
Metabolic Flexibility Is a Determinant of Breast Cancer Heterogeneity and Progression


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
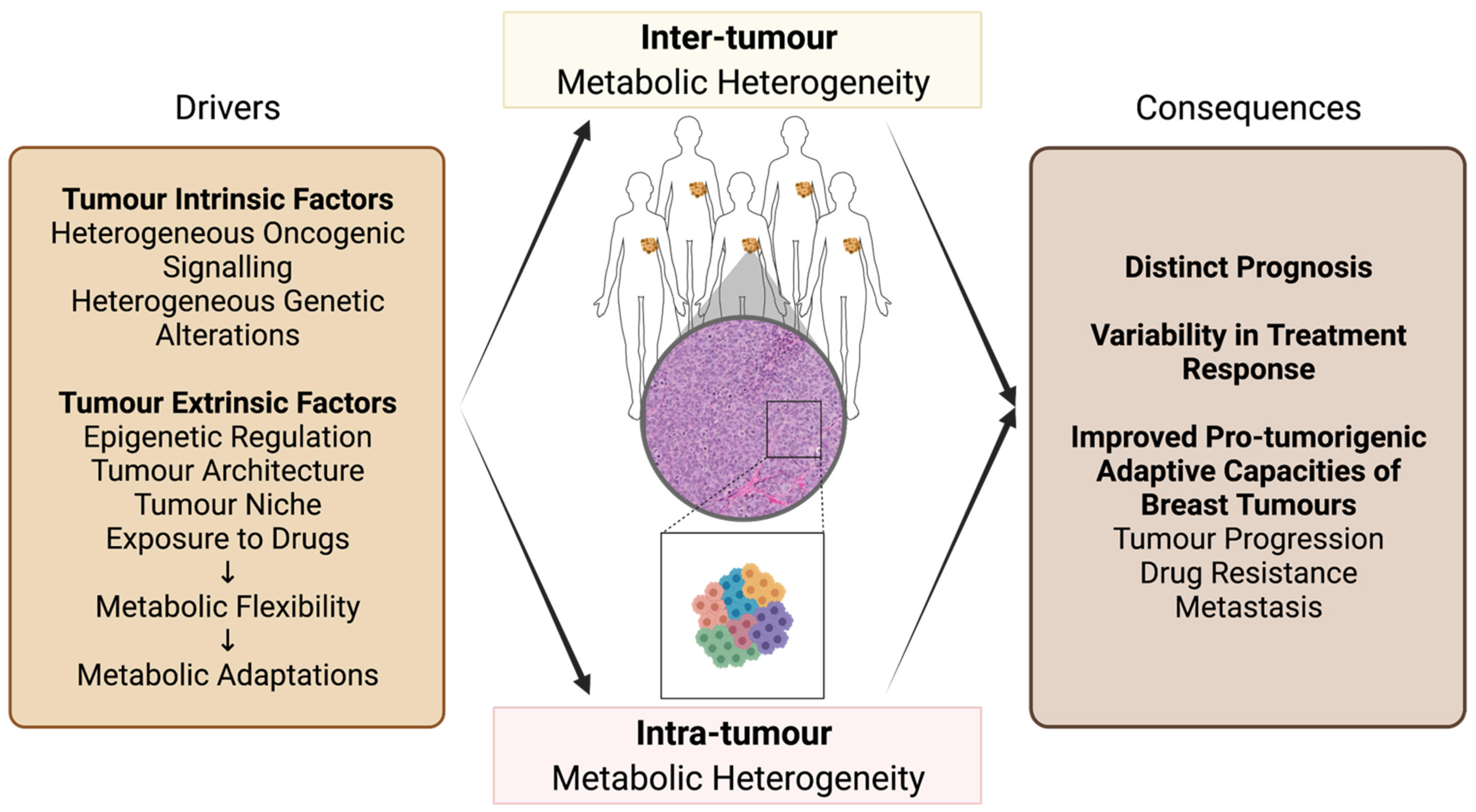
2. Metabolic Heterogeneity of Breast Cancer
2.1. Inter-Tumour Metabolic Heterogeneity of Breast Cancer
2.2. Inter-Tumour Metabolic Heterogeneity within Breast Cancer Subtypes
2.3. Intra-Tumour Metabolic Heterogeneity of Breast Cancer
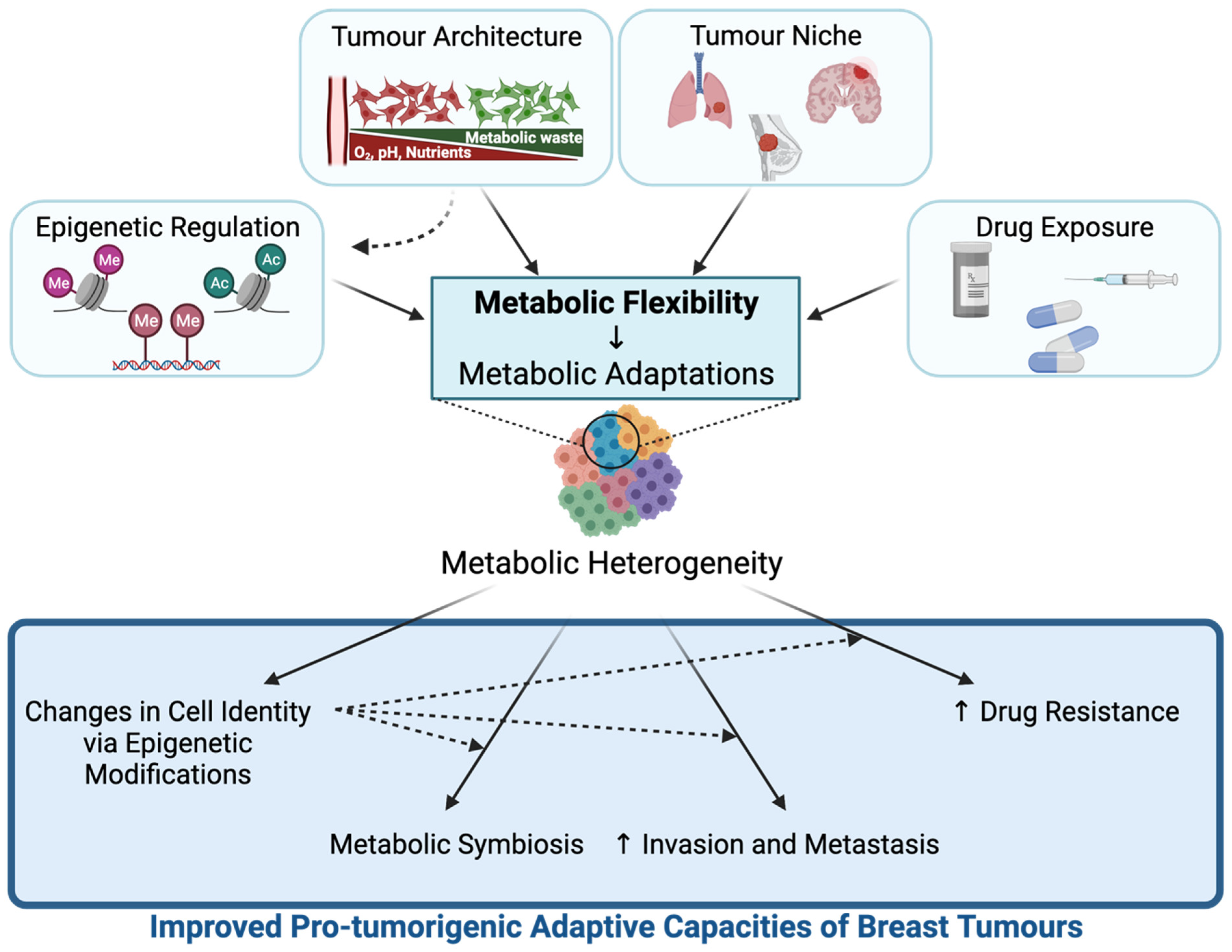
3. Metabolic Flexibility Contributes to Breast Tumour Metabolic Heterogeneity and Pro-Tumorigenic Adaptive Capacities of Breast Tumours
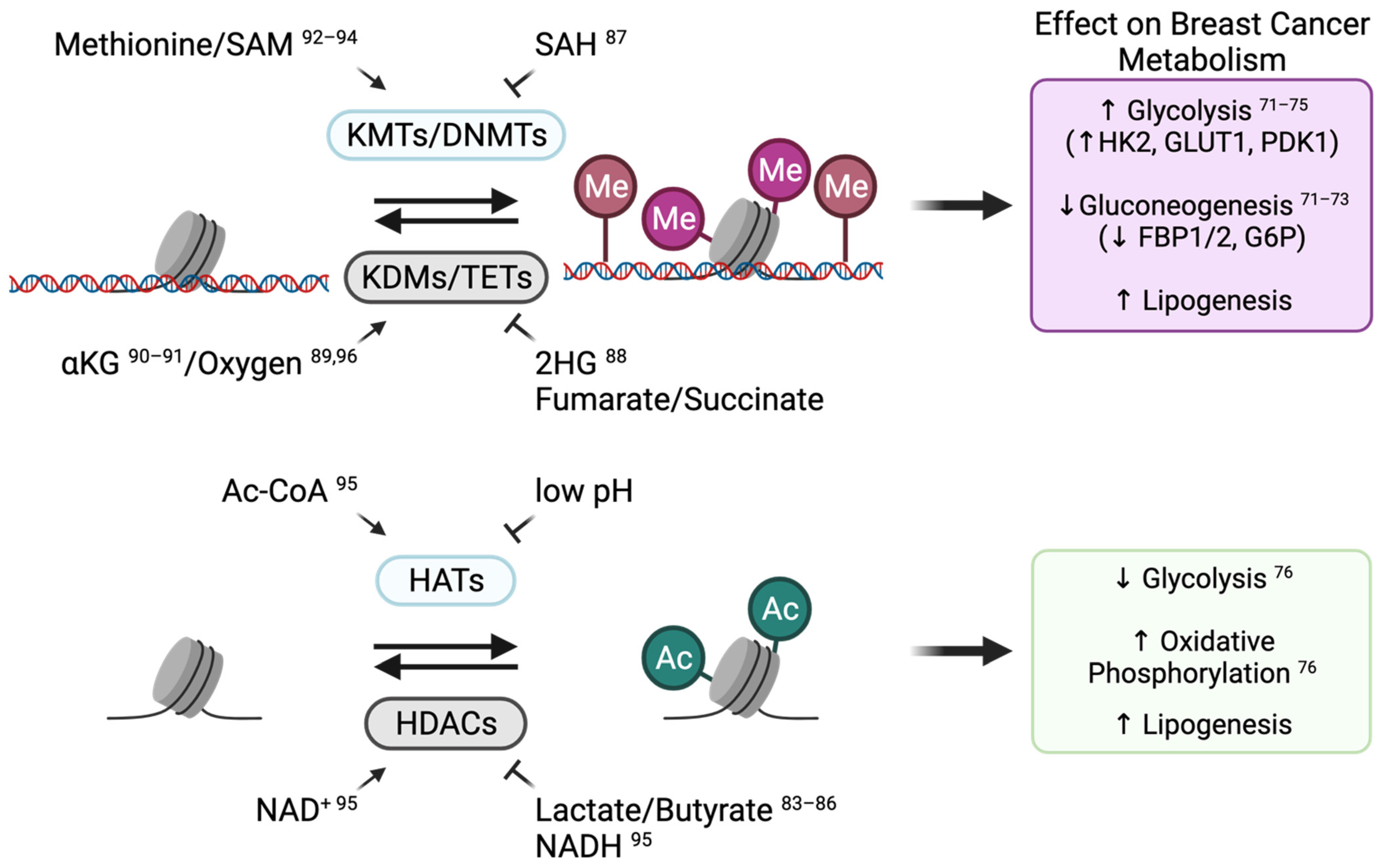
3.1. Epigenetic Regulation
3.1.1. Epigenetic Reprogramming Can Modulate Metabolic Gene Expression
3.1.2. Metabolic Adaptations Alter Breast Cancer Cell Identity by Modulating Chromatin-Modifying Enzyme Activity
3.2. Tumour Architecture and Microenvironment
3.2.1. 3D Spatial Organization Affects Metabolic Profiles in Breast Tumours
3.2.2. Metabolic Symbiosis within the Tumour Microenvironment Can Contribute to Breast Tumour Development and Progression
3.3. Metastasis
3.3.1. Tumour Niche Provides Specific Microenvironments That Modulate Metabolic Adaptations in Breast Cancer Cells
3.3.2. Metabolic Flexibility of Breast Cancer Cells Contributes to Enhanced Invasion and Metastasis
3.4. Breast Cancer Treatment and Metabolic Adaptations
3.4.1. Exposure to Anticancer Drugs Can Induce Metabolic Adaptations in Breast Cancer Cells
3.4.2. Metabolic Flexibility Contributes to the Development of Drug Resistance in Breast Tumours
4. Targeting Metabolic Adaptations as a Therapeutic Approach for Breast Cancer Patients
4.1. Challenges in Developing a Therapy Targeting Breast Cancer Metabolism
4.2. Targeting Metabolic Adaptations of Breast Cancer Using Combination Therapy
4.2.1. Using Drugs Targeting Tumour Metabolism as Part of Combination Therapies
4.2.2. Strategies Targeting Cancer Metabolism and Epigenetic-Modifying Enzymes
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghoncheh, M.; Pournamdar, Z.; Salehiniya, H. Incidence and Mortality and Epidemiology of Breast Cancer in the World. Asian Pac. J. Cancer Prev. 2016, 17, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Riggio, A.I.; Varley, K.E.; Welm, A.L. The lingering mysteries of metastatic recurrence in breast cancer. Br. J. Cancer 2021, 124, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Sims, A.H.; Howell, A.; Howell, S.J.; Clarke, R.B. Origins of breast cancer subtypes and therapeutic implications. Nat. Clin. Pract. Oncol. 2007, 4, 516–525. [Google Scholar] [CrossRef]
- Allison, K.H. Molecular pathology of breast cancer: What a pathologist needs to know. Am. J. Clin. Pathol. 2012, 138, 770–780. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehuédé, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res. 2016, 76, 5201–5208. [Google Scholar] [CrossRef] [Green Version]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabariès, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787.e5. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- Deblois, G.; Smith, H.W.; Tam, I.S.; Gravel, S.-P.; Caron, M.; Savage, P.; Labbé, D.P.; Bégin, L.R.; Tremblay, M.L.; Park, M.; et al. ERRα mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat. Commun. 2016, 7, 12156. [Google Scholar] [CrossRef]
- Deblois, G.; Tonekaboni, S.A.M.; Grillo, G.; Martinez, C.; Kao, Y.I.; Tai, F.; Ettayebi, I.; Fortier, A.-M.; Savage, P.; Fedor, A.N.; et al. Epigenetic Switch-Induced Viral Mimicry Evasion in Chemotherapy-Resistant Breast Cancer. Cancer Discov. 2020, 10, 1312–1329. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, M.; Wang, M.; Yu, X.; Guo, J.; Sun, T.; Li, X.; Yao, L.; Dong, H.; Xu, Y. Metabolic Reprogramming in Triple-Negative Breast Cancer. Front. Oncol. 2020, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, Q.; Dong, C. Metabolic reprogramming in triple-negative breast cancer. Cancer Biol. Med. 2020, 17, 44–59. [Google Scholar] [CrossRef]
- Mishra, P.; Ambs, S. Metabolic Signatures of Human Breast Cancer. Mol. Cell. Oncol. 2015, 2, e992217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.-P.; Li, X.-N.; Zhang, F. Targeting metabolism in breast cancer: How far we can go? World J. Clin. Oncol. 2016, 7, 122–130. [Google Scholar] [CrossRef]
- Serrano-Carbajal, E.A.; Espinal-Enríquez, J.; Hernández-Lemus, E. Targeting Metabolic Deregulation Landscapes in Breast Cancer Subtypes. Front. Oncol. 2020, 10, 97. [Google Scholar] [CrossRef] [Green Version]
- Willmann, L.; Schlimpert, M.; Halbach, S.; Erbes, T.; Stickeler, E.; Kammerer, B. Metabolic profiling of breast cancer: Differences in central metabolism between subtypes of breast cancer cell lines. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 1000, 95–104. [Google Scholar] [CrossRef]
- Hussien, R.; Brooks, G.A. Mitochondrial and plasma membrane lactate transporter and lactate dehydrogenase isoform expression in breast cancer cell lines. Physiol. Genom. 2011, 43, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, H.-N.; Marks, J.R.; Chi, J.-T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Kim, D.H.; Jung, W.H.; Koo, J.S. Metabolic interaction between cancer cells and stromal cells according to breast cancer molecular subtype. Breast Cancer Res. 2013, 15, R78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, K.J.; Koivunen, P.; Cao, S.; Backus, K.M.; Olenchock, B.A.; Patel, H.; Zhang, Q.; Signoretti, S.; Gerfen, G.J.; Richardson, A.L.; et al. Paracrine Induction of HIF by Glutamate in Breast Cancer: EglN1 Senses Cysteine. Cell 2016, 166, 126–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, E.; Lord, S.; Harris, A.L.; Buffa, F.M. Gene expression and hypoxia in breast cancer. Genome Med. 2011, 3, 55. [Google Scholar] [CrossRef]
- Koboldt, D.C.F.R.; Fulton, R.; McLellan, M.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.; Fulton, L.; Dooling, D.; Ding, L.; Mardis, E.; et al. The Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, M.D.; Lamichhane, S.; Lundgren, S.; Bofin, A.; Fjøsne, H.; Giskeødegård, G.F.; Bathen, T.F. Metabolic characterization of triple negative breast cancer. BMC Cancer 2014, 14, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van ’t Veer, L.J.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef] [Green Version]
- Castagnoli, L.; Iorio, E.; Dugo, M.; Koschorke, A.; Faraci, S.; Canese, R.; Casalini, P.; Nanni, P.; Vernieri, C.; Di Nicola, M.; et al. Intratumor lactate levels reflect HER2 addiction status in HER2-positive breast cancer. J. Cell. Physiol. 2019, 234, 1768–1779. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, W.; Holloway, P.A.M. Targeting mTOR and Glycolysis in HER2-Positive Breast Cancer. Cancers 2021, 13, 2922. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, Z.; Desai, S.; Zhao, Y.; Liu, H.; Pannell, L.K.; Yi, H.; Wright, E.R.; Owen, L.B.; Dean-Colomb, W.; et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat. Commun. 2012, 3, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lee, Y.; Koo, J.S. Differential expression of lipid metabolism-related proteins in different breast cancer subtypes. PLoS ONE 2015, 10, e0119473. [Google Scholar] [CrossRef] [Green Version]
- Jin, Q.; Yuan, L.X.; Boulbes, D.; Baek, J.M.; Wang, Y.N.; Gomez-Cabello, D.; Hawke, D.H.; Yeung, S.C.; Lee, M.H.; Hortobagyi, G.N.; et al. Fatty acid synthase phosphorylation: A novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 2010, 12, R96. [Google Scholar] [CrossRef] [Green Version]
- Poudel, P.; Nyamundanda, G.; Patil, Y.; Cheang, M.C.U.; Sadanandam, A. Heterocellular gene signatures reveal luminal-A breast cancer heterogeneity and differential therapeutic responses. NPJ Breast Cancer 2019, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Bediaga, N.G.; Beristain, E.; Calvo, B.; Viguri, M.A.; Gutierrez-Corres, B.; Rezola, R.; Ruiz-Diaz, I.; Guerra, I.; De Pancorbo, M.M. Luminal B breast cancer subtype displays a dicotomic epigenetic pattern. Springerplus 2016, 5, 623. [Google Scholar] [CrossRef] [Green Version]
- Godoy-Ortiz, A.; Sanchez-Muñoz, A.; Chica Parrado, M.R.; Álvarez, M.; Ribelles, N.; Rueda Dominguez, A.; Alba, E. Deciphering HER2 Breast Cancer Disease: Biological and Clinical Implications. Front. Oncol. 2019, 9, 1124. [Google Scholar] [CrossRef]
- Craze, M.L.; Cheung, H.; Jewa, N.; Coimbra, N.D.M.; Soria, D.; El-Ansari, R.; Aleskandarany, M.A.; Wai Cheng, K.; Diez-Rodriguez, M.; Nolan, C.C.; et al. MYC regulation of glutamine-proline regulatory axis is key in luminal B breast cancer. Br. J. Cancer 2018, 118, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Shi, W.; Natowicz, R.; Ononye, S.N.; Wali, V.B.; Kluger, Y.; Pusztai, L.; Hatzis, C. Statistical measures of transcriptional diversity capture genomic heterogeneity of cancer. BMC Genom. 2014, 15, 876. [Google Scholar] [CrossRef] [Green Version]
- Elia, I.; Schmieder, R.; Christen, S.; Fendt, S.-M. Organ-Specific Cancer Metabolism and Its Potential for Therapy. Handb. Exp. Pharmacol. 2016, 233, 321–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortmayr, K.; Dubuis, S.; Zampieri, M. Metabolic profiling of cancer cells reveals genome-wide crosstalk between transcriptional regulators and metabolism. Nat. Commun. 2019, 10, 1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Ji, P.; Yang, Y.-S.; Xie, S.; Yu, T.-J.; Xiao, Y.; Jin, M.-L.; Ma, D.; Guo, L.-W.; Pei, Y.-C.; et al. Metabolic-Pathway-Based Subtyping of Triple-Negative Breast Cancer Reveals Potential Therapeutic Targets. Cell Metab. 2021, 33, 51–64.e9. [Google Scholar] [CrossRef] [PubMed]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Ba-Alawi, W.; Deblois, G.; Cruickshank, J.; Duan, S.; Lima-Fernandes, E.; Haight, J.; Tonekaboni, S.A.M.; Fortier, A.-M.; Kuasne, H.; et al. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat. Commun. 2020, 11, 4205. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Argüello, R.J.; Combes, A.J.; Char, R.; Gigan, J.-P.; Baaziz, A.I.; Bousiquot, E.; Camosseto, V.; Samad, B.; Tsui, J.; Yan, P.; et al. SCENITH: A Flow Cytometry-Based Method to Functionally Profile Energy Metabolism with Single-Cell Resolution. Cell Metab. 2020, 32, 1063–1075.e7. [Google Scholar] [CrossRef]
- Rappez, L.; Stadler, M.; Triana, S.; Gathungu, R.M.; Ovchinnikova, K.; Phapale, P.; Heikenwalder, M.; Alexandrov, T. SpaceM reveals metabolic states of single cells. Nat. Methods 2021, 18, 799–805. [Google Scholar] [CrossRef]
- Hartmann, F.J.; Mrdjen, D.; McCaffrey, E.; Glass, D.R.; Greenwald, N.F.; Bharadwaj, A.; Khair, Z.; Verberk, S.G.S.; Baranski, A.; Baskar, R.; et al. Single-cell metabolic profiling of human cytotoxic T cells. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef]
- Luskin, M.R.; Murakami, M.A.; Manalis, S.R.; Weinstock, D.M. Targeting minimal residual disease: A path to cure? Nat. Rev. Cancer 2018, 18, 255–263. [Google Scholar] [CrossRef]
- Caswell, D.R.; Swanton, C. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med. 2017, 15, 133. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Glöckner, S.; Buurman, H.; Kleeberger, W.; Lehmann, U.; Kreipe, H. Marked intratumoral heterogeneity of c-myc and cyclinD1 but not of c-erbB2 amplification in breast cancer. Lab. Investig. 2002, 82, 1419–1426. [Google Scholar] [CrossRef] [Green Version]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [Green Version]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Warita, K.; Warita, T.; Faeder, J.R.; Lee, R.E.C.; Sant, S.; Oltvai, Z.N. Shift from Stochastic to Spatially-Ordered Expression of Serine-Glycine Synthesis Enzymes in 3D Microtumors. Sci. Rep. 2018, 8, 9388. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Ratcliffe, C.D.H.; Hooper, S.; Ellis, J.; MacRae, J.I.; Hennequart, M.; Dunsby, C.W.; Anderson, K.I.; Sahai, E. Single-cell resolved imaging reveals intra-tumor heterogeneity in glycolysis, transitions between metabolic states, and their regulatory mechanisms. Cell Rep. 2021, 34, 108750. [Google Scholar] [CrossRef] [PubMed]
- Wein, L.; Loi, S. Mechanisms of resistance of chemotherapy in early-stage triple negative breast cancer (TNBC). Breast 2017, 34 (Suppl. 1), S27–S30. [Google Scholar] [CrossRef]
- Allison, K.H.; Sledge, G.W. Heterogeneity and cancer. Oncology 2014, 28, 772–778. [Google Scholar]
- Hoadley, K.A.; Siegel, M.B.; Kanchi, K.L.; Miller, C.A.; Ding, L.; Zhao, W.; He, X.; Parker, J.S.; Wendl, M.C.; Fulton, R.S.; et al. Tumor Evolution in Two Patients with Basal-like Breast Cancer: A Retrospective Genomics Study of Multiple Metastases. PLoS Med. 2016, 13, e1002174. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, Y.; Li, Q.; Cao, L.; Sun, Z.; Jin, J.; Fang, H.; Zhu, A.; Li, Y.; Zhang, W.; et al. Intratumor heterogeneity predicts metastasis of triple-negative breast cancer. Carcinogenesis 2017, 38, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Reid, M.A.; Kong, M.; Locasale, J.W. Metabolic interactions with cancer epigenetics. Mol. Asp. Med. 2017, 54, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A. Hiding in Plain Sight: Epigenetic Plasticity in Drug-Induced Tumor Evolution. Epigenet. Insights 2019, 12, 2516865719870760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [Green Version]
- Gayther, S.A.; Batley, S.J.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.F.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.M.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.M.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, W. Epithelial-mesenchymal transition in human cancer: Comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol. Ther. 2015, 150, 33–46. [Google Scholar] [CrossRef]
- Pan, D.; Mao, C.; Wang, Y.-X. Suppression of gluconeogenic gene expression by LSD1-mediated histone demethylation. PLoS ONE 2013, 8, e66294. [Google Scholar] [CrossRef]
- Luo, W.; Chang, R.; Zhong, J.; Pandey, A.; Semenza, G.L. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc. Natl. Acad. Sci. USA 2012, 109, E3367–E3376. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Serra, P.; Marcilla, M.; Villanueva, A.; Ramos-Fernandez, A.; Palau, A.; Leal, L.; Wahi, J.E.; Setien-Baranda, F.; Szczesna, K.; Moutinho, C.; et al. A DERL3-associated defect in the degradation of SLC2A1 mediates the Warburg effect. Nat. Commun. 2014, 5, 3608. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.F.; Carvalho, É.; Pezzuto, P.; Rumjanek, F.D.; Amoêdo, N.D. Reciprocal modulation of histone deacetylase inhibitors sodium butyrate and trichostatin A on the energy metabolism of breast cancer cells. J. Cell. Biochem. 2015, 116, 797–808. [Google Scholar] [CrossRef] [PubMed]
- da Motta, L.L.; Ledaki, I.; Purshouse, K.; Haider, S.; De Bastiani, M.A.; Baban, D.; Morotti, M.; Steers, G.; Wigfield, S.; Bridges, E.; et al. The BET Inhibitor JQ1 Selectively Impairs Tumour Response to Hypoxia and Downregulates CA9 and Angiogenesis in Triple Negative Breast Cancer. Oncogene 2017, 36, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrieu, G.P.; Shafran, J.S.; Deeney, J.T.; Bharadwaj, K.R.; Rangarajan, A.; Denis, G.V. BET proteins in abnormal metabolism, inflammation, and the breast cancer microenvironment. J. Leukoc. Biol. 2018, 104, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Tonini, C.; Colardo, M.; Colella, B.; Di Bartolomeo, S.; Berardinelli, F.; Caretti, G.; Pallottini, V.; Segatto, M. Inhibition of Bromodomain and Extraterminal Domain (BET) Proteins by JQ1 Unravels a Novel Epigenetic Modulation to Control Lipid Homeostasis. Int. J. Mol. Sci. 2020, 21, 1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, T.Q.; Lowman, X.H.; Kong, M. Molecular Pathways: Metabolic Control of Histone Methylation and Gene Expression in Cancer. Clin. Cancer Res. 2017, 23, 4004–4009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.; Ramesh, V.; Locasale, J.W. The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 2020, 21, 737–753. [Google Scholar] [CrossRef]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012, 40, 4794–4803. [Google Scholar] [CrossRef] [Green Version]
- Wagner, W.; Ciszewski, W.M.; Kania, K.D. L- and D-lactate enhance DNA repair and modulate the resistance of cervical carcinoma cells to anticancer drugs via histone deacetylase inhibition and hydroxycarboxylic acid receptor 1 activation. Cell Commun. Signal. 2015, 13, 36. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Lisanti, M.P.; Sotgia, F. Ketone bodies and two-compartment tumor metabolism: Stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 2012, 11, 3956–3963. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef] [Green Version]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terunuma, A.; Putluri, N.; Mishra, P.; Mathé, E.A.; Dorsey, T.H.; Yi, M.; Wallace, T.A.; Issaq, H.J.; Zhou, M.; Killian, J.K.; et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Investig. 2014, 124, 398–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prickaerts, P.; Adriaens, M.E.; Van Den Beucken, T.; Koch, E.; Dubois, L.; Dahlmans, V.E.H.; Gits, C.; Evelo, C.T.A.; Chan-Seng-Yue, M.; Wouters, B.G.; et al. Hypoxia increases genome-wide bivalent epigenetic marking by specific gain of H3K27me3. Epigenet. Chromatin 2016, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef]
- Ishak Gabra, M.B.; Yang, Y.; Li, H.; Senapati, P.; Hanse, E.A.; Lowman, X.H.; Tran, T.Q.; Zhang, L.; Doan, L.T.; Xu, X.; et al. Dietary glutamine supplementation suppresses epigenetically-activated oncogenic pathways to inhibit melanoma tumour growth. Nat. Commun. 2020, 11, 3326. [Google Scholar] [CrossRef]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Gómez Padilla, P.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Mentch, S.J.; Gao, X.; Nichenametla, S.N.; Locasale, J.W. Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat. Commun. 2018, 9, 1955. [Google Scholar] [CrossRef]
- Gao, X.; Sanderson, S.M.; Dai, Z.; Reid, M.A.; Cooper, D.E.; Lu, M.; Richie, J.P., Jr.; Ciccarella, A.; Calcagnotto, A.; Mikhael, P.G.; et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 2019, 572, 397–401. [Google Scholar] [CrossRef]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.-F.; Lim, H.-W.; Liu, S.; et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Van Den Beucken, T.; Koch, E.; Chu, K.; Rupaimoole, R.; Prickaerts, P.; Adriaens, M.; Voncken, J.W.; Harris, A.L.; Buffa, F.M.; Haider, S.; et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat. Commun. 2014, 5, 5203. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sharife, H.; Kreisel, T.; Mogilevsky, M.; Bar-Lev, L.; Grunewald, M.; Aizenshtein, E.; Karni, R.; Paldor, I.; Shlomi, T.; et al. Intra-Tumoral Metabolic Zonation and Resultant Phenotypic Diversification Are Dictated by Blood Vessel Proximity. Cell Metab. 2019, 30, 201–211.e6. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisarsky, L.; Bill, R.; Fagiani, E.; Dimeloe, S.; Goosen, R.W.; Hagmann, J.; Hess, C.; Christofori, G. Targeting Metabolic Symbiosis to Overcome Resistance to Anti-angiogenic Therapy. Cell Rep. 2016, 15, 1161–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.N.; Zheng, G.; Tchou, J.; Nioka, S.; Li, L.Z. Characterizing the metabolic heterogeneity in human breast cancer xenografts by 3D high resolution fluorescence imaging. SpringerPlus 2013, 2, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; Saedeleer, C.J.D.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.-N.; Vidal, N.; Berthezène, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Miéville, P.; Warren, C.M.; Saghafinia, S.; Li, L.; Peng, M.-W.; Hanahan, D. Metabolic Symbiosis Enables Adaptive Resistance to Anti-angiogenic Therapy that Is Dependent on mTOR Signaling. Cell Rep. 2016, 15, 1144–1160. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, E.C.; Van Houten, B. Metabolic symbiosis in cancer: Refocusing the Warburg lens. Mol. Carcinog. 2013, 52, 329–337. [Google Scholar] [CrossRef]
- Kennedy, K.M.; Scarbrough, P.M.; Ribeiro, A.; Richardson, R.; Yuan, H.; Sonveaux, P.; Landon, C.D.; Chi, J.-T.; Pizzo, S.; Schroeder, T.; et al. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS ONE 2013, 8, e75154. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Bio. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; Van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reggiani, F.; Bertolini, F. Roles of obesity in the development and progression of breast cancer. Discov. Med. 2017, 24, 183–190. [Google Scholar] [PubMed]
- Zahid, H.; Simpson, E.R.; Brown, K.A. Inflammation, dysregulated metabolism and aromatase in obesity and breast cancer. Curr. Opin. Pharmacol. 2016, 31, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef] [Green Version]
- Pollari, S.; Käkönen, S.-M.; Edgren, H.; Wolf, M.; Kohonen, P.; Sara, H.; Guise, T.; Nees, M.; Kallioniemi, O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 2011, 125, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemma, S.; Di Pompo, G.; Porporato, P.E.; Sboarina, M.; Russell, S.; Gillies, R.J.; Baldini, N.; Sonveaux, P.; Avnet, S. MDA-MB-231 breast cancer cells fuel osteoclast metabolism and activity: A new rationale for the pathogenesis of osteolytic bone metastases. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3254–3264. [Google Scholar] [CrossRef]
- Ngo, B.; Kim, E.; Osorio-Vasquez, V.; Doll, S.; Bustraan, S.; Liang, R.J.; Luengo, A.; Davidson, S.M.; Ali, A.; Ferraro, G.B.; et al. Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer Discov. 2020, 10, 1352–1373. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, G.B.; Ali, A.; Luengo, A.; Kodack, D.P.; Deik, A.; Abbott, K.L.; Bezwada, D.; Blanc, L.; Prideaux, B.; Jin, X.; et al. Fatty acid synthesis is required for breast cancer brain metastasis. Nat Cancer 2021, 2, 414–428. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, N.; Parbin, S.; Kar, S.; Das, L.; Kirtana, R.; Suma Seshadri, G.; Sengupta, D.; Deb, M.; Kausar, C.; Patra, S.K. Epigenetic silencing of genes enhanced by collective role of reactive oxygen species and MAPK signaling downstream ERK/Snail axis: Ectopic application of hydrogen peroxide repress CDH1 gene by enhanced DNA methyltransferase activity in human breast cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1651–1665. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Spinelli, J.B.; Asara, J.M.; Toker, A. Adaptive Reprogramming of De Novo Pyrimidine Synthesis Is a Metabolic Vulnerability in Triple-Negative Breast Cancer. Cancer Discov. 2017, 7, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.P.; Mockabee-Macias, A.; Jiang, C.; Falzone, A.; Prieto-Farigua, N.; Stone, E.; Harris, I.S.; DeNicola, G.M. Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab. 2021, 33, 174–189.e7. [Google Scholar] [CrossRef]
- Liu, Y.; Hyde, A.S.; Simpson, M.A.; Barycki, J.J. Emerging regulatory paradigms in glutathione metabolism. Adv. Cancer Res. 2014, 122, 69–101. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; Ledoux, S.P.; Wilson, G.L.; Voellmy, R.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011, 71, 4585–4597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol. Cancer 2010, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, E.C.; Lyssiotis, C.A.; Juvekar, A.; Hu, H.; Asara, J.M.; Cantley, L.C.; Toker, A. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 2016, 18, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Pommey, S.; Smyth, M.J.; Stagg, J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res. 2013, 19, 5626–5635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Masri, M.; Paliotti, K.; Tran, R.; Halaoui, R.; Lelarge, V.; Chatterjee, S.; Wang, L.-T.; Moraes, C.; McCaffrey, L. Architectural control of metabolic plasticity in epithelial cancer cells. Commun. Biol. 2021, 4, 371. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension: How 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [Green Version]
- Wulftange, W.J.; Rose, M.A.; Garmendia-Cedillos, M.; da Silva, D.; Poprawski, J.E.; Srinivasachar, D.; Sullivan, T.; Lim, L.; Bliskovsky, V.V.; Hall, M.D.; et al. Spatial control of oxygen delivery to three-dimensional cultures alters cancer cell growth and gene expression. J. Cell. Physiol. 2019, 234, 20608–20622. [Google Scholar] [CrossRef]
- Hickman, J.A.; Graeser, R.; De Hoogt, R.; Vidic, S.; Brito, C.; Gutekunst, M.; Van Der Kuip, H.; IMI PREDECT Consortium. Three-dimensional models of cancer for pharmacology and cancer cell biology: Capturing tumor complexity In Vitro/ex vivo. Biotechnol. J. 2014, 9, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.; Pacis, A.; Kuasne, H.; Liu, L.; Lai, D.; Wan, A.; Dankner, M.; Martinez, C.; Muñoz-Ramos, V.; Pilon, V.; et al. Chemogenomic profiling of breast cancer patient-derived xenografts reveals targetable vulnerabilities for difficult-to-treat tumors. Commun. Biol. 2020, 3, 310. [Google Scholar] [CrossRef]
- Savage, P.; Blanchet-Cohen, A.; Revil, T.; Badescu, D.; Saleh, S.M.I.; Wang, Y.-C.; Zuo, D.; Liu, L.; Bertos, N.R.; Munoz-Ramos, V.; et al. A Targetable EGFR-Dependent Tumor-Initiating Program in Breast Cancer. Cell Rep. 2017, 21, 1140–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhute, V.J.; Ma, Y.; Bao, X.; Palecek, S.P. The Poly (ADP-Ribose) Polymerase Inhibitor Veliparib and Radiation Cause Significant Cell Line Dependent Metabolic Changes in Breast Cancer Cells. Sci. Rep. 2016, 6, 36061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emberley, E.D.; Bennett, M.; Chen, J.; Gross, M.; Huang, T.; Makkouk, A.; Marguier, G.; Pan, A.; Spurlock, S.M.; Steggerda, S.; et al. The glutaminase inhibitor CB-839 synergizes with CDK4/6 and PARP inhibitors in pre-clinical tumor models. Cancer Res. 2018, 78, 3509. [Google Scholar] [CrossRef]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Braas, D.; Go, J.; Graeber, T.G.; Parlati, F.; Demo, S.; Li, R.; et al. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell 2018, 33, 905–921.e5. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863–35876. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Magyar, C.; Braas, D.; Graeber, T.; Jackson, N.J.; Czernin, J.; Emberley, E.; et al. Targeted Inhibition of EGFR and Glutaminase Induces Metabolic Crisis in EGFR Mutant Lung Cancer. Cell Rep. 2017, 18, 601–610. [Google Scholar] [CrossRef]
- Zaal, E.A.; Berkers, C.R. The Influence of Metabolism on Drug Response in Cancer. Front. Oncol. 2018, 8, 500. [Google Scholar] [CrossRef]
- Desbats, M.A.; Giacomini, I.; Prayer-Galetti, T.; Montopoli, M. Metabolic Plasticity in Chemotherapy Resistance. Front. Oncol. 2020, 10, 281. [Google Scholar] [CrossRef]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenetics 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Heidenreich, D.; Zhou, S.; Ackloo, S.; Krämer, A.; Nakka, K.; Lima-Fernandes, E.; Deblois, G.; Duan, S.; Vellanki, R.N.; et al. A chemical toolbox for the study of bromodomains and epigenetic signaling. Nat. Commun. 2019, 10, 1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.-S.; Little, J.B.; Yuan, Z.-M. Glycolytic metabolism influences global chromatin structure. Oncotarget 2015, 6, 4214–4225. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukano, M.; Park, M.; Deblois, G. Metabolic Flexibility Is a Determinant of Breast Cancer Heterogeneity and Progression. Cancers 2021, 13, 4699. https://doi.org/10.3390/cancers13184699
Fukano M, Park M, Deblois G. Metabolic Flexibility Is a Determinant of Breast Cancer Heterogeneity and Progression. Cancers. 2021; 13(18):4699. https://doi.org/10.3390/cancers13184699
Chicago/Turabian StyleFukano, Marina, Morag Park, and Geneviève Deblois. 2021. "Metabolic Flexibility Is a Determinant of Breast Cancer Heterogeneity and Progression" Cancers 13, no. 18: 4699. https://doi.org/10.3390/cancers13184699