity across the samples was performed in Cluster 3.0 (C Clustering Library 1.59 for Mac OS X, Tokyo, Japan) and visualised using Java Tree View (v.1.2.0, Boston, MA, USA).
2.7. Kinase Inhibitor Drug Screen
Kinase inhibitor library (Inhibitor SelectTM Protein Kinase Inhibitor Library I (cat. no. 539744, Calbiochem Germany), Library II (cat. no. 539745), and Library III (cat. no. 539746) used were kindly gifted by Professor Michael Givskov. Inhibitors were at a 1 mM stock solution in dimethyl sulfoxide(DMSO). Working stock solutions were prepared as follows: dilute to 100 μM in 10% DMSO and 90% PBS. For each library, 10 μL of drug was then aliquoted into 1 ‘working’ drug plate per repeat. At time of use, 90 μL of 2% serum media was added to each drug to make a 10 μM solution in 1% DMSO, 9% PBS, and 90% media. A unit of 20 μL of each drug at 10 μM was then added to 180 μL of media and cells (2% FBS) with a final drug concentration of 1 μM.
2.7.1. Primary Lung Fibroblast and mCAF1 Kinase Inhibitor Drug Screen
Primary lung FDM and mCAF1 FDM was prepared as previously described in Corning® high-content-imaging 96-well imaging plates (cat. no. CLS4580-10). 15,000 cells per well were seeded. After the fibroblasts were denuded, the lung FDMs were placed in PBS while the 4T1-H2B-GFP cells were prepared. A total of 3000 4T1-H2B-GFP cells were added to each well in 180 μL 2% FBS complete media, and after 16 h the kinase inhibitor drug libraries were added. After 3 days, plates were fixed and imaged. Each plate of the drug screen within each repeat had a DMSO control (in triplicate) present. In addition, edge wells were not used for screening to limit edge effects and were filled with 200 μL PBS.
2.7.2. Matrigel and Gelatin Kinase Inhibitor Drug Screen
Corning® high-content-imaging 96-well imaging plates (cat. no. CLS4580-10) were coated with Matrigel overnight (Cultrex 3-D culture matrix cat. no. 3445-005-01, lot no. 1519214,), which was diluted in serum-free media (1:1000) to obtain a 15 μg/mL solution at 4 °C. Plates were also coated in 0.2% gelatin (Sigma-Aldrich, Gelatin solution, Type B, 2% in H2O, G1393-100 mL) overnight at 4 °C. Before use, coatings were washed in PBS and then 3000 4T1-H2B-GFP cells were seeded, and kinase inhibitor library added as above.
2.7.3. Cell Viability
To perform cell viability assays, 4T1 cells were added to FDMs or plastic in 2% FBS medium. RealTime-Glo (Promega, cat. no. G9712) was then added at ×5 concentration (stock ×1000) and incubated for 60 min at 37 °C, after which luminescence (578 nm) was read using the SpectraMax Paradigm multi-mode detection platform. Luminescence reading was then taken every 24 h for a total of 72 h.
2.8. Statistical Analysis
All statistical analyses were performed in GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA) unless otherwise stated. Data were tested for normal distribution, following which the appropriate statistical analysis was chosen. Drug inhibition was tested using a 2-way ANOVA with multiple comparisons (corrected using Šídák’s hypothesis testing). Adjusted p-values were reported with a confidence level of 0.05 (95% confidence interval). Simple linear regression analysis was performed to find the best-fit value of the slope and intercept.
2.9. Generating Protein Interaction Networks
ECM proteins that were identified in lung FDM, kinases that were activated by lung FDM, and targets of drugs that reduced 4T1 proliferation on lung FDM were assembled. Interaction networks were generated using Cytoscape (version 3.8.2, San Diego, CA, USA) [
35] and Genemania (version 3.5.2, Toronto, Canada) [
36] as previously described [
37]. Lines between nodes (proteins) represent reported physical protein–protein interactions, and up to 100 additional interacting proteins were automatically added to assist with hypothesis generation. Finally, proteins with no interacting partners were removed and the yFiles organic layout [
38] was applied.
4. Discussion
Knowing the important role that the ECM plays during metastatic progression of breast cancer, we adapted a cell-based approach to derive in vitro native ECM and investigated kinase signalling and drug response in a model of metastatic breast cancer. We used normal fibroblasts from known metastatic organs in breast cancer (lymph nodes and lungs) as well as the primary site (mammary gland). These FDMs closely resemble in vivo the ECM components of decellularised lungs vs. mammary, and we show by mass spectrometry that the FDMs generated are complex and abundant in ECM constituents.
Cancer–ECM interactions are critical during metastatic progression. In order for cancer cells at the primary tumour site to travel to local and distant organs, they must come in contact with and move through the ECM within and surrounding the primary tumour (invasion), followed by breaching of the basement membrane (intravasation), and then the reverse at the secondary site (extravasation) [
49]. At all times, the ECM is providing biophysical support and biochemical cues that affect the ability of cancer cells to proliferate and invade, which could be targeted by therapeutics [
50]. The ECM during cancer progression is often altered in terms of stiffness, structure, arrangement, and composition [
4] and as such, provides a unique microenvironment in which cancer cells can activate intracellular signalling pathways necessary for proliferation and migration. We also see that our mammary FDMs have significantly increased collagen I levels compared to the lung FDM, which recapitulates the in vivo situation, as our previous proteomics also shows an increase in collagen I. Increasingly, ‘simpler’ ECM components have also been the focus of cancer studies to include a relevant tumour microenvironment. However, they are often limited in their ability to recapitulate these cancer niche properties as products such as Matrigel and gelatin are only composed of a limited number of ECM components and lack resemblance to native tissue architecture and biophysical properties [
51]. The presence of the ECM is also critical when studying cancer cell behaviour in vitro to aid in increasing the efficacy of drugs tested in vitro to patients, as current studies suggest fewer than 5% of all drugs tested make it to the clinic [
52,
53]. To our knowledge, using FDMs to investigate global kinase signalling and for use as a high throughput drug screening platform has not been previously performed.
We show that irrespective of which organ-specific FDM the 4T1 breast cancer cells are in contact with, overall, there are only minor changes in kinase activation and de-activation. Cancer cells bind to the ECM through integrins and glycoproteins, which transduce signals from the tumour microenvironment into the cell. Integrins are versatile transmembrane proteins that are able to bind to different ECM components such as collagen, laminins, and fibronectin [
54]. We therefore posit that if there are a range of different ECM components present surrounding the cancer cells, integrin-mediated kinase signalling can be induced. Further in-depth investigations looking at which specific ECM components (or redundancy in the system) induce key signalling pathways, and through which receptors signalling occurs, will be very beneficial to better understand the ECM-cancer cell interaction in breast cancer. While not directly studied here, the role of specific, important signalling pathways, such as those related to TGFβ, are also likely to be altered and contribute to the results we see. TGFβ has been shown to activate fibroblasts within the tumour microenvironment and therefore induce the production of lysyl oxidase, via SMAD, leading to increased collagen crosslinking and metastasis in breast cancer [
55]. We see that both in the mammary and lung FDM, lox is significantly increased in lung vs. mammary. Furthermore, it would be beneficial in this system to further explore whether other functions of the cancers cells are altered via looking at migration and invasion. Multiple studies have shown [
56] that FDMs can be used to perform migration studies as they produce the necessary collagen structures to allow this [
13,
57].
The organ-specific fibroblasts that we used in this study were derived from resident fibroblasts that are present within the organ of healthy mice. While this gives us a good opportunity to look at differences in inter-organ composition during tumour progression (when cancer cells arrive at a new organ) and kinase signalling, during breast cancer progression, cancer cells are in contact with CAFs as well as resident fibroblasts. CAFs have been shown to remodel and reorganise the ECM, provide tracks for cell migration, and physically pull cancer cells through the tumour microenvironment, aiding in cancer dissemination [
7]. We see that when on the mCAF1 FDM, kinase signalling is different to the other FDMs and observe that numerous kinases were de-activated on mCAF1 FDM compared to plastic. Furthermore, we saw that there were no mCAF1 FDM-specific inhibitors. Our study therefore highlights the need for further investigations including the difference between CAF and normally secreted FDM in order to provide better in vitro drug testing and kinase signalling platforms. We also hypothesise that the ECM deposited by the mCAF1s leads to lowered kinase activation levels across many kinases that are important for cell proliferation (such as cyclin-dependent kinases). When these levels are ‘lowered’, drugs that target kinases which are normally highly activated in cancer cells are no longer as efficacious. It would therefore be of interest to see whether the use of combination treatments of kinases inhibitors and PP2A (Protein Phosphatase 2) activators such as SMAP (small molecule activator of PP2A), that allows the re-activation of kinase signalling pathways [
58], could lead to re-sensitisation to certain kinase inhibitors and increased efficacy.
We have shown that 4T1 cells on different ECM types display only subtle changes in cell response, despite differences in ECM composition. However, we observe very large differences in both kinase signalling and, more importantly, drug response when 4T1 cells are seeded on plastic. We show that distinctive kinase signalling pathways are activated in 4T1 breast cancer cells and 67NRs depending on whether they are on ECM or plastic, while comparatively minor signalling changes were observed in response to FDMs of different composition. Kinases that could be used as targets at the metastatic lung site would might be EphA5, Tyro3, Ron, TRKB, CK2alpha, and Pim3; however, these kinases display lower changes in activation in the lung compared to mammary. Kinases that would be of interest, as targeting possibilities for metastasis in general in breast cancer would be those that are activated on the FDMs vs. plastic, and in the 4T1, such as ITK, IRR, PKCα/δ/γ/φ/ε, PKG1/2, Pim1, PKD1, RSK1, IKKα/β, ANP, CK1, MAPKAPK2/3, Akt1/2, CamK4, p7056kβ, SGK, PDGFRα/β, FGFR, pKA, and PRKX. These could also be further investigated as biomarkers in breast cancer, as changed expression levels may correlate with metastasis-free survival, particularly to the lungs.
Conventional drug screening platforms performed in vitro are often 2D and use plastic as a substrate. Increasingly, pre-clinical cancer research is moving towards using models which are 3D and encompass the greater complexities of the tumour microenvironment to better model diseases and test drugs. However, progress in this area is hindered as platforms such as these are difficult to ‘scale-up’. Matrigel-based approaches to drug screening are common in organoid cultures, where primary cells from patient tumours are embedded within 3D Matrigel. While these cultures often maintain the genomic properties of the tumours, and encompass cell heterogeneity, they have can be difficult to maintain [
59]. While other Matrigel-based cultures such as MBOC (Matrigel bilayer organoid culture) developed for gynaecological tumours [
60] have been shown to improve the viability of cell cultures and allow for drug screening, they yet do not encompass the complex mix of ECM proteins that are deposited by fibroblasts. FDMs, while they provide a relevant tumour microenvironment, are easily manipulated, which limits their versatility. Furthermore, fibroblasts in culture change their phenotype (such as an increase in αSMA expression [
61]) to varying degrees, which requires careful phenotypic and molecular monitoring. A good test of our drug screening platform would be to see whether a clinically effective kinase therapy was effective on our FDMs but not on other 2D substrates. This would demonstrate the further clinical relevance of FDMs. Here, we show that we can use FDMs to perform high-throughput drug screening and highlight that using an ECM substrate during this process is of critical importance.
In our drug screens, we treated 4T1 cells with kinase inhibitors at one concentration (1 μM) for a set time (3 days). The inhibitory capabilities of these inhibitors differ when 4T1 cells are on plastic or ECM. There are drugs that show efficacy when 4T1s are on plastic but not on ECM, irrespective of the matrix substrate (gelatin, Matrigel, or lung FDM), which we hypothesise have less clinical relevance, as they do not encompass the complex protein composition seen in vivo. We know the ECM can act as a barrier to drug delivery to cancer cells [
62] and, as such, is a vital element in drug testing. The differences that we see in drug response to kinase inhibition are also validated as the global kinase signalling that we have performed clearly shows that activation of kinases differs greatly between 4T1 cells on ECM and plastic.
Here, we present a novel method of performing large-scale drug screening of kinase inhibitor, which encompasses the ECM by using FDMs. While we provide several ECM substrates that could be used to represent the ECM for drug screening, we observe that the FDMs contain the complex mixture of ECM proteins, in addition to other biophysical cues, that might best represent the in vivo situation. We also show that using matrices from different metastatic organs in breast cancer can elucidate subtle changes in kinase signalling pathways that could be targeted. Moreover, in this study, we highlight that there is a real need to incorporate the ECM into in vitro drug screening as results from screening on plastic alone do not replicate those of 4T1 cells on ECM. We therefore stress the importance of including ECM during in vitro breast cancer metastasis investigations, including drug screening.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}