
Proteomic Landscape of Prostate Cancer: The View Provided by Quantitative Proteomics, Integrative Analyses, and Protein Interactomes

Abstract
:Simple Summary
Abstract
1. Introduction
2. Proteomes of Clinical Prostate Cancer Samples
2.1. Large-Scale Proteomics of Primary Cancer of the Prostate
2.1.1. Functional Pathways’ Surfacing Based on Proteomics Studies of Primary Prostate Cancer
2.1.2. Utility of Large-Scale Proteomics in Patient Stratification and Personalized Medicine
2.1.3. Proteomics in Assessing Heterogeneity in Prostate Cancer
2.2. Large-Scale Proteomics of Advanced Prostate Cancer
2.3. Integrative Studies Comparing Genomic, Transcriptomic, and Proteomic Alterations in Clinical Prostate Cancer
3. Large-Scale Proteomes of Prostate Cancer Models Provide Mechanistic Insights
4. Large-Scale Proteomics in Protein Dynamics: Functional Interactomes and Subcellular Localization Patterns in Prostate Cancer
4.1. Interactomes of AR wt and Mutants
4.2. Other Prostate Cancer-Relevant Protein Interactomes
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Filson, C.P.; Marks, L.S.; Litwin, M.S. Expectant management for men with early stage prostate cancer. CA Cancer J. Clin. 2015, 65, 265–282. [Google Scholar] [CrossRef] [Green Version]
- Litwin, M.S.; Tan, H.J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef]
- Artibani, W.; Porcaro, A.B.; De Marco, V.; Cerruto, M.A.; Siracusano, S. Management of Biochemical Recurrence after Primary Curative Treatment for Prostate Cancer: A Review. Urol. Int. 2018, 100, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [Green Version]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Epstein, J.I.; Egevad, L.; Amin, M.B.; Delahunt, B.; Srigley, J.R.; Humphrey, P.A.; Grading Committee. The 2014 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma: Definition of Grading Patterns and Proposal for a New Grading System. Am. J. Surg. Pathol. 2016, 40, 244–252. [Google Scholar] [CrossRef]
- Pierorazio, P.M.; Walsh, P.C.; Partin, A.W.; Epstein, J.I. Prognostic Gleason grade grouping: Data based on the modified Gleason scoring system. BJU Int. 2013, 111, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Cyll, K.; Ersvær, E.; Vlatkovic, L.; Pradhan, M.; Kildal, W.; Avranden Kjær, M.; Kleppe, A.; Hveem, T.S.; Carlsen, B.; Gill, S.; et al. Tumour heterogeneity poses a significant challenge to cancer biomarker research. Br. J. Cancer 2017, 117, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Dehm, S.M.; Tindall, D.J. Molecular regulation of androgen action in prostate cancer. J. Cell Biochem. 2006, 99, 333–344. [Google Scholar] [CrossRef]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocr. Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Park, K.; Tomlins, S.A.; Mudaliar, K.M.; Chiu, Y.L.; Esgueva, R.; Mehra, R.; Suleman, K.; Varambally, S.; Brenner, J.C.; MacDonald, T.; et al. Antibody-based detection of ERG rearrangement-positive prostate cancer. Neoplasia 2010, 12, 590–598. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.V.; Tomavo, N.; Huot, L.; Flourens, A.; Bonnelye, E.; Flajollet, S.; Hot, D.; Leroy, X.; de Launoit, Y.; Duterque-Coquillaud, M. Identification of novel TMPRSS2:ERG mechanisms in prostate cancer metastasis: Involvement of MMP9 and PLXNA2. Oncogene 2014, 33, 2204–2214. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Dobi, A.; Mohamed, A.; Li, H.; Thangapazham, R.L.; Furusato, B.; Shaheduzzaman, S.; Tan, S.H.; Vaidyanathan, G.; Whitman, E.; et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene 2008, 27, 5348–5353. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Liu, W.; Roberts, W.; Hooker, S.; Fedor, H.; DeMarzo, A.; Isaacs, W.; Kittles, R.A. 8q24 allelic imbalance and MYC gene copy number in primary prostate cancer. Prostate Cancer Prostatic Dis. 2010, 13, 238–243. [Google Scholar] [CrossRef] [Green Version]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Sarker, D.; Reid, A.H.; Yap, T.A.; de Bono, J.S. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin. Cancer Res. 2009, 15, 4799–4805. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Dean, J.L.; Knudsen, K.E. The role of tumor suppressor dysregulation in prostate cancer progression. Curr. Drug Targets 2013, 14, 460–471. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Rubin, M.A.; Demichelis, F. The Genomics of Prostate Cancer: A Historic Perspective. Cold Spring Harb. Perspect. Med. 2019, 9, a034942. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Haffner, M.C.; Zwart, W.; Roudier, M.P.; True, L.D.; Nelson, W.G.; Epstein, J.I.; De Marzo, A.M.; Nelson, P.S.; Yegnasubramanian, S. Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 2021, 18, 79–92. [Google Scholar] [CrossRef]
- Hahn, W.C.; Bader, J.S.; Braun, T.P.; Califano, A.; Clemons, P.A.; Druker, B.J.; Ewald, A.J.; Fu, H.; Jagu, S.; Kemp, C.J.; et al. An expanded universe of cancer targets. Cell 2021, 184, 1142–1155. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantsiou, A.; Vlahou, A.; Zoidakis, J. Tissue proteomics studies in the investigation of prostate cancer. Expert Rev. Proteom. 2018, 15, 593–611. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, A.; Pasic, M.D.; Yousef, G.M. Proteomics and peptidomics: Moving toward precision medicine in urological malignancies. Oncotarget 2016, 7, 52460–52474. [Google Scholar] [CrossRef] [Green Version]
- Tanase, C.P.; Codrici, E.; Popescu, I.D.; Mihai, S.; Enciu, A.M.; Necula, L.G.; Preda, A.; Ismail, G.; Albulescu, R. Prostate cancer proteomics: Current trends and future perspectives for biomarker discovery. Oncotarget 2017, 8, 18497–18512. [Google Scholar] [CrossRef] [Green Version]
- Tonry, C.; Finn, S.; Armstrong, J.; Pennington, S.R. Clinical proteomics for prostate cancer: Understanding prostate cancer pathology and protein biomarkers for improved disease management. Clin. Proteom. 2020, 17, 41. [Google Scholar] [CrossRef]
- Flores-Morales, A.; Iglesias-Gato, D. Quantitative Mass Spectrometry-Based Proteomic Profiling for Precision Medicine in Prostate Cancer. Front. Oncol. 2017, 7, 267. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Ni, J.; Beretov, J.; Cozzi, P.; Willcox, M.; Wasinger, V.; Walsh, B.; Graham, P.; Li, Y. Urinary biomarkers in prostate cancer detection and monitoring progression. Crit. Rev. Oncol. Hematol. 2017, 118, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Jedinak, A.; Loughlin, K.R.; Moses, M.A. Approaches to the discovery of non-invasive urinary biomarkers of prostate cancer. Oncotarget 2018, 9, 32534–32550. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Shi, T.; Srivastava, S.; Kagan, J.; Liu, T.; Rodland, K.D. Proteomic Analysis of Exosomes for Discovery of Protein Biomarkers for Prostate and Bladder Cancer. Cancers 2020, 12, 2335. [Google Scholar] [CrossRef] [PubMed]
- Khoo, A.; Liu, L.Y.; Nyalwidhe, J.O.; Semmes, O.J.; Vesprini, D.; Downes, M.R.; Boutros, P.C.; Liu, S.K.; Kislinger, T. Proteomic discovery of non-invasive biomarkers of localized prostate cancer using mass spectrometry. Nat. Rev. Urol. 2021, 1–18. [Google Scholar] [CrossRef]
- Zhu, Y.; Weiss, T.; Zhang, Q.; Sun, R.; Wang, B.; Yi, X.; Wu, Z.; Gao, H.; Cai, X.; Ruan, G.; et al. High-throughput proteomic analysis of FFPE tissue samples facilitates tumor stratification. Mol. Oncol. 2019, 13, 2305–2328. [Google Scholar] [CrossRef] [PubMed]
- Mantsiou, A.; Makridakis, M.; Fasoulakis, K.; Katafigiotis, I.; Constantinides, C.A.; Zoidakis, J.; Roubelakis, M.G.; Vlahou, A.; Lygirou, V. Proteomics Analysis of Formalin Fixed Paraffin Embedded Tissues in the Investigation of Prostate Cancer. J. Proteome Res. 2020, 19, 2631–2642. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Gato, D.; Wikström, P.; Tyanova, S.; Lavallee, C.; Thysell, E.; Carlsson, J.; Hägglöf, C.; Cox, J.; Andrén, O.; Stattin, P.; et al. The Proteome of Primary Prostate Cancer. Eur. Urol. 2016, 69, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.S.; von Lersner, A.K.; Sang, Q.X. Proteomic Upregulation of Fatty Acid Synthase and Fatty Acid Binding Protein 5 and Identification of Cancer- and Race-Specific Pathway Associations in Human Prostate Cancer Tissues. J. Cancer 2016, 7, 1452–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staunton, L.; Tonry, C.; Lis, R.; Espina, V.; Liotta, L.; Inzitari, R.; Bowden, M.; Fabre, A.; O’Leary, J.; Finn, S.P.; et al. Pathology-Driven Comprehensive Proteomic Profiling of the Prostate Cancer Tumor Microenvironment. Mol. Cancer Res. 2017, 15, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Li, L.; Zhong, Q.; Rupp, N.J.; Charmpi, K.; Wong, C.E.; Wagner, U.; Rueschoff, J.H.; Jochum, W.; Fankhauser, C.D.; et al. Multi-region proteome analysis quantifies spatial heterogeneity of prostate tissue biomarkers. Life Sci. Alliance 2018, 1, e201800042. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, R.; Recuero, S.; Nogueira, F.C.S.; Domont, G.B.; Leite, K.R.M.; Srougi, M.; Thaysen-Andersen, M.; Palmisano, G. Tissue Proteome Signatures Associated with Five Grades of Prostate Cancer and Benign Prostatic Hyperplasia. Proteomics 2019, 19, e1900174. [Google Scholar] [CrossRef]
- Zhou, B.; Yan, Y.; Wang, Y.; You, S.; Freeman, M.R.; Yang, W. Quantitative proteomic analysis of prostate tissue specimens identifies deregulated protein complexes in primary prostate cancer. Clin. Proteom. 2019, 16, 15. [Google Scholar] [CrossRef] [Green Version]
- Sinha, A.; Huang, V.; Livingstone, J.; Wang, J.; Fox, N.S.; Kurganovs, N.; Ignatchenko, V.; Fritsch, K.; Donmez, N.; Heisler, L.E.; et al. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell 2019, 35, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Latosinska, A.; Davalieva, K.; Makridakis, M.; Mullen, W.; Schanstra, J.P.; Vlahou, A.; Mischak, H.; Frantzi, M. Molecular Changes in Tissue Proteome during Prostate Cancer Development: Proof-of-Principle Investigation. Diagnostics 2020, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, J.; Sethi, A.; Li, Q.K.; Chen, L.; Collins, B.; Gillet, L.C.; Wollscheid, B.; Zhang, H.; Aebersold, R. Glycoproteomic analysis of prostate cancer tissues by SWATH mass spectrometry discovers N-acylethanolamine acid amidase and protein tyrosine kinase 7 as signatures for tumor aggressiveness. Mol. Cell Proteom. 2014, 13, 1753–1768. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.M.; Paull, E.O.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Latonen, L.; Afyounian, E.; Jylhä, A.; Nättinen, J.; Aapola, U.; Annala, M.; Kivinummi, K.K.; Tammela, T.T.L.; Beuerman, R.W.; Uusitalo, H.; et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat. Commun. 2018, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.K.; Föll, M.; Heckelmann, B.; Kiefer, S.; Werner, M.; Schilling, O.; Biniossek, M.L.; Jilg, C.A.; Drendel, V. Proteomic Characterization of Prostate Cancer to Distinguish Nonmetastasizing and Metastasizing Primary Tumors and Lymph Node Metastases. Neoplasia 2018, 20, 140–151. [Google Scholar] [CrossRef]
- Iglesias-Gato, D.; Thysell, E.; Tyanova, S.; Crnalic, S.; Santos, A.; Lima, T.S.; Geiger, T.; Cox, J.; Widmark, A.; Bergh, A.; et al. The Proteome of Prostate Cancer Bone Metastasis Reveals Heterogeneity with Prognostic Implications. Clin. Cancer Res. 2018, 24, 5433–5444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, O.K.; Ha, Y.S.; Na, A.Y.; Chun, S.Y.; Kwon, T.G.; Lee, J.N.; Lee, S. Identification of Novel Prognosis and Prediction Markers in Advanced Prostate Cancer Tissues Based on Quantitative Proteomics. Cancer Genom. Proteom. 2020, 17, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Furusato, B.; Fang, X.; He, F.; Mohamed, A.A.; Griner, N.B.; Sood, K.; Saxena, S.; Katta, S.; Young, D.; et al. Evaluation of ERG responsive proteome in prostate cancer. Prostate 2014, 74, 70–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberhuber, M.; Pecoraro, M.; Rusz, M.; Oberhuber, G.; Wieselberg, M.; Haslinger, P.; Gurnhofer, E.; Schlederer, M.; Limberger, T.; Lagger, S.; et al. STAT3-dependent analysis reveals PDK4 as independent predictor of recurrence in prostate cancer. Mol. Syst. Biol. 2020, 16, e9247. [Google Scholar] [CrossRef]
- Ramberg, H.; Richardsen, E.; de Souza, G.A.; Rakaee, M.; Stensland, M.E.; Braadland, P.R.; Nygård, S.; Ögren, O.; Guldvik, I.J.; Berge, V.; et al. Proteomic analyses Identify Major Vault Protein as a Prognostic Biomarker for Fatal Prostate Cancer. Carcinogenesis 2021, 42, 685–693. [Google Scholar] [CrossRef]
- Cheng, L.C.; Tan, V.M.; Ganesan, S.; Drake, J.M. Integrating phosphoproteomics into the clinical management of prostate cancer. Clin. Transl. Med. 2017, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Ramroop, J.R.; Stein, M.N.; Drake, J.M. Impact of Phosphoproteomics in the Era of Precision Medicine for Prostate Cancer. Front. Oncol. 2018, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Macklin, A.; Khan, S.; Kislinger, T. Recent advances in mass spectrometry based clinical proteomics: Applications to cancer research. Clin. Proteom. 2020, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Venkadakrishnan, V.B.; Ben-Salem, S.; Heemers, H.V. AR-dependent phosphorylation and phospho-proteome targets in prostate cancer. Endocr. Relat. Cancer 2020, 27, R193–R210. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Freeman, M.R.; Kyprianou, N. Personalization of prostate cancer therapy through phosphoproteomics. Nat. Rev. Urol. 2018, 15, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Magi-Galluzzi, C.; Xu, X.; Hlatky, L.; Hahnfeldt, P.; Kaplan, I.; Hsiao, P.; Chang, C.; Loda, M. Heterogeneity of androgen receptor content in advanced prostate cancer. Mod. Pathol. 1997, 10, 839–845. [Google Scholar]
- Shah, R.B.; Bentley, J.; Jeffery, Z.; DeMarzo, A.M. Heterogeneity of PTEN and ERG expression in prostate cancer on core needle biopsies: Implications for cancer risk stratification and biomarker sampling. Hum. Pathol. 2015, 46, 698–706. [Google Scholar] [CrossRef]
- Boutros, P.C.; Fraser, M.; Harding, N.J.; de Borja, R.; Trudel, D.; Lalonde, E.; Meng, A.; Hennings-Yeomans, P.H.; McPherson, A.; Sabelnykova, V.Y.; et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet. 2015, 47, 736–745. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schüffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.W.B.; Zhao, Y.; Sue, A.C.; Guo, T.; Wong, L. Proteomic investigation of intra-tumor heterogeneity using network-based contextualization—A case study on prostate cancer. J. Proteom. 2019, 206, 103446. [Google Scholar] [CrossRef] [PubMed]
- Bonollo, F.; Thalmann, G.N.; Kruithof-de Julio, M.; Karkampouna, S. The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis. Cancers 2020, 12, 1887. [Google Scholar] [CrossRef]
- Shen, S.; Li, J.; Huo, S.; Ma, M.; Zhu, X.; Rasam, S.; Duan, X.; Qu, M.; Titus, M.A.; Qu, J. Parallel, High-Quality Proteomic and Targeted Metabolomic Quantification Using Laser Capture Microdissected Tissues. Anal. Chem. 2021, 93, 8711–8718. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, E.V.; Pereira, B.A.; Lawrence, M.G.; Ma, X.; Rebello, R.J.; Chan, H.; Niranjan, B.; Wu, Y.; Ellem, S.; Guan, X.; et al. Proteomic Profiling of Human Prostate Cancer-associated Fibroblasts (CAF) Reveals LOXL2-dependent Regulation of the Tumor Microenvironment. Mol. Cell Proteom. 2019, 18, 1410–1427. [Google Scholar] [CrossRef] [Green Version]
- McDonnell, L.A.; Corthals, G.L.; Willems, S.M.; van Remoortere, A.; van Zeijl, R.J.; Deelder, A.M. Peptide and protein imaging mass spectrometry in cancer research. J. Proteom. 2010, 73, 1921–1944. [Google Scholar] [CrossRef] [PubMed]
- Cazares, L.H.; Troyer, D.A.; Wang, B.; Drake, R.R.; Semmes, O.J. MALDI tissue imaging: From biomarker discovery to clinical applications. Anal. Bioanal. Chem. 2011, 401, 17–27. [Google Scholar] [CrossRef]
- Flatley, B.; Malone, P.; Cramer, R. MALDI mass spectrometry in prostate cancer biomarker discovery. Biochim. Biophys. Acta 2014, 1844, 940–949. [Google Scholar] [CrossRef]
- Kurreck, A.; Vandergrift, L.A.; Fuss, T.L.; Habbel, P.; Agar, N.Y.R.; Cheng, L.L. Prostate cancer diagnosis and characterization with mass spectrometry imaging. Prostate Cancer Prostatic Dis. 2018, 21, 297–305. [Google Scholar] [CrossRef]
- Randall, E.C.; Zadra, G.; Chetta, P.; Lopez, B.G.C.; Syamala, S.; Basu, S.S.; Agar, J.N.; Loda, M.; Tempany, C.M.; Fennessy, F.M.; et al. Molecular Characterization of Prostate Cancer with Associated Gleason Score Using Mass Spectrometry Imaging. Mol. Cancer Res. 2019, 17, 1155–1165. [Google Scholar] [CrossRef]
- Andersen, M.K.; Høiem, T.S.; Claes, B.S.R.; Balluff, B.; Martin-Lorenzo, M.; Richardsen, E.; Krossa, S.; Bertilsson, H.; Heeren, R.M.A.; Rye, M.B.; et al. Spatial differentiation of metabolism in prostate cancer tissue by MALDI-TOF MSI. Cancer Metab. 2021, 9, 9. [Google Scholar] [CrossRef]
- Morse, N.; Jamaspishvili, T.; Simon, D.; Patel, P.G.; Ren, K.Y.M.; Wang, J.; Oleschuk, R.; Kaufmann, M.; Gooding, R.J.; Berman, D.M. Reliable identification of prostate cancer using mass spectrometry metabolomic imaging in needle core biopsies. Lab. Investig. 2019, 99, 1561–1571. [Google Scholar] [CrossRef]
- Cazares, L.H.; Troyer, D.; Mendrinos, S.; Lance, R.A.; Nyalwidhe, J.O.; Beydoun, H.A.; Clements, M.A.; Drake, R.R.; Semmes, O.J. Imaging mass spectrometry of a specific fragment of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase 2 discriminates cancer from uninvolved prostate tissue. Clin. Cancer Res. 2009, 15, 5541–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angel, P.M.; Spruill, L.; Jefferson, M.; Bethard, J.R.; Ball, L.E.; Hughes-Halbert, C.; Drake, R.R. Zonal regulation of collagen-type proteins and posttranslational modifications in prostatic benign and cancer tissues by imaging mass spectrometry. Prostate 2020, 80, 1071–1086. [Google Scholar] [CrossRef] [PubMed]
- Flores-Morales, A.; Bergmann, T.B.; Lavallee, C.; Batth, T.S.; Lin, D.; Lerdrup, M.; Friis, S.; Bartels, A.; Kristensen, G.; Krzyzanowska, A.; et al. Proteogenomic Characterization of Patient-Derived Xenografts Highlights the Role of REST in Neuroendocrine Differentiation of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 595–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Sinha, C.; Kalyana-Sundaram, S.; Chinnaiyan, A.M. Landscape of gene fusions in epithelial cancers: Seq and ye shall find. Genome Med. 2015, 7, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaravilli, M.; Koivukoski, S.; Latonen, L. Androgen-Driven Fusion Genes and Chimeric Transcripts in Prostate Cancer. Front. Cell Dev. Biol. 2021, 9, 623809. [Google Scholar] [CrossRef] [PubMed]
- Cima, I.; Schiess, R.; Wild, P.; Kaelin, M.; Schüffler, P.; Lange, V.; Picotti, P.; Ossola, R.; Templeton, A.; Schubert, O.; et al. Cancer genetics-guided discovery of serum biomarker signatures for diagnosis and prognosis of prostate cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 3342–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, N.; Hjorth-Jensen, K.; Hekmat, O.; Iglesias-Gato, D.; Kruse, T.; Wang, C.; Wei, W.; Ke, B.; Yan, B.; Niu, Y.; et al. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene 2015, 34, 2764–2776. [Google Scholar] [CrossRef]
- Höti, N.; Shah, P.; Hu, Y.; Yang, S.; Zhang, H. Proteomics analyses of prostate cancer cells reveal cellular pathways associated with androgen resistance. Proteomics 2017, 17, 1600228. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zheng, C.; Yao, S.; Wang, Z.; Xu, L.; Yang, R.; Meng, X.; Wu, J.; Zhou, L.; Sun, Z. Proteomic analysis of human prostate cancer PC-3M-1E8 cells and PC-3M-2B4 cells of same origin but with different metastatic potential. PLoS ONE 2018, 13, e0206139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, D.; Li, M.; Wei, X.; Liu, S.; Zhao, M.; Liu, C.; Wang, X.; Jiang, X.; Li, X.; et al. Quantitative Proteomics of TRAMP Mice Combined with Bioinformatics Analysis Reveals That PDGF-B Regulatory Network Plays a Key Role in Prostate Cancer Progression. J. Proteome Res. 2018, 17, 2401–2411. [Google Scholar] [CrossRef] [PubMed]
- Katsogiannou, M.; Boyer, J.B.; Valdeolivas, A.; Remy, E.; Calzone, L.; Audebert, S.; Rocchi, P.; Camoin, L.; Baudot, A. Integrative proteomic and phosphoproteomic profiling of prostate cell lines. PLoS ONE 2019, 14, e0224148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, O.K.; Ha, Y.S.; Lee, J.N.; Kim, S.; Lee, H.; Chun, S.Y.; Kwon, T.G.; Lee, S. Comparative Proteome Profiling and Mutant Protein Identification in Metastatic Prostate Cancer Cells by Quantitative Mass Spectrometry-based Proteogenomics. Cancer Genom. Proteom. 2019, 16, 273–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, W.; Yuan, J.; Li, L.; Wang, Y. Parallel-Reaction-Monitoring-Based Proteome-Wide Profiling of Differential Kinase Protein Expression during Prostate Cancer Metastasis in Vitro. Anal. Chem. 2019, 91, 9893–9900. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.N.; Sharma, N. Quantitative SWATH-Based Proteomic Profiling for Identification of Mechanism-Driven Diagnostic Biomarkers Conferring in the Progression of Metastatic Prostate Cancer. Front. Oncol. 2020, 10, 493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kim, S.; Li, L.; Kemp, C.J.; Jiang, C.; Lü, J. Proteomic and transcriptomic profiling of Pten gene-knockout mouse model of prostate cancer. Prostate 2020, 80, 588–605. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, C.; Malik, A.; Abeysinghe, P.; Clements, J.; Batra, J. SWATH-MS Based Proteomic Profiling of Prostate Cancer Cells Reveals Adaptive Molecular Mechanisms in Response to Anti-Androgen Therapy. Cancers 2021, 13, 715. [Google Scholar] [CrossRef] [PubMed]
- Glen, A.; Evans, C.A.; Gan, C.S.; Cross, S.S.; Hamdy, F.C.; Gibbins, J.; Lippitt, J.; Eaton, C.L.; Noirel, J.; Wright, P.C.; et al. Eight-plex iTRAQ analysis of variant metastatic human prostate cancer cells identifies candidate biomarkers of progression: An exploratory study. Prostate 2010, 70, 1313–1332. [Google Scholar] [CrossRef]
- Qin, W.; Cho, K.F.; Cavanagh, P.E.; Ting, A.Y. Deciphering molecular interactions by proximity labeling. Nat. Methods 2021, 18, 133–143. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Stelloo, S.; Bergman, A.M.; Zwart, W. Androgen receptor enhancer usage and the chromatin regulatory landscape in human prostate cancers. Endocr. Relat. Cancer 2019, 26, R267–R285. [Google Scholar] [CrossRef] [Green Version]
- Zaman, N.; Giannopoulos, P.N.; Chowdhury, S.; Bonneil, E.; Thibault, P.; Wang, E.; Trifiro, M.; Paliouras, M. Proteomic-coupled-network analysis of T877A-androgen receptor interactomes can predict clinical prostate cancer outcomes between White (non-Hispanic) and African-American groups. PLoS ONE 2014, 9, e113190. [Google Scholar] [CrossRef] [PubMed]
- Paliouras, M.; Zaman, N.; Lumbroso, R.; Kapogeorgakis, L.; Beitel, L.K.; Wang, E.; Trifiro, M. Dynamic rewiring of the androgen receptor protein interaction network correlates with prostate cancer clinical outcomes. Integr. Biol. 2011, 3, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.J.; Smits, M.M.; Ng, B.H.; Lee, J.; Wright, M.E. Discovery Proteomics Identifies a Molecular Link between the Coatomer Protein Complex I and Androgen Receptor-dependent Transcription. J. Biol. Chem. 2016, 291, 18818–18842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lempiäinen, J.K.; Niskanen, E.A.; Vuoti, K.M.; Lampinen, R.E.; Göös, H.; Varjosalo, M.; Palvimo, J.J. Agonist-specific Protein Interactomes of Glucocorticoid and Androgen Receptor as Revealed by Proximity Mapping. Mol. Cell Proteom. 2017, 16, 1462–1474. [Google Scholar] [CrossRef] [Green Version]
- Vélot, L.; Lessard, F.; Bérubé-Simard, F.A.; Tav, C.; Neveu, B.; Teyssier, V.; Boudaoud, I.; Dionne, U.; Lavoie, N.; Bilodeau, S.; et al. Proximity-dependent mapping of the Androgen Receptor identifies Kruppel-Like Factor 4 as a functional partner. Mol. Cell Proteom. 2021, 20, 100064. [Google Scholar] [CrossRef]
- Lempiäinen, J.K.; Manjur, A.B.M.K.; Malinen, M.; Ketola, K.; Niskanen, E.A.; Palvimo, J.J. BCOR-coupled H2A monoubiquitination represses a subset of androgen receptor target genes regulating prostate cancer proliferation. Oncogene 2020, 39, 2391–2407. [Google Scholar] [CrossRef]
- Paltoglou, S.; Das, R.; Townley, S.L.; Hickey, T.E.; Tarulli, G.A.; Coutinho, I.; Fernandes, R.; Hanson, A.R.; Denis, I.; Carroll, J.S.; et al. Novel Androgen Receptor Coregulator GRHL2 Exerts Both Oncogenic and Antimetastatic Functions in Prostate Cancer. Cancer Res. 2017, 77, 3417–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelloo, S.; Nevedomskaya, E.; Kim, Y.; Hoekman, L.; Bleijerveld, O.B.; Mirza, T.; Wessels, L.F.A.; van Weerden, W.M.; Altelaar, A.F.M.; Bergman, A.M.; et al. Endogenous androgen receptor proteomic profiling reveals genomic subcomplex involved in prostate tumorigenesis. Oncogene 2018, 37, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Launonen, K.M.; Paakinaho, V.; Sigismondo, G.; Malinen, M.; Sironen, R.; Hartikainen, J.M.; Laakso, H.; Visakorpi, T.; Krijgsveld, J.; Niskanen, E.A.; et al. Chromatin-directed proteomics-identified network of endogenous androgen receptor in prostate cancer cells. Oncogene 2021, 40, 4567–4579. [Google Scholar] [CrossRef]
- Agrawal, P.; Yu, K.; Salomon, A.R.; Sedivy, J.M. Proteomic profiling of Myc-associated proteins. Cell Cycle 2010, 9, 4908–4921. [Google Scholar] [CrossRef] [Green Version]
- Dingar, D.; Kalkat, M.; Chan, P.K.; Srikumar, T.; Bailey, S.D.; Tu, W.B.; Coyaud, E.; Ponzielli, R.; Kolyar, M.; Jurisica, I.; et al. BioID identifies novel c-MYC interacting partners in cultured cells and xenograft tumors. J. Proteom. 2015, 118, 95–111. [Google Scholar] [CrossRef]
- Kalkat, M.; Resetca, D.; Lourenco, C.; Chan, P.K.; Wei, Y.; Shiah, Y.J.; Vitkin, N.; Tong, Y.; Sunnerhagen, M.; Done, S.J.; et al. MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol. Cell 2018, 72, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Rubin, M.A.; Bristow, R.G.; Thienger, P.D.; Dive, C.; Imielinski, M. Impact of Lineage Plasticity to and from a Neuroendocrine Phenotype on Progression and Response in Prostate and Lung Cancers. Mol. Cell 2020, 80, 562–577. [Google Scholar] [CrossRef]
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Investig. 2019, 129, 3924–3940. [Google Scholar] [CrossRef] [PubMed]
- Cyrta, J.; Augspach, A.; De Filippo, M.R.; Prandi, D.; Thienger, P.; Benelli, M.; Cooley, V.; Bareja, R.; Wilkes, D.; Chae, S.S.; et al. Role of specialized composition of SWI/SNF complexes in prostate cancer lineage plasticity. Nat. Commun 2020, 11, 5549. [Google Scholar] [CrossRef]
- Smith, S.L.; Pitt, A.R.; Spickett, C.M. Approaches to Investigating the Protein Interactome of PTEN. J. Proteome Res. 2021, 20, 60–77. [Google Scholar] [CrossRef]
- Kaikkonen, E.; Takala, A.; Pursiheimo, J.P.; Wahlström, G.; Schleutker, J. The interactome of the prostate-specific protein Anoctamin 7. Cancer Biomark. 2020, 28, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Rais, Y.; Bismar, T.A.; Hyndman, M.E.; Le, X.C.; Drabovich, A.P. Mapping Isoform Abundance and Interactome of the Endogenous TMPRSS2-ERG Fusion Protein by Orthogonal Immunoprecipitation-Mass Spectrometry Assays. Mol. Cell Proteom. 2021, 20, 100075. [Google Scholar] [CrossRef] [PubMed]
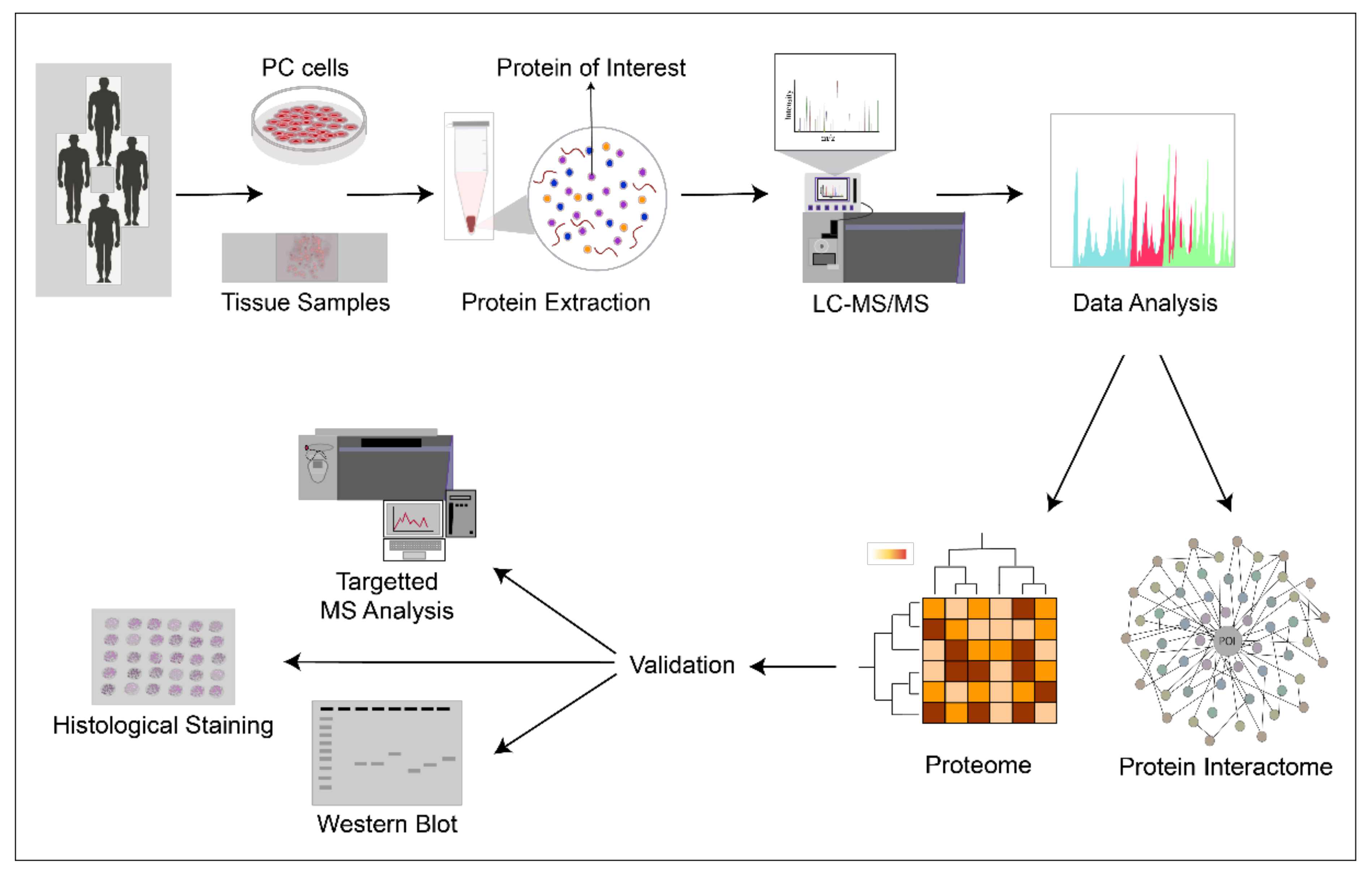


{kind=link}
{kind=link}
| Publication | Number of High-Confidence Proteins Present in All Samples | MS *** Method | Patient Samples Included in Proteomics | Additional Information | Reference |
|---|---|---|---|---|---|
| Primary prostate cancer | |||||
| Iglesias-Gato et al., 2016 | 4900 * (in >50% of samples) | Super-SILAC spike-in | adjacent non-malignant (n = 8), PCa (n = 28; n = 16 high-risk PCa, n = 12 low-risk PCa) | [46] | |
| Myers et al., 2016 | 1500 * (in total) | label-free LC-MS/MS | matched non-malignant tissue (n = 9), PCa (n = 14) | comparing patient samples of African American and Caucasian American men | [47] |
| Staunton et al., 2017 | 2000 | label-free LC-MS/MS | 8 PCa (GG3 n = 4), GG4 n = 4) | Micro-dissected epithelium and stroma | [48] |
| Guo et al., 2018 | 3700 | pressure cycling technology, SWATH | adjacent benign (n = 3), PCa (n = 3) | assessment of tissue heterogeneity with 3–6 spatially distinct samples/tissue type/patient (n = 60 in total) | [49] |
| Kawahara et al., 2019 | 2100 * (in total) | label-free LC-MS/MS | BPH (n = 5), PCa (n = 50; n = 10 in each ISUP grade group) | [50] | |
| Zhou et al., 2019 | 3600 | tandem mass tagging-SPS-MS3 | adjacent normal (n = 9), PCa (n = 18; GS6 n = 9, GS8–9 n = 9) | [51] | |
| Sinha et al., 2019 | 3400 | label-free LC-MS/MS | localized PCa (n = 75; ISUP1 n = 4, ISUP2 n = 58, ISUP3 n = 13) | integrative with WGS (n = 74), methylome (n = 72), H3K27Ac (n = 35), transcriptome (n = 55) | [52] |
| Latosinska et al., 2020 | 1400 * (in total) | label-free LC-MS/MS | BPH (n = 5), PCa (n = 17) | [53] | |
| Primary and advanced prostate cancer | |||||
| Liu et al., 2014 | 2100 ** N-glycosite peptides | SPEG for N-linked glycopeptides; SWATH-MS | non-malignant (n = 10), PCa (n = 40; non-aggressive n = 24, aggressive n = 16) | large-scale proteomics performed on pooled samples of each sample group | [54] |
| Drake et al., 2016 | 8300 ** phosphor-ylated peptides | IP for pS/T and pY phospho-proteomics, LC-MS/MS | treatment-naïve PCa (n = 11), metastatic CRPC (n = 16) | integrative with genomic and transcriptomic data | [55] |
| Latonen et al., 2018 | 3400 | SWATH-MS | BPH (n = 10), PCa (n = 17), CRPC (n = 11) | integrative with WGS, methylome, and transcriptome data | [56] |
| Müller et al., 2018 | 1200 * (present in at least four cases of each sample type) | label-free LC/MS-MS | PCa (n = 5), metastasizing PCa (n = 5), LNM (n = 5) | non-metastasizing vs. metastasizing primary tumors; primary tumors vs. matched LNM | [57] |
| Iglesias-Gato et al., 2018 | 5000 *(per sample, on average) | Super-SILAC spike-in | bone distal metastases (n = 22, of which 16 CRPC) | transcriptome for certain samples; compared with non-malignant and PCa reported earlier (Iglesias-Gato et al., 2016) | [58] |
| Kwon et al., 2020 | 1600 * (in total) | TMT-LC-MS/MS | non-malignant (n = 10), PCa (n = 8), metastatic PCa (n = 2) | cancer samples according to T-stage (T2 n = 4, T3 n = 4, T3-N1 n = 2) | [59] |
| Publication | Models Included | Specifications | References |
|---|---|---|---|
| Cima et al., 2011 | Pten−/− mice | glycoproteomes of wt and Pten−/− prostates | [90] |
| Tan et al., 2014 | VCaP cell line | proteomic changes with downregulation of ERG with siRNA; phosphoproteomics; in comparison with clinical samples | [60] |
| Jiang et al., 2015 | LNCaP xenografts | phosphoproteomes in intact and castrated mice | [91] |
| Drake et al., 2016 | cell lines and xenografts (22Rv1, LNCaP) | phosphoproteomics, integrative with genomic and transcriptomic data, in comparison with clinical tumor samples (see Table 1) | [55] |
| Höti et al., 2017 | LNCaP, LNCaP-95 | proteomic changes in androgen resistance | [92] |
| Zhang S. et al., 2018 | cell lines PC-3M-2B4, PC-3M-1E8 | proteomes associated with different metastatic ability | [93] |
| Zhang Y. et al., 2018 | TRAMP mice | proteomes of wt and TRAMP prostates | [94] |
| Katsogiannou et al., 2019 | cell lines PNT1A8, LNCaP, DU145, PC-3 | differences in proteins and phosphosites between cell lines | [95] |
| Kwon et al., 2019 | cell lines LNCaP, PC-3 LCaP-LN3, PC-3M | comparison of parental to more metastatic sub-lines | [96] |
| Miao et al., 2019 | cell lines PC-3, PC-3MLN4 | kinome associated with increased metastasis | [97] |
| Singh and Sharma, 2020 | cell lines PC-3, LNCaP | proteomic changes in TGF-ß-induced EMT | [98] |
| Zhang et al., 2020 | Pten−/− mice | proteomes of wt and Pten−/− prostates | [99] |
| Liynage et al., 2021 | cell lines LNCaP, C4-2B | proteomic changes stimulated by anti-androgens bicalutamide and enzalutamide | [100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadeesh, N.; Scaravilli, M.; Latonen, L. Proteomic Landscape of Prostate Cancer: The View Provided by Quantitative Proteomics, Integrative Analyses, and Protein Interactomes. Cancers 2021, 13, 4829. https://doi.org/10.3390/cancers13194829
Sadeesh N, Scaravilli M, Latonen L. Proteomic Landscape of Prostate Cancer: The View Provided by Quantitative Proteomics, Integrative Analyses, and Protein Interactomes. Cancers. 2021; 13(19):4829. https://doi.org/10.3390/cancers13194829
Chicago/Turabian StyleSadeesh, Nithin, Mauro Scaravilli, and Leena Latonen. 2021. "Proteomic Landscape of Prostate Cancer: The View Provided by Quantitative Proteomics, Integrative Analyses, and Protein Interactomes" Cancers 13, no. 19: 4829. https://doi.org/10.3390/cancers13194829