
Tumor Microenvironment of Esophageal Cancer

 , , ,
, , , 

Abstract
:Simple Summary
Abstract
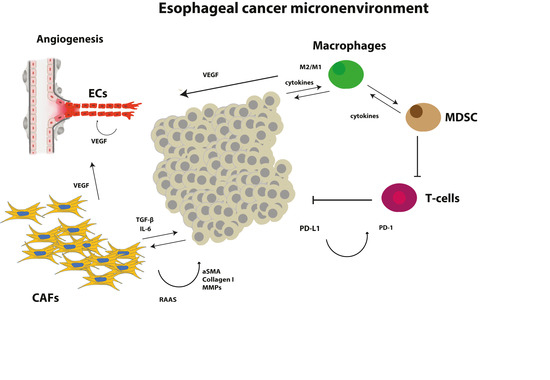
1. Introduction
2. Endothelial Cells and Tumor Angiogenesis
3. Cancer-Associated Fibroblasts
4. Tumor-associated Macrophages
5. T-Cells and Myeloid-Derived Suppressor Cells, Immunotherapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lagergren, J.; Smyth, E.; Cunningham, D.; Lagergren, P. Oesophageal cancer. Lancet 2017, 390, 2383–2396. [Google Scholar] [CrossRef] [Green Version]
- Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; Wolfe, C.; et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef]
- Al-Batran, S.-E.; Hofheinz, R.D.; Pauligk, C.; Kopp, H.-G.; Haag, G.M.; Luley, K.B.; Meiler, J.; Homann, N.; Lorenzen, S.; Schmalenberg, H.; et al. Histopathological regression after neoadjuvant docetaxel, oxaliplatin, fluorouracil, and leucovorin versus epirubicin, cisplatin, and fluorouracil or capecitabine in patients with resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4-AIO): Results from the phase 2 part of a multicentre, open-label, randomised phase 2/3 trial. Lancet Oncol. 2016, 17, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Al-Batran, S.-E.; Homann, N.; Pauligk, C.; Goetze, T.O.; Meiler, J.; Kasper, S.; Kopp, H.-G.; Mayer, F.; Haag, G.M.; Luley, K.; et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): A randomised, phase 2/3 trial. Lancet 2019, 393, 1948–1957. [Google Scholar] [CrossRef]
- Shapiro, J.; van Lanschot, J.J.B.; Hulshof, M.C.C.M.; van Hagen, P.; van Berge Henegouwen, M.I.; Wijnhoven, B.P.L.; van Laarhoven, H.W.M.; Nieuwenhuijzen, G.A.P.; Hospers, G.A.P.; Bonenkamp, J.J.; et al. CROSS study group Neoadjuvant chemoradiotherapy plus surgery versus surgery alone for oesophageal or junctional cancer (CROSS): Long-term results of a randomised controlled trial. Lancet Oncol. 2015, 16, 1090–1098. [Google Scholar] [CrossRef]
- Lorenzen, S.; Thuss-Patience, P.; Al-Batran, S.E.; Lordick, F.; Haller, B.; Schuster, T.; Pauligk, C.; Luley, K.; Bichev, D.; Schumacher, G.; et al. Impact of pathologic complete response on disease-free survival in patients with esophagogastric adenocarcinoma receiving preoperative docetaxel-based chemotherapy. Ann. Oncol. 2013, 24, 2068–2073. [Google Scholar] [CrossRef] [PubMed]
- Mönig, S.P.; Schiffmann, L.M. Resection of advanced esophagogastric adenocarcinoma: Extended indications. Chirurg 2016, 87, 398–405. [Google Scholar] [CrossRef]
- Homann, N.; Pauligk, C.; Luley, K.; Kraus, T.W.; Bruch, H.-P.; Atmaca, A.; Noack, F.; Altmannsberger, H.-M.; Jäger, E.; Al-Batran, S.-E. Pathological complete remission in patients with oesophagogastric cancer receiving preoperative 5-fluorouracil, oxaliplatin and docetaxel. Int. J. Cancer 2011, 130, 1706–1713. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-M.; Hong, P.; Xu, W.W.; He, Q.-Y.; Li, B. Advances in targeted therapy for esophageal cancer. Signal Transduct. Target. Ther. 2020, 5, 229. [Google Scholar] [CrossRef]
- Vivaldi, C.; Catanese, S.; Massa, V.; Pecora, I.; Salani, F.; Santi, S.; Lencioni, M.; Vasile, E.; Falcone, A.; Fornaro, L. Immune Checkpoint Inhibitors in Esophageal Cancers: Are We Finally Finding the Right Path in the Mist? Int. J. Mol. Sci. 2020, 21, 1658. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; Wong, A.H.-K.; Jain, R.K. Vascular Normalization as a Therapeutic Strategy for Malignant and Nonmalignant Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006486. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Duda, D.G.; Clark, J.W.; Loeffler, J.S. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat. Clin. Pr. Oncol. 2006, 3, 24–40. [Google Scholar] [CrossRef]
- Fukumura, D.; Jain, R.K. Tumor microenvironment abnormalities: Causes, consequences, and strategies to normalize. J. Cell. Biochem. 2007, 101, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F.; Kozin, S.V.; Tong, R.T.; Chae, S.-S.; Booth, M.F.; Garkavtsev, I.; Xu, L.; Hicklin, D.J.; Fukumura, D.; di Tomaso, E.; et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation. Cancer Cell 2004, 6, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Wang, X.; Lu, J.; Salfenmoser, M.; Wirsik, N.M.; Schleussner, N.; Imle, A.; Valls, A.F.; Radhakrishnan, P.; Liang, J.; et al. Reduction of Liver Metastasis Stiffness Improves Response to Bevacizumab in Metastatic Colorectal Cancer. Cancer Cell 2020, 37, 800–817. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Shimada, H.; Takeda, A.; Nabeya, Y.; Okazumi, S.-I.; Matsubara, H.; Funami, Y.; Hayashi, H.; Gunji, Y.; Kobayashi, S.; Suzuki, T.; et al. Clinical significance of serum vascular endothelial growth factor in esophageal squamous cell carcinoma. Cancer 2001, 92, 663–669. [Google Scholar] [CrossRef]
- Nienhüser, H.; Schmidt, T. Angiogenesis and Anti-Angiogenic Therapy in Gastric Cancer. Int. J. Mol. Sci. 2017, 19, 43. [Google Scholar] [CrossRef] [Green Version]
- Ohtsu, A.; Shah, M.A.; Van Cutsem, E.; Rha, S.Y.; Sawaki, A.; Park, S.R.; Lim, H.Y.; Yamada, Y.; Wu, J.; Langer, B.; et al. Bevacizumab in Combination with Chemotherapy As First-Line Therapy in Advanced Gastric Cancer: A Randomized, Double-Blind, Placebo-Controlled Phase III Study. J. Clin. Oncol. 2011, 29, 3968–3976. [Google Scholar] [CrossRef]
- Shen, L.; Li, J.; Xu, J.; Pan, H.; Dai, G.; Qin, S.; Wang, L.; Wang, J.; Yang, Z.; Shu, Y.; et al. Bevacizumab plus capecitabine and cisplatin in Chinese patients with inoperable locally advanced or metastatic gastric or gastroesophageal junction cancer: Randomized, double-blind, phase III study (AVATAR study). Gastric Cancer 2015, 18, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.J.; Gagnon-Bartsch, J.A.; Tan, I.B.; Earle, S.; Ruff, L.; Pettinger, K.; Ylstra, B.; Van Grieken, N.; Rha, S.Y.; Chung, H.C.; et al. Signatures of tumour immunity distinguish Asian and non-Asian gastric adenocarcinomas. Gut 2015, 64, 1721–1731. [Google Scholar] [CrossRef]
- Cunningham, D.; Stenning, S.P.; Smyth, E.C.; Okines, A.F.; Allum, W.H.; Rowley, S.; Stevenson, L.; Grabsch, H.I.; Alderson, D.; Crosby, T.; et al. Peri-operative chemotherapy with or without bevacizumab in operable oesophagogastric adenocarcinoma (UK Medical Research Council ST03): Primary analysis results of a multicentre, open-label, randomised phase 2–3 trial. Lancet Oncol. 2017, 18, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Santos, L.V.D.; Aprile, G.; Ferry, D.R.; et al. REGARD Trial Investigators Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.-C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.-Y.; et al. RAINBOW Study Group Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Smyth, E.C.; Verheij, M.; Allum, W.; Cunningham, D.; Cervantes, A.; Arnold, D. ESMO Guidelines Committee Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v38–v49. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, L.M.; Brunold, M.; Liwschitz, M.; Goede, V.; Loges, S.; Wroblewski, M.; Quaas, A.; Alakus, H.; Stippel, D.; Bruns, C.J.; et al. A combination of low-dose bevacizumab and imatinib enhances vascular normalisation without inducing extracellular matrix deposition. Br. J. Cancer 2017, 116, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Coutelle, O.; Schiffmann, L.M.; Liwschitz, M.; Brunold, M.; Goede, V.; Hallek, M.; Kashkar, H.; Hacker, U.T. Dual targeting of Angiopoetin-2 and VEGF potentiates effective vascular normalisation without inducing empty basement membrane sleeves in xenograft tumours. Br. J. Cancer 2015, 112, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffmann, L.M.; Fritsch, M.; Gebauer, F.; Günther, S.D.; Stair, N.R.; Seeger, J.M.; Thangarajah, F.; Dieplinger, G.; Bludau, M.; Alakus, H.; et al. Tumour-infiltrating neutrophils counteract anti-VEGF therapy in metastatic colorectal cancer. Br. J. Cancer 2019, 120, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebos, J.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated Metastasis after Short-Term Treatment with a Potent Inhibitor of Tumor Angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Valls, A.F.; Knipper, K.; Giannakouri, E.; Sarachaga, V.; Hinterkopf, S.; Wuehrl, M.; Shen, Y.; Radhakrishnan, P.; Klose, J.; Ulrich, A.; et al. VEGFR1 + Metastasis–Associated Macrophages Contribute to Metastatic Angiogenesis and Influence Colorectal Cancer Patient Outcome. Clin. Cancer Res. 2019, 25, 5674–5685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacker, U.T.; Escalona-Espinosa, L.; Consalvo, N.; Goede, V.; Schiffmann, L.; Scherer, S.J.; Hedge, P.; Van Cutsem, E.; Coutelle, O.; Buning, H. Evaluation of Angiopoietin-2 as a biomarker in gastric cancer: Results from the randomised phase III AVAGAST trial. Br. J. Cancer 2016, 114, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Goede, V.; Coutelle, O.; Neuneier, J.; Reinacher-Schick, A.; Schnell, R.; Koslowsky, T.C.; Weihrauch, M.R.; Cremer, B.; Kashkar, H.; Odenthal, M.; et al. Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br. J. Cancer 2010, 103, 1407–1414. [Google Scholar] [CrossRef] [Green Version]
- Dreikhausen, L.; Blank, S.; Sisic, L.; Heger, U.; Weichert, W.; Jäger, D.; Bruckner, T.; Giese, N.; Grenacher, L.; Falk, C.; et al. Association of angiogenic factors with prognosis in esophageal cancer. BMC Cancer 2015, 15, 121. [Google Scholar] [CrossRef] [Green Version]
- Nienhüser, H.; Crnovrsanin, N.; Nerz, D.; Heckler, M.; Sisic, L.; Lasitschka, F.; Schneider, M.; Schmidt, T. Expression of Angiogenic Proteins in Tumor and Stroma Affects Survival in Patients with Gastric Cancer. J. Surg. Res. 2020, 255, 172–180. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.-C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.-A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- Conradi, L.-C.; Brajic, A.; Cantelmo, A.R.; Bouché, A.; Kalucka, J.; Pircher, A.; Brüning, U.; Teuwen, L.-A.; Vinckier, S.; Ghesquière, B.; et al. Tumor vessel disintegration by maximum tolerable PFKFB3 blockade. Angiogenesis 2017, 20, 599–613. [Google Scholar] [CrossRef]
- Schiffmann, L.M.; Werthenbach, J.P.; Heintges-Kleinhofer, F.; Seeger, J.M.; Fritsch, M.; Günther, S.D.; Willenborg, S.; Brodesser, S.; Lucas, C.; Jüngst, C.; et al. Mitochondrial respiration controls neoangiogenesis during wound healing and tumour growth. Nat. Commun. 2020, 11, 1231. [Google Scholar] [CrossRef]
- Coutelle, O.; Hornig-Do, H.; Witt, A.; Andree, M.; Schiffmann, L.M.; Piekarek, M.; Brinkmann, K.; Seeger, J.M.; Liwschitz, M.; Miwa, S.; et al. Embelin inhibits endothelial mitochondrial respiration and impairs neoangiogenesis during tumor growth and wound healing. EMBO Mol. Med. 2014, 6, 624–639. [Google Scholar] [CrossRef]
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; Treps, L.; Li, X.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis Updated. Circ. Res. 2020, 127, 310–329. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumura, D.; Xavier, R.; Sugiura, T.; Chen, Y.; Park, E.-C.; Lu, N.; Selig, M.; Nielsen, G.; Taksir, T.; Jain, R.K.; et al. Tumor Induction of VEGF Promoter Activity in Stromal Cells. Cell 1998, 94, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Manousopoulou, A.; Hayden, A.; Mellone, M.; Baquero, D.G.; White, C.; Noble, F.; Lopez, M.; Thomas, G.J.; Underwood, T.J.; Garbis, S.D. Quantitative proteomic profiling of primary cancer-associated fibroblasts in oesophageal adenocarcinoma. Br. J. Cancer 2018, 118, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.-L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551. [Google Scholar] [CrossRef] [Green Version]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow–derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef] [Green Version]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Schoppmann, S.F.; Jesch, B.; Riegler, M.F.; Maroske, F.; Schwameis, K.; Jomrich, G.; Birner, P. Podoplanin expressing cancer associated fibroblasts are associated with unfavourable prognosis in adenocarcinoma of the esophagus. Clin. Exp. Metastasis 2012, 30, 441–446. [Google Scholar] [CrossRef]
- Underwood, T.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef]
- Hanley, C.J.; Mellone, M.; Ford, K.; Thirdborough, S.M.; Mellows, T.; Frampton, S.J.; Smith, D.M.; Harden, E.; Szyndralewiez, C.; Bullock, M.; et al. Targeting the Myofibroblastic Cancer-Associated Fibroblast Phenotype through Inhibition of NOX4. J. Natl. Cancer Inst. 2018, 110, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Galván, J.A.; Wiprächtiger, J.; Slotta-Huspenina, J.; Feith, M.; Ott, K.; Kröll, D.; Seiler, C.A.; Langer, R. Immunohistochemical analysis of the expression of cancer-associated fibroblast markers in esophageal cancer with and without neoadjuvant therapy. Virchows Archiv 2019, 476, 725–734. [Google Scholar] [CrossRef] [PubMed]
- van Pelt, G.; Krol, J.; Lips, I.; Peters, F.; van Klaveren, D.; Boonstra, J.; de Steur, W.; Tollenaar, R.; Sarasqueta, A.F.; Mesker, W.; et al. The value of tumor-stroma ratio as predictor of pathologic response after neoadjuvant chemoradiotherapy in esophageal cancer. Clin. Transl. Radiat. Oncol. 2020, 20, 39–44. [Google Scholar] [CrossRef]
- Steins, A.; Ebbing, E.A.; Creemers, A.; Van Der Zalm, A.P.; Jibodh, R.A.; Waasdorp, C.; Meijer, S.; Van Delden, O.M.; Krishnadath, K.K.; Hulshof, M.C.; et al. Chemoradiation induces epithelial-to-mesenchymal transition in esophageal adenocarcinoma. Int. J. Cancer 2019, 145, 2792–2803. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Fang, H.-Y.; Mohammad-Shahi, D.; Ingermann, J.; Baumeister, T.; Strangmann, J.; Schmid, R.M.; Wang, T.C.; Quante, M. Elimination of NF-κB signaling in Vimentin+ stromal cells attenuates tumorigenesis in a mouse model of Barrett’s Esophagus. Carcinogenesis 2021, 42, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef] [Green Version]
- Karakasheva, T.A.; Lin, E.W.; Tang, Q.; Qiao, E.; Waldron, T.J.; Soni, M.; Klein-Szanto, A.J.; Sahu, V.; Basu, D.; Ohashi, S.; et al. IL-6 Mediates Cross-Talk between Tumor Cells and Activated Fibroblasts in the Tumor Microenvironment. Cancer Res. 2018, 78, 4957–4970. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Xu, Y.; Wu, S. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. 2017, 8, e2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Xie, C.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Wang, Y.; Wu, S. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFβ1 signaling loop in esophageal squamous cell carcinoma. Mol. Carcinog. 2017, 56, 1150–1163. [Google Scholar] [CrossRef]
- Higashino, N.; Koma, Y.-I.; Hosono, M.; Takase, N.; Okamoto, M.; Kodaira, H.; Nishio, M.; Shigeoka, M.; Kakeji, Y.; Yokozaki, H. Fibroblast activation protein-positive fibroblasts promote tumor progression through secretion of CCL2 and interleukin-6 in esophageal squamous cell carcinoma. Lab. Investig. 2019, 99, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Miyata, H.; Sugimura, K.; Fukuda, S.; Kanemura, T.; Yamashita, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. miR-27 is associated with chemoresistance in esophageal cancer through transformation of normal fibroblasts to cancer-associated fibroblasts. Carcinogenesis 2015, 36, 894–903. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Cao, P.; Hu, S.; Kong, K.; Deng, Y.; Zhao, B.; Li, F. Esophageal Microenvironment: From Precursor Microenvironment to Premetastatic Niche. Cancer Manag. Res. 2020, 12, 5857–5879. [Google Scholar] [CrossRef]
- Palumbo, J.A.; Da Costa, N.M.; Pontes, B.; De Oliveira, F.L.; Codeço, M.L.; Pinto, L.F.R.; Nasciutti, L.E. Esophageal Cancer Development: Crucial Clues Arising from the Extracellular Matrix. Cells 2020, 9, 455. [Google Scholar] [CrossRef] [Green Version]
- Bratlie, S.O.; Edebo, A.; Casselbrant, A.; Helander, H.F.; Fändriks, L. The renin-angiotensin system in Barrett’s esophagus. Scand J. Gastroenterol. 2016, 51, 1037–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Sweigert, P.; Eguia, E.; Varsnik, M.A.; Renz, C.R.; Terrasse, W.A.; Gauthier, M.; Aranha, G.; Knab, L.M.; Abood, G. Impact of angiotensin system inhibitors on esophageal cancer survival. Surg. Open Sci. 2020, 3, 34–38. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate Partners for Tumor Cell Migration, Invasion, and Metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.K.; Natoli, G. Molecular control of activation and priming in macrophages. Nat. Immunol. 2016, 17, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deyell, M.; Garris, C.S.; Laughney, A.M. Cancer metastasis as a non-healing wound. Br. J. Cancer 2021, 124, 1491–1502. [Google Scholar] [CrossRef]
- Zhou, G.; Peng, K.; Song, Y.; Yang, W.; Shu, W.; Yu, T.; Yu, L.; Lin, M.; Weigang, S.; Chen, C.; et al. CD177+ neutrophils suppress epithelial cell tumourigenesis in colitis-associated cancer and predict good prognosis in colorectal cancer. Carcinogenesis 2018, 39, 272–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, W.; Charles, K.A.; Baracos, V.E.; Clarke, S.J. Neutrophil/lymphocyte ratio predicts chemotherapy outcomes in patients with advanced colorectal cancer. Br. J. Cancer 2011, 104, 1288–1295. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xie, Y.; Wang, X.; Li, F.; Li, S.; Li, M.; Peng, H.; Yang, L.; Liu, C.; Pang, L.; et al. Prognostic impact of tumor-associated macrophage infiltration in esophageal cancer: A meta-analysis. Futur. Oncol. 2019, 15, 2303–2317. [Google Scholar] [CrossRef]
- Sugimura, K.; Miyata, H.; Tanaka, K.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; Nakajima, K.; Takiguchi, S.; Mori, M.; Doki, Y. High infiltration of tumor-associated macrophages is associated with a poor response to chemotherapy and poor prognosis of patients undergoing neoadjuvant chemotherapy for esophageal cancer. J. Surg. Oncol. 2015, 111, 752–759. [Google Scholar] [CrossRef]
- Blank, S.; Nienhüser, H.; Dreikhausen, L.; Sisic, L.; Heger, U.; Ott, K.; Schmidt, T. Inflammatory cytokines are associated with response and prognosis in patients with esophageal cancer. Oncotarget 2017, 8, 47518–47532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G.H.; Schetter, A.J.; Chou, D.; Bowman, E.D.; Zhao, R.; Hawkes, J.E.; Mathe, E.; Kumamoto, K.; Zhao, Y.; Budhu, A.; et al. Inflammatory and MicroRNA Gene Expression as Prognostic Classifier of Barrett’s-Associated Esophageal Adenocarcinoma. Clin. Cancer Res. 2010, 16, 5824–5834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Peters, J.H.; Nieman, D.R.; Sharma, M.; Watson, T.J.; Yu, J. Macrophage subtype predicts lymph node metastasis in oesophageal adenocarcinoma and promotes cancer cell invasion in vitro. Br. J. Cancer 2015, 113, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Miyashita, T.; Tajima, H.; Shah, F.A.; Oshima, M.; Makino, I.; Nakagawara, H.; Kitagawa, H.; Fujimura, T.; Harmon, J.W.; Ohta, T. Impact of Inflammation–Metaplasia–Adenocarcinoma Sequence and Inflammatory Microenvironment in Esophageal Carcinogenesis Using Surgical Rat Models. Ann. Surg. Oncol. 2014, 21, 2012–2019. [Google Scholar] [CrossRef]
- Brana, I.; Calles, A.; Lorusso, P.M.; Yee, L.K.; Puchalski, T.A.; Seetharam, S.; Zhong, B.; De Boer, C.J.; Tabernero, J.; Calvo, E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: An open-label, multicenter phase 1b study. Target. Oncol. 2014, 10, 111–123. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Burg, J.M.; Mick, R.; Trosko, J.A.; Li, D.; Shaik, M.N.; Tolcher, A.W.; Hamid, O. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. OncoImmunology 2013, 2, e23033. [Google Scholar] [CrossRef] [Green Version]
- Razak, A.R.; Cleary, J.M.; Moreno, V.; Boyer, M.; Aller, E.C.; Edenfield, W.; Tie, J.; Harvey, R.D.; Rutten, A.; Shah, M.A.; et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J. Immunother. Cancer 2020, 8, e001006. [Google Scholar] [CrossRef]
- Teng, M.; Swann, J.B.; Koebel, C.M.; Schreiber, R.D.; Smyth, M. Immune-mediated dormancy: An equilibrium with cancer. J. Leukoc. Biol. 2008, 84, 988–993. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karwacz, K.; Bricogne, C.; MacDonald, D.; Arce, F.; Bennett, C.L.; Collins, M.; Escors, D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol. Med. 2011, 3, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Ohigashi, Y.; Sho, M.; Yamada, Y.; Tsurui, Y.; Hamada, K.; Ikeda, N.; Mizuno, T.; Yoriki, R.; Kashizuka, H.; Yane, K.; et al. Clinical Significance of Programmed Death-1 Ligand-1 and Programmed Death-1 Ligand-2 Expression in Human Esophageal Cancer. Clin. Cancer Res. 2005, 11, 2947–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, T.; Shah, M.A.; Muro, K.; Francois, E.; Adenis, A.; Hsu, C.-H.; Doi, T.; Moriwaki, T.; Kim, S.-B.; Lee, S.-H.; et al. KEYNOTE-181 Investigators Randomized Phase III KEYNOTE-181 Study of Pembrolizumab Versus Chemotherapy in Advanced Esophageal Cancer. J. Clin. Oncol. 2020, 38, 4138–4148. [Google Scholar] [CrossRef]
- Shitara, K.; Özgüroglu, M.; Bang, Y.-J.; Di Bartolomeo, M.; Mandalà, M.; Ryu, M.-H.; Fornaro, L.; Olesinski, T.; Caglevic, C.; Chung, H.; et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): A randomised, open-label, controlled, phase 3 trial. Lancet 2018, 392, 123–133. [Google Scholar] [CrossRef]
- Cho, B.; Kato, K.; Takahashi, M.; Okada, M.; Lin, C.-Y.; Chin, K.; Kadowaki, S.; Ahn, M.-J.; Hamamoto, Y.; Doki, Y.; et al. Nivolumab versus chemotherapy in advanced esophageal squamous cell carcinoma (ESCC): The phase III ATTRACTION-3 study. Ann. Oncol. 2019, 30, v873–v874. [Google Scholar] [CrossRef]
- Schumacher, K.; Haensch, W.; Röefzaad, C.; Schlag, P.M. Prognostic significance of activated CD8(+) T cell infiltrations within esophageal carcinomas. Cancer Res. 2001, 61, 3932–3936. [Google Scholar] [PubMed]
- Thompson, E.D.; Zahurak, M.; Murphy, A.; Cornish, T.; Cuka, N.; Abdelfatah, E.; Yang, S.; Duncan, M.; Ahuja, N.; Taube, J.M.; et al. Patterns of PD-L1 expression and CD8 T cell infiltration in gastric adenocarcinomas and associated immune stroma. Gut 2017, 66, 794–801. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [Green Version]
- Karakasheva, T.; Waldron, T.J.; Eruslanov, E.; Kim, S.-B.; Lee, J.-S.; O’Brien, S.; Hicks, P.; Basu, D.; Singhal, S.; Malavasi, F.; et al. CD38-Expressing Myeloid-Derived Suppressor Cells Promote Tumor Growth in a Murine Model of Esophageal Cancer. Cancer Res. 2015, 75, 4074–4085. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Chen, Z.; Han, Y.; Han, L.; Zou, X.; Zhou, B.; Hu, R.; Hao, J.; Bai, S.; Xiao, H.; et al. Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment. Nat. Commun. 2020, 11, 6268. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Drug | Targeted Mechanism | Stage Towards Clinical Application, Reference | Including EAC/ESCC |
|---|---|---|---|
| Carlumab | CCL2 Inhibition | Phase I [89] | + |
| Vanucizumab | VEGF/ANG-2 Inhibition | Phase I, NCT02665416 | − |
| CP-870,893 | CD40 Agonism | Phase I [90] | − |
| AMG820 | CSF-1R Inhibition | Phase I [91] | − |
| LY3022855 | CSF-1R Inhibition | Phase I NCT02718911 | + |
| EF-022 (Efranat) | Modified vitamin-D-binding protein (macrophage-activating factor) | Phase I NCT02052492 | + |
| PLX7486 | CSF-1R Inhibition | Phase I NCT01804530 | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiffmann, L.M.; Plum, P.S.; Fuchs, H.F.; Babic, B.; Bruns, C.J.; Schmidt, T. Tumor Microenvironment of Esophageal Cancer. Cancers 2021, 13, 4678. https://doi.org/10.3390/cancers13184678
Schiffmann LM, Plum PS, Fuchs HF, Babic B, Bruns CJ, Schmidt T. Tumor Microenvironment of Esophageal Cancer. Cancers. 2021; 13(18):4678. https://doi.org/10.3390/cancers13184678
Chicago/Turabian StyleSchiffmann, Lars M., Patrick S. Plum, Hans F. Fuchs, Benjamin Babic, Christiane J. Bruns, and Thomas Schmidt. 2021. "Tumor Microenvironment of Esophageal Cancer" Cancers 13, no. 18: 4678. https://doi.org/10.3390/cancers13184678
APA StyleSchiffmann, L. M., Plum, P. S., Fuchs, H. F., Babic, B., Bruns, C. J., & Schmidt, T. (2021). Tumor Microenvironment of Esophageal Cancer. Cancers, 13(18), 4678. https://doi.org/10.3390/cancers13184678