Simple Summary
The incidence rate of endometrial cancer is rising globally. The molecular subtypes of endometrial cancer are independent of histology and have strong prognostic value in high-risk cancer. However, molecular profiling has not made it to clinical practice in Aotearoa, New Zealand. Therefore, we aimed to explore the feasibility of molecular profiling to examine the distribution of endometrial cancer subtypes and identify areas of need for implementation.
Abstract
Background: Aotearoa, New Zealand, has one of the fastest-rising rates of endometrial cancer (EC) worldwide, increasing particularly in younger Māori and Pasifika women. There is a move towards using molecular profiling to direct treatment for each EC subtype. Aim: This study aimed to explore the molecular profiling of primary EC tissue in Aotearoa. Methods: We used the PORTEC guidelines for the molecular subtyping of 90 patients’ samples into four categories: POLE-mutated, p53 abnormal, mismatch repair deficient (MMRd) and no specific molecular profile (NSMP). The CTNNB1 mutation and L1CAM expression were also included in the analysis. POLE and CTNNB1 mutations were analysed using targeted next-generation sequencing (NGS). Novel mutations were assessed using VarSome. MMRd, L1CAM and p53 abnormalities were analysed using immunohistochemistry. Results: In total, 15 samples were MMRd, 9 were p53 abnormal, 8 were POLE-mutated and the rest (56) were NSMP. Eleven samples had exon 3 CTNNB1 mutations and eleven novel POLE mutations were described. Conclusion: Surrogate markers for POLE mutations should be investigated. The validation of POLE variants and CTNNB1 mutations as part of an Aotearoa-based molecular panel is warranted.
1. Introduction
Incidence rates of endometrial cancer (EC) are rising globally; however, Aotearoa, New Zealand, has one of the fastest-rising rates of EC. The majority of Aotearoa’s population is of European descent (70.2%), with the indigenous Māori being the largest minority (16.5%), followed by people of Asian ethnicity (15.1%), and Pacific Islanders (8.1%) (per the 2018 census). There are persistent and stark inequities in access to healthcare and health outcomes, the impacts of which vary with ethnicity and socioeconomic status [1,2]. EC patients are subject to the effects of these inequities; women who identify as Māori or Pasifika experience the greatest burden from this disease and the rapid increase in diagnosis is observed primarily in younger age groups [3,4]. Whilst the scientific literature continues to characterise cancers by genomic classification, clinical translation of these advances is not a reality in Aotearoa. In 2013, the Cancer Genome Atlas (TCGA) project identified four molecular classifications of EC [5], which were then simplified into the Proactive Molecular Risk Classifier for EC (ProMisE) guidelines for ease of clinical testing [6]. These guidelines distinguished: mismatch repair deficient (MMRd), p53 abnormal (p53abn), POLE-mutated (POLE) and non-specified molecular profile (NSMP) subtypes. To determine most of these profiles, immunohistochemistry (IHC) can be used. However, POLE mutations still need to be confirmed via DNA sequencing [7].
There has been increased interest driving the use of molecular classification in clinical practice [8] since the PORTEC-3 trial results showed that molecular classification has a strong prognostic value in high-risk EC [9]. Women with p53abn tumours had significantly improved survival rates when treated with adjuvant chemo-radiation therapy, whilst women with POLE-mutated EC had incredibly good prognoses regardless of treatment arm (chemotherapy or chemo-radiation therapy). However, whilst MMRd IHC has been in routine clinical practice in Aotearoa since 2017 (mainly for Lynch syndrome), p53 IHC and POLE testing have not been routinely available.
The pathogenicity of POLE mutations has been confirmed in the most common variants of EC; however, challenges around interpreting novel variants arise when this test is applied to clinical use. Authors Leon-Castillo et al. [10] set out to provide guidance around classifying novel somatic mutations based on a tumour mutation burden (TMB) score, thereby facilitating the implementation of POLE testing. This may not be feasible for clinical use as only hotspot mutations will be investigated.
Before changes to patient treatment pathways can be made, we firstly need to understand the molecular profile of EC in Aotearoa and then, secondly, determine whether testing is feasible and relevant. There are no surrogate markers of POLE mutation status; though some researchers have attempted to histologically identify differences in immune components such as tumour infiltrating lymphocytes (TILs) to indicate POLE status (evidence has suggested that an increase in these populations can indeed indicate POLE status) [11,12]. Therefore, this paper aims to pilot the molecular classification of EC in New Zealand women as compared to PORTEC3 data.
2. Materials and Methods
2.1. Cohort
This retrospective study was approved by the Health and Disability Ethics Committee (HDEC 19CEN146). Formalin-fixed paraffin-embedded (FFPE) tissue blocks of 90 ECs diagnosed between 2015 and 2017 were retrieved from a New Zealand tertiary hospital. The inclusion criterion was adenocarcinoma of any histology, and equal samples were sourced from Māori, Pasifika, and NZ–European ethnicities. Curetting/pipelle biopsy samples were prioritised due to their better fixation for IHC and higher clinical relevance; however, if these were not available, a block of the resected primary tumour was chosen. Tumour stage and grade were taken from the diagnosis at the time of resection.
2.2. Molecular Profiling
IHC staining for p53 and MMR proteins (MLH1, MSH2, MSH6 and PMS2) was performed at Southern Community Laboratories (SCL). IHC was performed on the Ventana Benchmark Ultra; the detection system used was a 3-step polymer horseradish peroxidase (HRP) DAB kit (OptiView DAB) from Roche (760–700)—two tests (MSH2 and PMS2) also required tyramide signal amplification (860–099) steps. Heat-induced epitope retrieval was performed using CC1 (950–224), a high-pH Tris-based solution. All antibodies were incubated at 36 degrees Celsius and diluted in Roche antibody diluent ADB250 (F17676Z). All samples were then counter-stained with hematoxylin II (790–2208) and bluing reagent (760–2037) for 4 min each. Antibody clones, dilution and the antigen retrieval method are outlined in Table S1. Being an antibody for research purposes, L1CAM was validated using known positive tissue (such as kidney and colorectal adenocarcinoma, mesothelioma and ovarian serous carcinoma), and optimised for working conditions of high pH antigen retrieval for 56 min and primary antibody incubation for 32 min. In some cases, IHC was repeated on multiple blocks from the same patient as some samples had insufficient staining, possibly due to autolysis (Figure S1).
DNA extraction was performed using the Illumina AmpliSeq Direct FFPE DNA kit (Illumina, San Diego, CA, USA, #20023378); however, the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA, USA, #56404) was used when samples were small/low quality. Whole sections (curettings) or tumour sections (primary resection) of up to three FFPE block sections (each 10 µm in size) were used. The final DNA concentration was assessed fluorometrically (Qubit DNA HS assay, Thermo Fisher, Waltham, MA, USA). According to manufacturer instructions, 10–100 ng of DNA is required for library preparation. For samples with high quantities, 100 ng was used for each primer pool. For lower-quantity samples, the maximum amount of DNA was used for each pool, which fell within the range of the required starting sample. The DNA libraries were prepared using the Illumina AmpliSeq Library PLUS custom panel (coverage: 94.8% POLE and 99% CTNNB1) and quality control (QC) of the amplified products was measured using the DNA 1000 BioAnalyzer Chip (Agilent Technologies, Waldbronn, Germany). Subsequent sequencing of the pooled libraries was performed on the iSeq 100 platform (Illumina, San Diego, CA, USA). Data analysis (checking alignment to the hg19 human reference genome and variant calling) was completed using the DNA Amplicon pipeline on Illumina BaseSpace software. The mean total of PF reads was 616,829, the mean percentage of on-target aligned reads was 93%, the mean target coverage percentage was 86% and the mean SNVs reported was 16. POLE mutations were considered pathogenic based on previous interpretations of exonuclease domains (ClinVar). However, for variants of uncertain significance, VarSome (Kopanos et al., 2018) was used to determine potential pathogenicity.
2.3. IHC Analysis
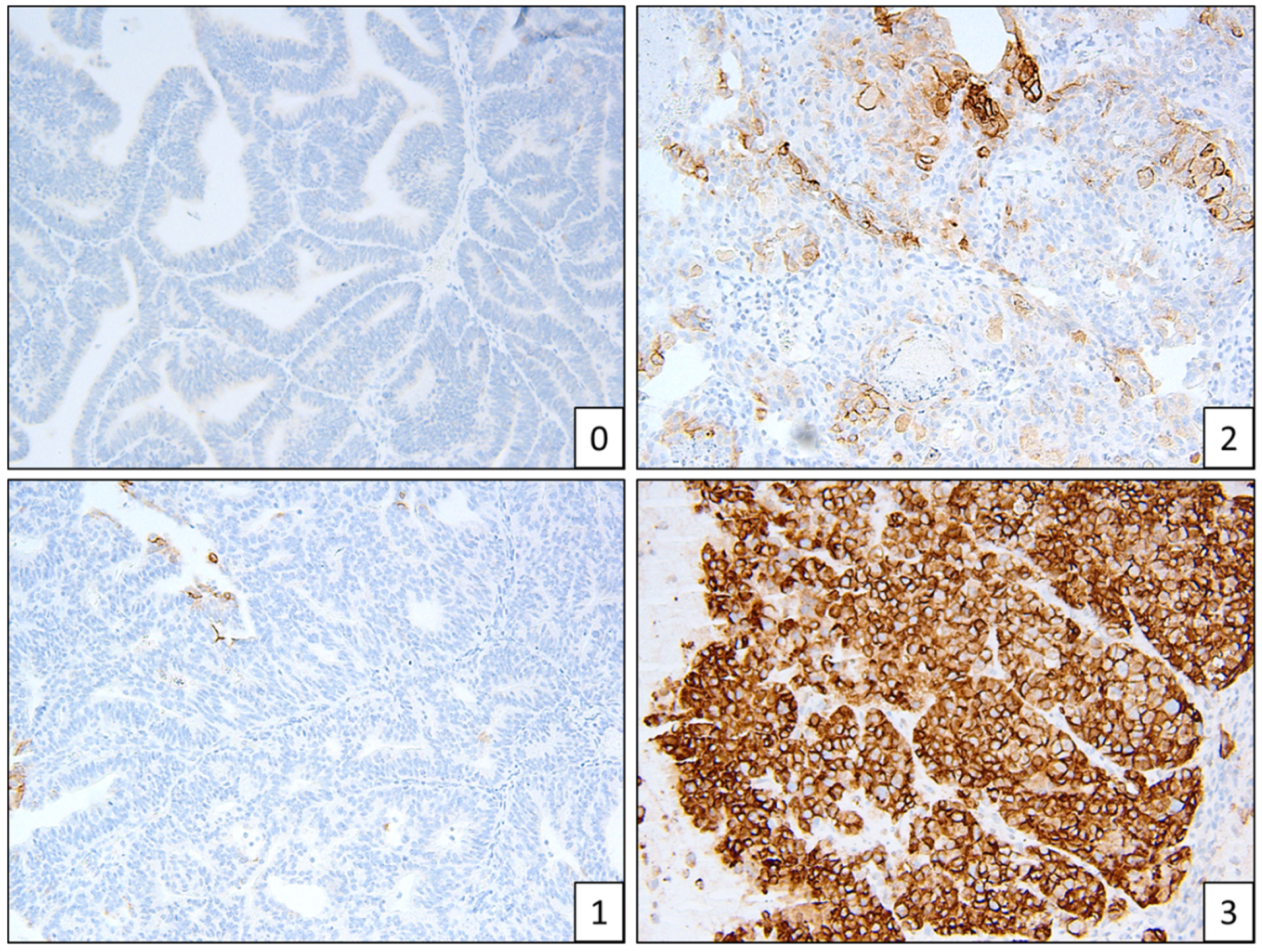
Two people (a gynaecologic histopathologist and a research scientist) assessed the IHC independently (Figure 1, Figure 2 and Figure 3). Staining was deemed sufficient if both external (control tissue) and internal controls (non-neoplastic cells such as lymphocytes) were positive. Any discordance was discussed until an agreement was reached. p53 wild-type tumours were characterised by a mixture of negative, weak and strong cells, whereas p53abn was characterised by either diffuse strong staining or a complete absence of staining [13]. MMRd tumours were characterised by a complete absence of staining. L1CAM scoring was assessed on a 0 (absence of staining) to 3 (diffuse strong staining) scale (Figure 2). As per PORTEC 3 criteria, samples with a pathogenic POLE mutation superseded p53 abnormality/MMR loss and were therefore classified as POLE, and samples with a combination of loss of MMR and abnormal p53 were classified as MMRd.
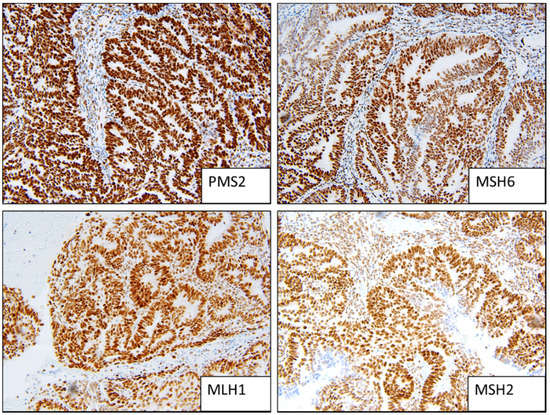
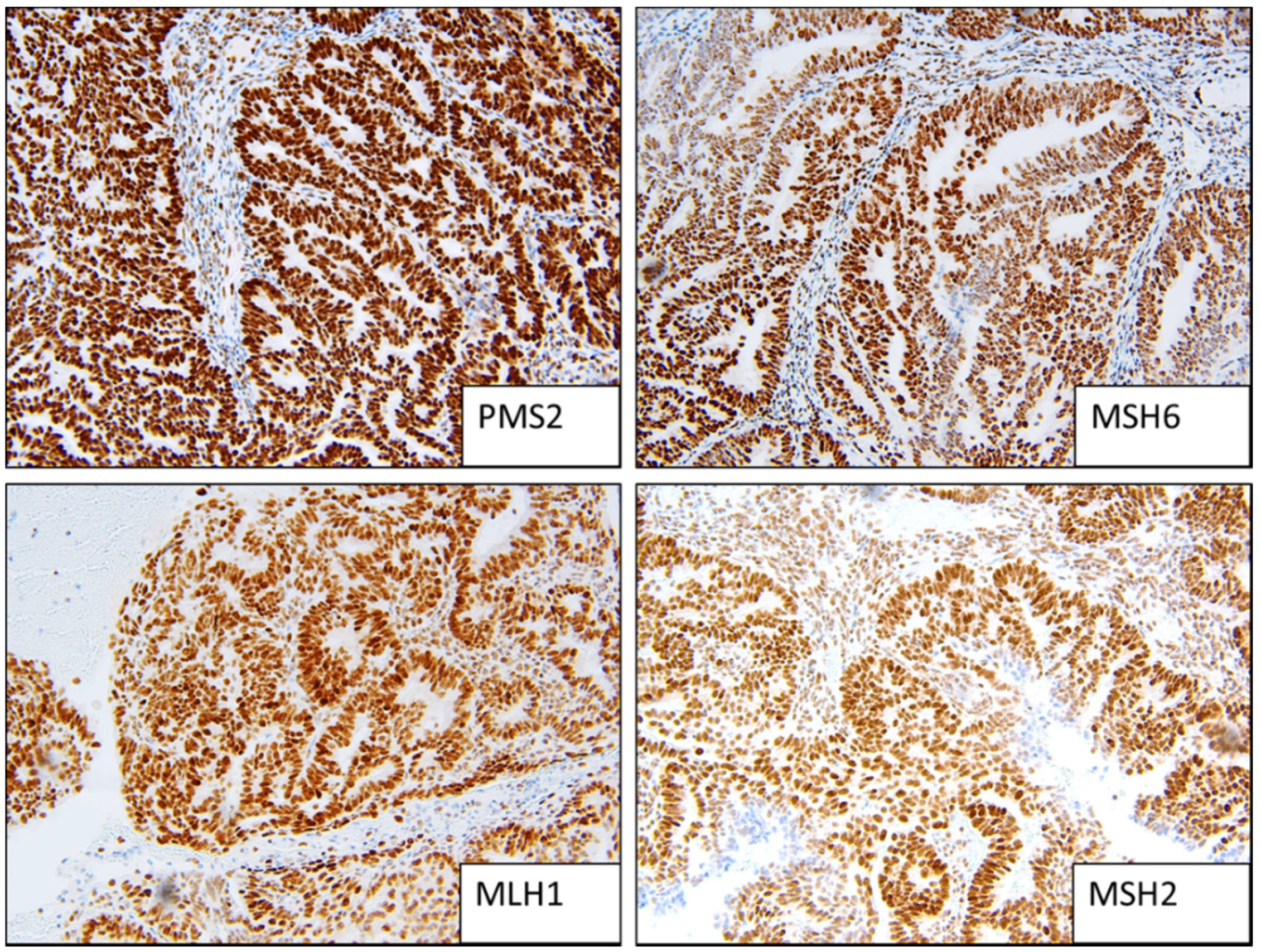
Figure 1.
Example of MMR stable staining patterns (strong positive).
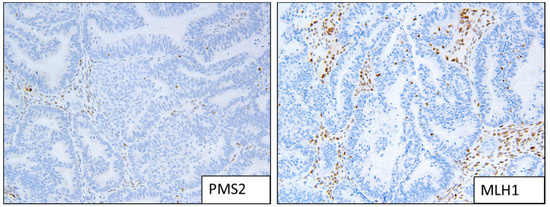
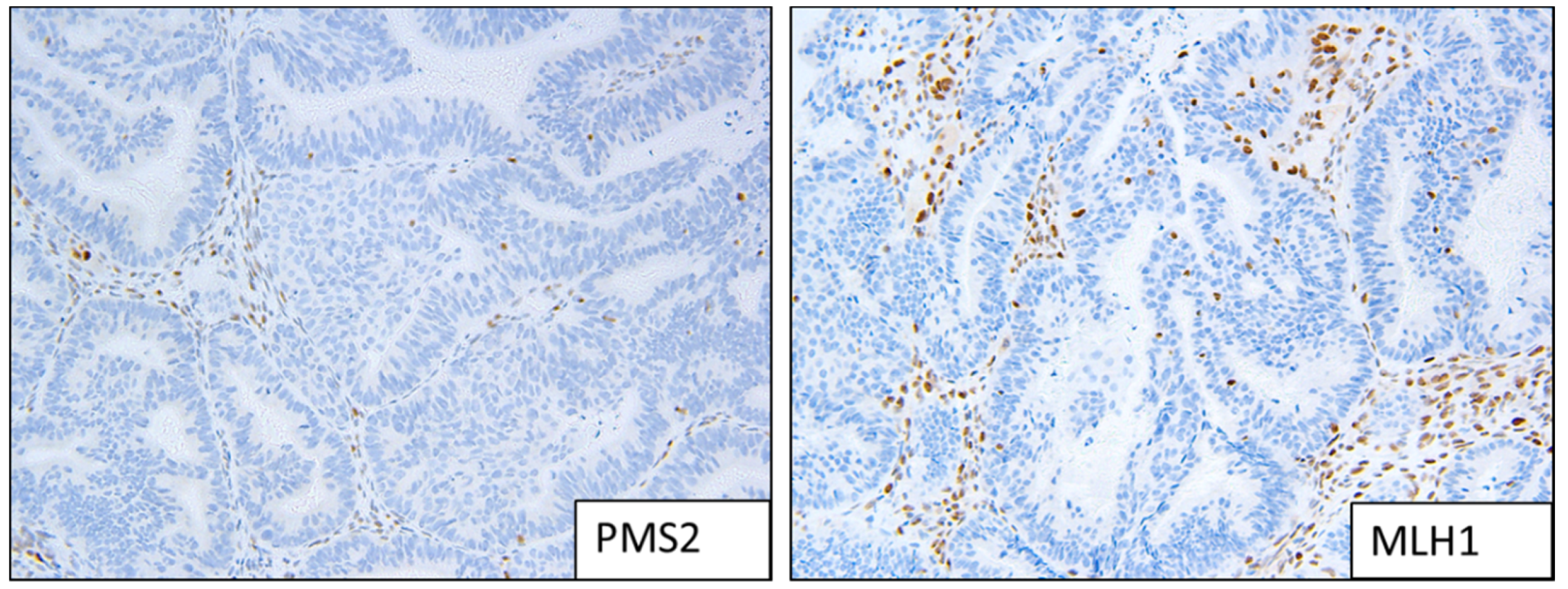
Figure 2.
Example of MMRd staining patterns; loss of expression of MLH1 and PMS2.
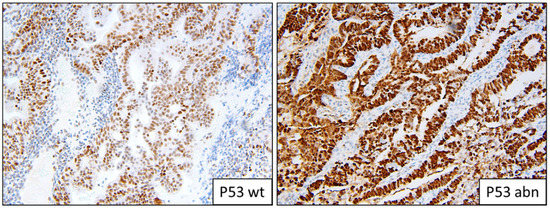
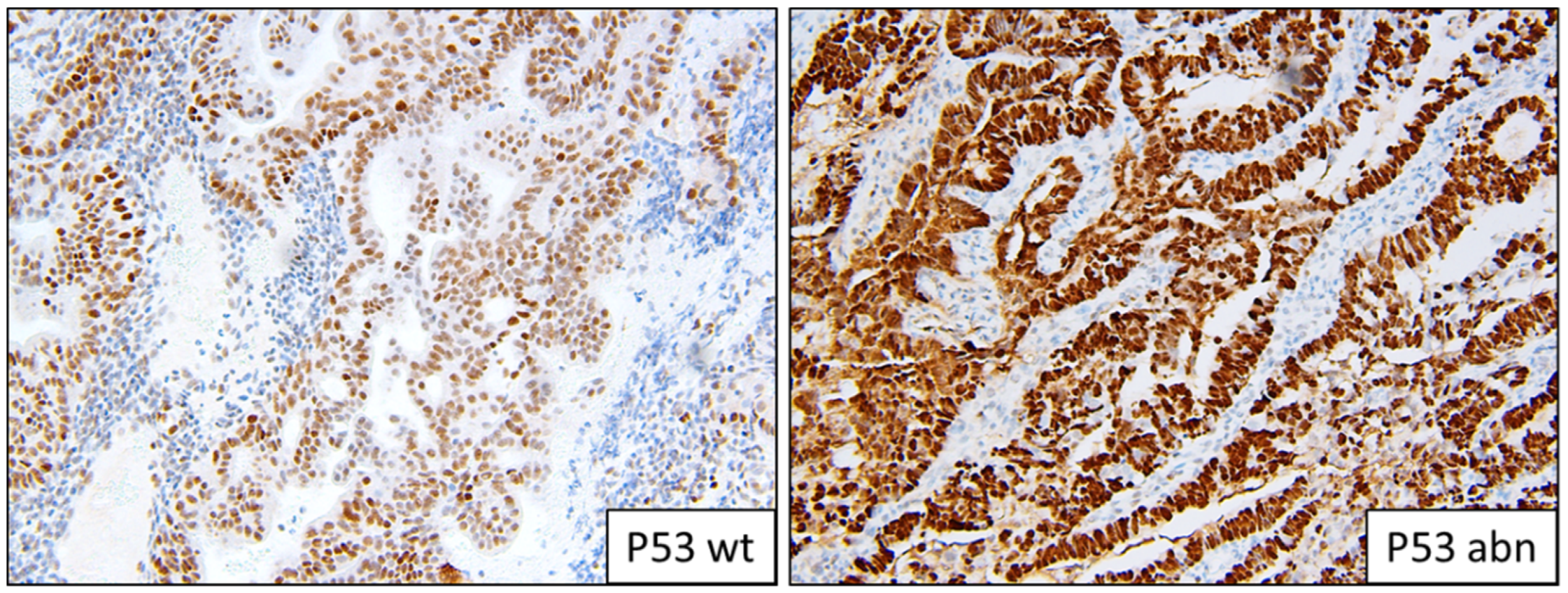
Figure 3.
Example of p53 staining patterns; wild-type and abnormal overexpression.
2.4. Datamining
Based on the theory of an activated T cell infiltrate, the immune microenvironment of POLE-mutated EC was interrogated using the integrated repository portal for tumour–immune system interactions (TISIDB) [14]. We firstly screened for all POLE mutations, then curated the list to only uterine corpus endometrial carcinoma subsets (UCEC). This gave us outputs of differentially expressed lymphocytes, immunomodulatory molecules and chemokines in POLE Mut vs. wild-type samples. The most significant genes were then investigated in the context of overall survival using cBioPortal.
3. Results
Molecular testing was successful in 88 samples. IHC was not performed on two samples due to poor quality; multiple blocks were sampled (different representations of the primary tumour/cassettes); however, due to autolysis and the age of the blocks, IHC was not possible (Figure S1). The patient cohort characteristics are described in Table 1 (outcomes are recorded in Table S3). POLE and CTNNB1 variants were classified as mutated if they were already reported as such in ClinVar. Variants that were likely pathogenic, or pathogenic as determined via VarSome, were documented. Benign or likely benign variants, determined by the type of mutation and location/pathogenicity algorithms, were excluded.

Table 1.
Cohort characteristics.
The 88 successful samples were then classified into one of four molecular subgroups (multiple-classifier ECs were identified and allocated a single molecular subgroup as previously described):
- MMRd: 15 (17%);
- P53abn: 9 (10%);
- POLEmut: 8 (9%);
- NSMP: 56 (64%).
Instances of multiple classifiers included three POLEmut samples that were also p53abn and two MMRd samples that also had pathogenic POLE mutations. We identified one sample that was stage IA, grade 1 and p53abn.
There were eight patients with high-grade, low-stage, high-risk endometrioid cancer (see Table 2). Of these, one was POLEmut, one was p53abn/POLEmut, four were p53abn and the other two were NSMP.
Table 2.
Molecular profile of high-risk EC (FIGO low stage, high grade).
CTNNB1 mutations seemed to be more frequent in Māori (n = 4) and Pasifika (n = 5) women, compared to NZ–European women (n = 2), although due to the small sample size this is not a significant difference (Table 3). All CTNNB1 mutations were found in exon 3 (Table S2). Hotspot POLE mutations are listed in Table 4. L1CAM staining was low overall, with 79 of 88 stained samples (90%) having a score of 0–1 (Figure 4). L1CAM correlated with p53 mutations—if p53 was abnormal, L1CAM was more strongly expressed. L1CAM was noted to have focal and patchy staining.
Table 3.
Pathogenic POLE and CTNNB1 mutations.
Table 4.
Hotspot POLE mutations.
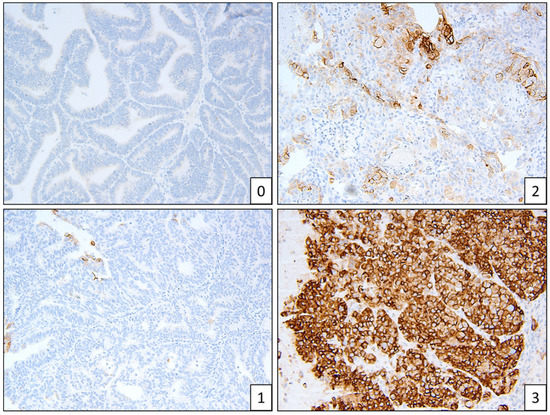
Figure 4.
Example of L1CAM staining patterns, scores 0–3.
POLE Morphology
The utility of POLE testing will be realised when a surrogate marker can be used instead of sequencing. Already, specific exons are targeted for NGS (9–14) as these mutations are well described. However, from our data, we identified an additional 11 variants of uncertain but potential pathogenic significance (Table 5). Therefore, like others, we aimed to investigate whether increased TIL and nuclear pleomorphism could characterise these samples.
Table 5.
Novel POLE mutations likely to be pathogenic.
Only 3 out of the 10 samples deemed to belong to the POLE subtype had classic morphological markers; that is, increased TIL and nuclear pleomorphism. Two samples that were originally noted as low grade (grade 1) were subsequently identified as belonging to grade 3.
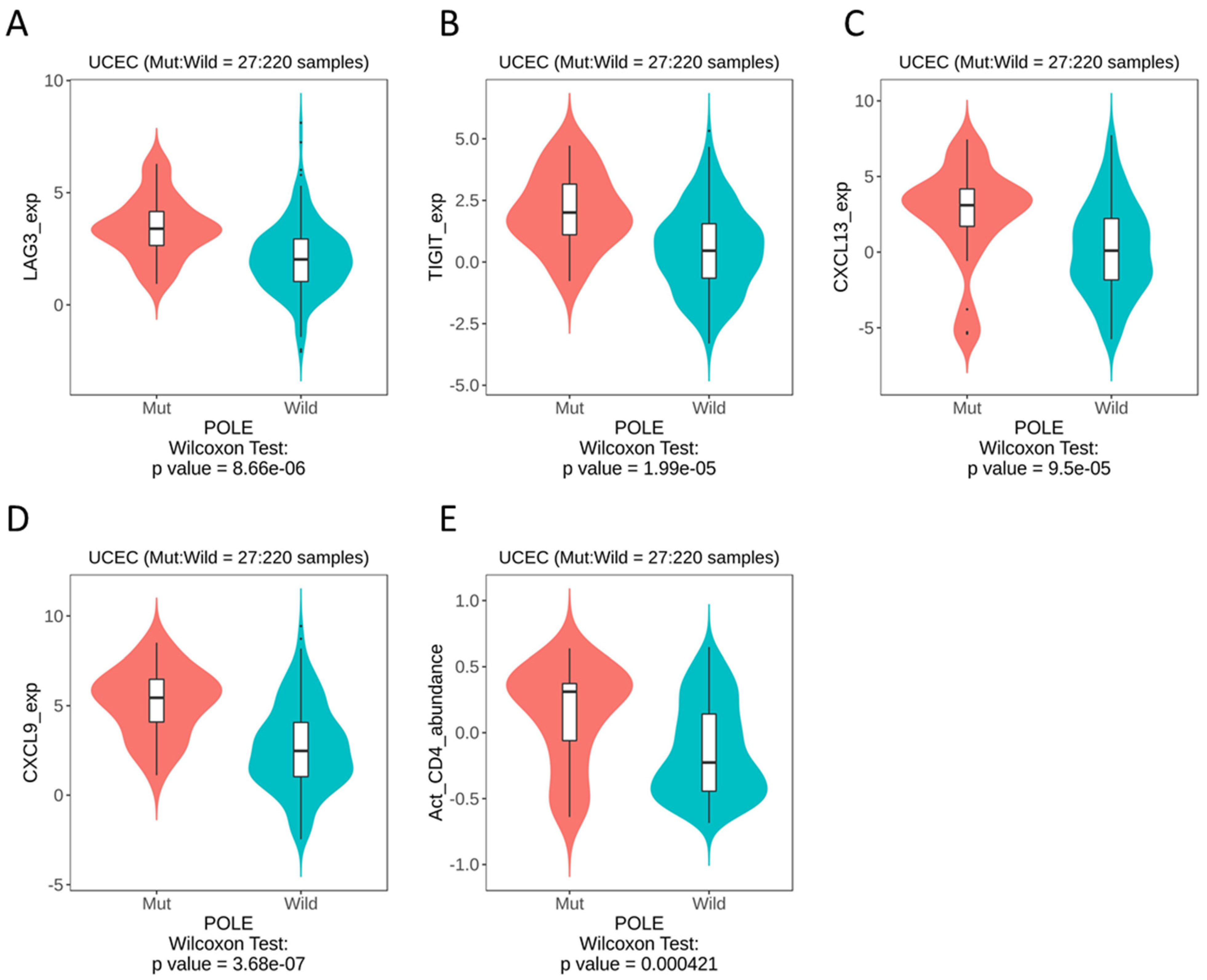
Therefore, we looked to publicly available databases to investigate surrogate markers of POLE, based on the theory of an activated T cell infiltrate. Whilst the data set was derived from a small sample size (with a mut:wt ratio of 27:220), we were able to identify the most significant differences in LAG3 (p = 8.66 × 10−6), TIGIT (p = 1.99 × 10−5), CXCL13 (p = 9.5 × 10−5) and CXCL9 (p = 3.68 × 10−7) (Figure 5A–D). As expected, our datamining also showed significant differences in activated CD4 T cells (Figure 5E). When these immune genes were interrogated using cBioPortal, patients with alterations in TIGIT showed significantly improved overall survival rates compared to those with alterations in LAG3, CXCL13 and CXCL9 (Figure S2).
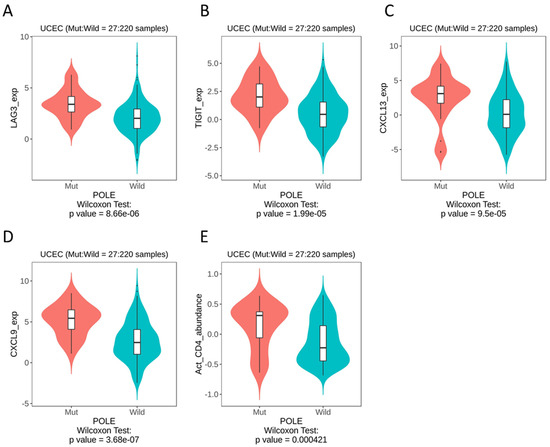
Figure 5.
TISIDB datamining in UCEC, POLE mutation vs. wild-type, immune gene expression. (A) LAG3. (B) TIGIT. (C) CXCL13. (D) CXCL9. (E) Activated CD4 T cells.
4. Discussion
This study was the first to explore molecular subtyping of EC in a New Zealand cohort. As expected for our cohort, most of the patients had a diagnosis of low-grade, low-stage endometrioid EC. We identified five samples with reportedly pathogenic POLE variants, and eleven potentially pathogenic variants; more research needs to be completed in order to validate these new variants. Interestingly, the only three classic-looking POLE samples (those showing increased TIL and nuclear pleomorphism) were those with variants of uncertain significance.
IHC was possible in 88 samples, with limitations caused by poor-quality staining on old and autolytic tissue. For the benefit of future molecular profiling, pipelle or curetting biopsies should be prioritised as these are not prone to poor fixation due to size, whereas large gynaecological samples are affected by this issue if they are not grossed in a timely manner. Failure to gross these large samples in a timely manner ultimately leads to tissue autolysis and poor staining and impacts on molecular typing. Furthermore, profiling on pre-surgical biopsies may influence the extent of surgical treatment, i.e., lymph node dissection, omental biopsy and/or washings for those with p53abn may be utilised, as appropriate. However, if DNA sequencing for POLE mutations is to remain in the profiling test, curettings may provide low-quantity material as there can be difficulty in getting sufficiently sized samples using a pipelle [15].
In this study, we included L1CAM and CTNNB1 in the biomarker panel. There was no strong evidence to include L1CAM in the molecular profiling; most samples exhibited low L1CAM staining, and when it was strong, it correlated to those that were p53abn, which can be detected using p53 IHC instead. CTNNB1 mutations were present in 11 samples, seemingly occurring more frequently in Māori or Pasifika women, although our sample size is too small to confirm this. CTNNB1 mutations in exon 3 are associated with enrichment of the Wnt signalling pathway, and may characterise a subset of aggressive tumours associated with younger age and earlier stage [16]. In their 2020 study, Bigby et al. [4] reported that even though Māori and Pasifika women did not present with higher stage or grade, they experienced worse survival outcomes compared to NZ–European women. In our cohort, cancers in women with CTNNB1 mutations did not have worse survival outcomes, but the average age of these women was less than that of the overall cohort (49 years compared to 59 years). Therefore, it may be worth validating CTNNB1 to assess whether it may assist in risk stratification and improve equity in outcomes through an NZ-based molecular profile panel.
Further research into identifying a surrogate marker for POLE mutations is warranted. Currently, if molecular subtyping is to be completed in Aotearoa, some DHBs send samples overseas (for example, to Australia) to complete hotspot analysis (i.e., only addressing a number of exons). Sending samples overseas for profiling may further increase inequity in personalised care in numerous ways, including cost and data sovereignty [17]. Personalised medicine risks compounding, rather than reducing, inequities in cancer care and therefore the use of genomics must be carefully considered [18]. If POLE sequencing is to become mainstream, we need to have the capacity to have it accredited in our laboratories, the funds to undertake the testing and a robust method of delineating novel variants of uncertain pathogenicity. We attempted to look at other immune markers that may triage POLE samples using online databases. We found significantly upregulated molecules that may be used as surrogate IHC markers and warrant further investigation to determine relevance to NZ patients.
Lymphocyte activation protein 3 (LAG3) is a cell surface molecule expressed on activated T cells. As such, it fits within the theory of increased TILs correlating with POLE tumours. LAG3 has been investigated as an immune checkpoint protein [19] and numerous checkpoint inhibitors targeting LAG3 are currently in clinical trials. In a meta-analysis, the high expression of LAG3 was associated with improved survival in multiple solid tumours [20]. LAG3 expression can be measured using IHC, as demonstrated across multiple tumour types, with positivity described as >15 LAG3 positive TIL per 40x magnification hot spot field [21].
T cell immunoglobulin and ITIM domain (TIGIT) is another immune checkpoint molecule that has recently garnered attention as an emerging target in cancer immunotherapy [22]. TIGIT is a cell receptor that regulates T-cell-mediated tumour recognition, and as such fits the theorised POLE-mutated phenotype. One study in endometrial cancer linked resident tumour NK cell expression of TIGIT to disease severity [23]. Our datamining showed that TIGIT RNA expression is upregulated in POLEmut tumours which, based on the literature, would indicate worse outcomes [24,25]. It may be possible that this is over-ridden by POLE mutation and therefore women with tumours that have POLE and high TIGIT expression still have a favourable outcome (this effect is seen with p53). Furthermore, our survival analysis showed that patients with TIGIT alterations had the same exceptionally positive outcomes as those with POLE alterations, indicating that this molecule may warrant further investigation in our NZ panel or provide evidence for anti-TIGIT therapy for the treatment of late-stage POLEmut tumours.
Over 40 chemokines have been identified in humans. We found CXCL13 and CXCL9 were the most upregulated in POLE-mutated EC. It is well established that these chemokines play key roles in immune responses, including inflammation in cancer [26,27]. CXCL13-positive TIL are associated with a high mutation load and, in uterine cancer, B cells were predominantly observed in large aggregations in the tumour and stroma in 92% of POLE samples, compared to 48% of Microsatellite Stable tumours [28]. In ovarian cancer, high IHC expression (measured on a 0–3 scale) of the tumour cell CXCL9 was associated with doubled overall survival rates [29].
The main limitation of this study is the small sample size of 90. Further investigation, particularly when breaking cohorts down to different ethnicities and/or molecular subtypes, is important. However, this was the first pilot of molecular profiling in Aotearoa and these data will be useful in developing further investigations and guidelines for the uptake of this testing in clinical practice. To date, there is only one other study that has aimed to pilot the use of molecular stratification in endometrial cancer: Oberndorfer et al. [30] in Austria retrospectively published the first “real-world data”, from a small sample size of 40. The distribution of subtypes within that study was comparable to that found in ours. Twenty-three of these had shifted risk grouping due to molecular profile results; four patients were upstaged and nineteen were downstaged, which would have led to a change in treatment regime for twelve patients.
5. Conclusions
There needs to be more research focussed on the possibility of changing EC treatment based on a molecular subtype; larger-scale clinical trials, developed from the PORTEC-3 data, can inform such research. We identified that 11/90 women (12%) had CTNNB1 mutations, suggesting that further investigation of this biomarker is needed in NZ women and that it may be appropriate to include it in an Aotearoa-specific genomic profiling panel. Overall, a large proportion of our cohort fell under the category of ‘non-specified molecular profile’. When breaking down subtypes and looking at treatment and outcomes, the major issues are around under-treating p53abn and early stage endometrioid tumours and over-treating those with POLE mutations. In our cohort, this accounted for around 8/90 patients (approx. 9%). Should it be, then, that POLE testing is only required for low-stage, high-grade tumours that harbour p53abn? In our cohort, this accounts for six women (7%). Based on NZ cancer statistics of newly registered uterine cases in 2018 (640), this would make a difference to an estimated 44 women each year. Further information in the form of prospective clinical trials is needed for these patients to safely receive de-escalated treatment based on their molecular profile.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/cancers13225641/s1, Figure S1: Example of insufficient staining patterns in autolysed endometrial tissue. Figure S2: Cbioportal output; Kaplan meier survival of altered immune genes identified in Figure 5; LAG3, TIGIT, CXCL13 and CXCL9. Table S1: IHC conditions. Table S2: CTNNB1 mutations. Table S3: Women who had recurrent endometrial cancer during the study period.
Author Contributions
Conceptualization, C.E.H., D.K., M.C.T. and S.K.F.; data curation, C.E.H. and E.J.O.; formal analysis, C.E.H., E.J.O. and D.K.; funding acquisition, C.E.H.; investigation, C.E.H. and K.P.; methodology, K.P., E.J.O. and D.K.; project administration, C.E.H., M.C.T. and S.K.F.; resources, K.P. and D.K.; supervision, S.K.F.; validation, K.P.; writing—original draft, C.E.H.; writing—review and editing, K.P., E.J.O., D.K., M.C.T. and S.K.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by RANZCOG and The Cancer Society Wellington.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Health and Disability Ethics Committee (HDEC 19CEN146), approved 25 October 2019.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not available.
Acknowledgments
We acknowledge Yee Syuen Low for her guidance on sequencing, the support of the Women’s Clinic, CCDHB, Wellington SCL and the department of Obstetrics, Gynaecology and Women’s Health at UOW.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Palmer, S.C.; Gray, H.; Huria, T.; Lacey, C.; Beckert, L.; Pitama, S.G. Reported Māori consumer experiences of health systems and programs in qualitative research: A systematic review with meta-synthesis. Int. J. Equity Health 2019, 18, 163. [Google Scholar] [CrossRef]
- Robson, B.; Cormack, D.; Purdie, G. Unequal Impact II: Māori and Non-Māori Cancer Statistics by Deprivation and Rural-Urban Status 2002–2006; Te Rōpū Rangahu Hauora a Eru Pōmare, University of Otago: Wellington, New Zealand, 2010. [Google Scholar]
- Scott, O.W.; Tin Tin, S.; Bigby, S.M.; Elwood, J.M. Rapid increase in endometrial cancer incidence and ethnic differences in New Zealand. Cancer Causes Control. 2019, 30, 121–127. [Google Scholar] [CrossRef]
- Bigby, S.M.; Tin Tin, S.; Eva, L.J.; Shirley, P.; Dempster-Rivett, K.; Elwood, M. Increasing incidence of endometrial carcinoma in a high-risk New Zealand community. Aust. N. Z. J. Obstet. Gynaecol. 2020, 60, 250–257. [Google Scholar] [CrossRef]
- Levine, D.A.; Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Kandoth, C.; Dooling, D.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Kommoss, S.; McConechy, M.K.; Kommoss, F.; Leung, S.; Bunz, A.; Magrill, J.; Britton, H.; Kommoss, F.; Grevenkamp, F.; Karnezis, A.; et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann. Oncol. 2018, 29, 1180–1188. [Google Scholar] [CrossRef]
- Imboden, S.; Nastic, D.; Ghaderi, M.; Rydberg, F.; Rau, T.T.; Mueller, M.D.; Epstein, E.; Carlson, J.W. Phenotype of POLE-mutated endometrial cancer. PLoS ONE 2019, 14, e0214318. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, A.; Bosse, T.; McAlpine, J.N. The emerging role of molecular pathology in directing the systemic treatment of endometrial cancer. Ther. Adv. Med. Oncol. 2021, 13, 17588359211035959. [Google Scholar] [CrossRef]
- León-Castillo, A.; de Boer, S.M.; Powell, M.E.; Mileshkin, L.R.; Mackay, H.J.; Leary, A.; Nijman, H.W.; Singh, N.; Pollock, P.M.; Bessette, P.; et al. Molecular Classification of the PORTEC-3 Trial for High-Risk Endometrial Cancer: Impact on Prognosis and Benefit From Adjuvant Therapy. J. Clin. Oncol. 2020, 38, 3388–3397. [Google Scholar] [CrossRef]
- León-Castillo, A.; Britton, H.; McConechy, M.K.; McAlpine, J.N.; Nout, R.; Kommoss, S.; Brucker, S.Y.; Carlson, J.W.; Epstein, E.; Rau, T.T.; et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J. Pathol. 2020, 250, 323–335. [Google Scholar] [CrossRef]
- Raffone, A.; Travaglino, A.; Raimondo, D.; Boccellino, M.P.; Maletta, M.; Borghese, G.; Casadio, P.; Insabato, L.; Mollo, A.; Zullo, F.; et al. Tumor-infiltrating lymphocytes and POLE mutation in endometrial carcinoma. Gynecol. Oncol. 2021, 161, 621–628. [Google Scholar] [CrossRef]
- Dong, D.; Lei, H.; Liu, D.; Bai, H.; Yang, Y.; Tang, B.; Li, K.; Liu, J.; Xu, G.; Xiao, X. POLE and Mismatch Repair Status, Checkpoint Proteins and Tumor-Infiltrating Lymphocytes in Combination, and Tumor Differentiation: Identify Endometrial Cancers for Immunotherapy. Front. Oncol. 2021, 11, 832. [Google Scholar] [CrossRef]
- Köbel, M.; Ronnett, B.M.; Singh, N.; Soslow, R.A.; Gilks, C.B.; McCluggage, W.G. Interpretation of P53 Immunohistochemistry in Endometrial Carcinomas: Toward Increased Reproducibility. Int. J. Gynecol. Pathol. 2019, 38 (Suppl. 1), S123–S131. [Google Scholar] [CrossRef]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor–immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef]
- Xie, B.; Qian, C.; Yang, B.; Ning, C.; Yao, X.; Du, Y.; Shi, Y.; Luo, X.; Chen, X. Risk Factors for Unsuccessful Office-Based Endometrial Biopsy: A Comparative Study of Office-Based Endometrial Biopsy (Pipelle) and Diagnostic Dilation and Curettage. J. Minim. Invasive Gynecol. 2018, 25, 724–729. [Google Scholar] [CrossRef]
- Liu, Y.; Patel, L.; Mills, G.B.; Lu, K.H.; Sood, A.K.; Ding, L.; Kucherlapati, R.; Mardis, E.R.; Levine, D.A.; Shmulevich, I.; et al. Clinical Significance of CTNNB1 Mutation and Wnt Pathway Activation in Endometrioid Endometrial Carcinoma. JNCI J. Natl. Cancer Inst. 2014, 106, dju245. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.; Kukutai, T.; Carroll, S.R.; Rodriguez-Lonebear, D. Indigenous Data Sovereignty and Policy; Taylor & Francis: London, UK, 2021. [Google Scholar]
- Robertson, S.P.; Hindmarsh, J.H.; Berry, S.; Cameron, V.A.; Cox, M.P.; Dewes, O.; Doughty, R.N.; Gray, G.; Jacobsen, J.C.; Laurence, A.; et al. Genomic medicine must reduce, not compound, health inequities: The case for hauora-enhancing genomic resources for New Zealand. N. Z. Med. J. 2018, 131, 81–89. [Google Scholar]
- Shan, C.; Li, X.; Zhang, J. Progress of immune checkpoint LAG-3 in immunotherapy. Oncol. Lett. 2020, 20, 207. [Google Scholar] [CrossRef]
- Saleh, R.R.; Peinado, P.; Fuentes-Antrás, J.; Pérez-Segura, P.; Pandiella, A.; Amir, E.; Ocaña, A. Prognostic Value of Lymphocyte-Activation Gene 3 (LAG3) in Cancer: A Meta-Analysis. Front. Oncol. 2019, 9, 1040. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Sherwood, T.; Costa, A.D.; Yee-Toy, N.; Lung, P.; Easton, A.; Sumrow, B.; Bonvini, E.; Moore, P.A. Immunohistochemistry analyses of LAG-3 expression across different tumor types and co-expression with PD-1. J. Clin. Oncol. 2020, 38, e15086. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Degos, C.; Heinemann, M.; Barrou, J.; Boucherit, N.; Lambaudie, E.; Savina, A.; Gorvel, L.; Olive, D. Endometrial Tumor Microenvironment Alters Human NK Cell Recruitment, and Resident NK Cell Phenotype and Function. Front. Immunol. 2019, 10, 877. [Google Scholar] [CrossRef]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseinkhani, N.; Shadbad, M.A.; Asghari Jafarabadi, M.; Karim Ahangar, N.; Asadzadeh, Z.; Mohammadi, S.M.; Lotfinejad, P.; Alizadeh, N.; Brunetti, O.; Fasano, R.; et al. A Systematic Review and Meta-Analysis on the Significance of TIGIT in Solid Cancers: Dual TIGIT/PD-1 Blockade to Overcome Immune-Resistance in Solid Cancers. Int. J. Mol. Sci. 2021, 22, 10389. [Google Scholar] [CrossRef] [PubMed]
- Kazanietz, M.G.; Durando, M.; Cooke, M. CXCL13 and Its Receptor CXCR5 in Cancer: Inflammation, Immune Response, and Beyond. Front. Endocrinol. 2019, 10, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.-J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Workel, H.H.; Lubbers, J.M.; Arnold, R.; Prins, T.M.; van der Vlies, P.; de Lange, K.; Bosse, T.; van Gool, I.C.; Eggink, F.A.; Wouters, M.C.A.; et al. A Transcriptionally Distinct CXCL13+ CD103+ CD8+ T-cell Population Is Associated with B-cell Recruitment and Neoantigen Load in Human Cancer. Cancer Immunol. Res. 2019, 7, 784–796. [Google Scholar] [CrossRef] [Green Version]
- Bronger, H.; Singer, J.; Windmüller, C.; Reuning, U.; Zech, D.; Delbridge, C.; Dorn, J.; Kiechle, M.; Schmalfeldt, B.; Schmitt, M.; et al. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br. J. Cancer 2016, 115, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Oberndorfer, F.; Moling, S.; Hagelkruys, L.A.; Grimm, C.; Polterauer, S.; Sturdza, A.; Aust, S.; Reinthaller, A.; Müllauer, L.; Schwameis, R. Risk Reclassification of Patients with Endometrial Cancer Based on Tumor Molecular Profiling: First Real World Data. J. Pers. Med. 2021, 11, 48. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).