
Oncogenic KRAS Requires Complete Loss of BAP1 Function for Development of Murine Intrahepatic Cholangiocarcinoma

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Conditional BAP1 Mice
2.2. Genotyping PCR
2.3. Histology and Immunohistochemistry
2.4. Microarray Analysis
2.5. Statistical Analysis
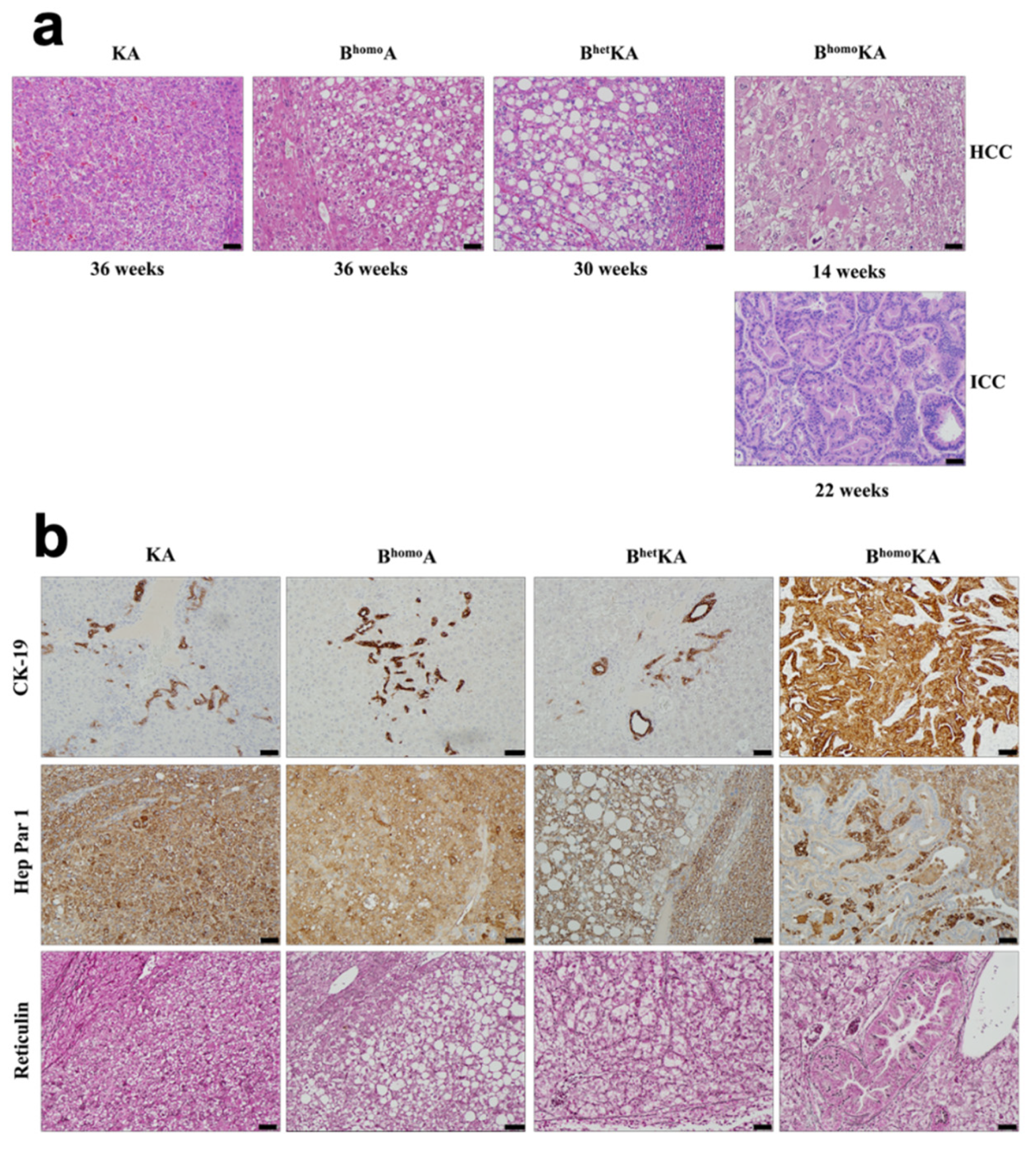
3. Results
3.1. Tissue-Specific Concomitant Expression of Oncogenic Kras and Bap1 Loss Results in Primary Liver Tumors and Decreased Survival
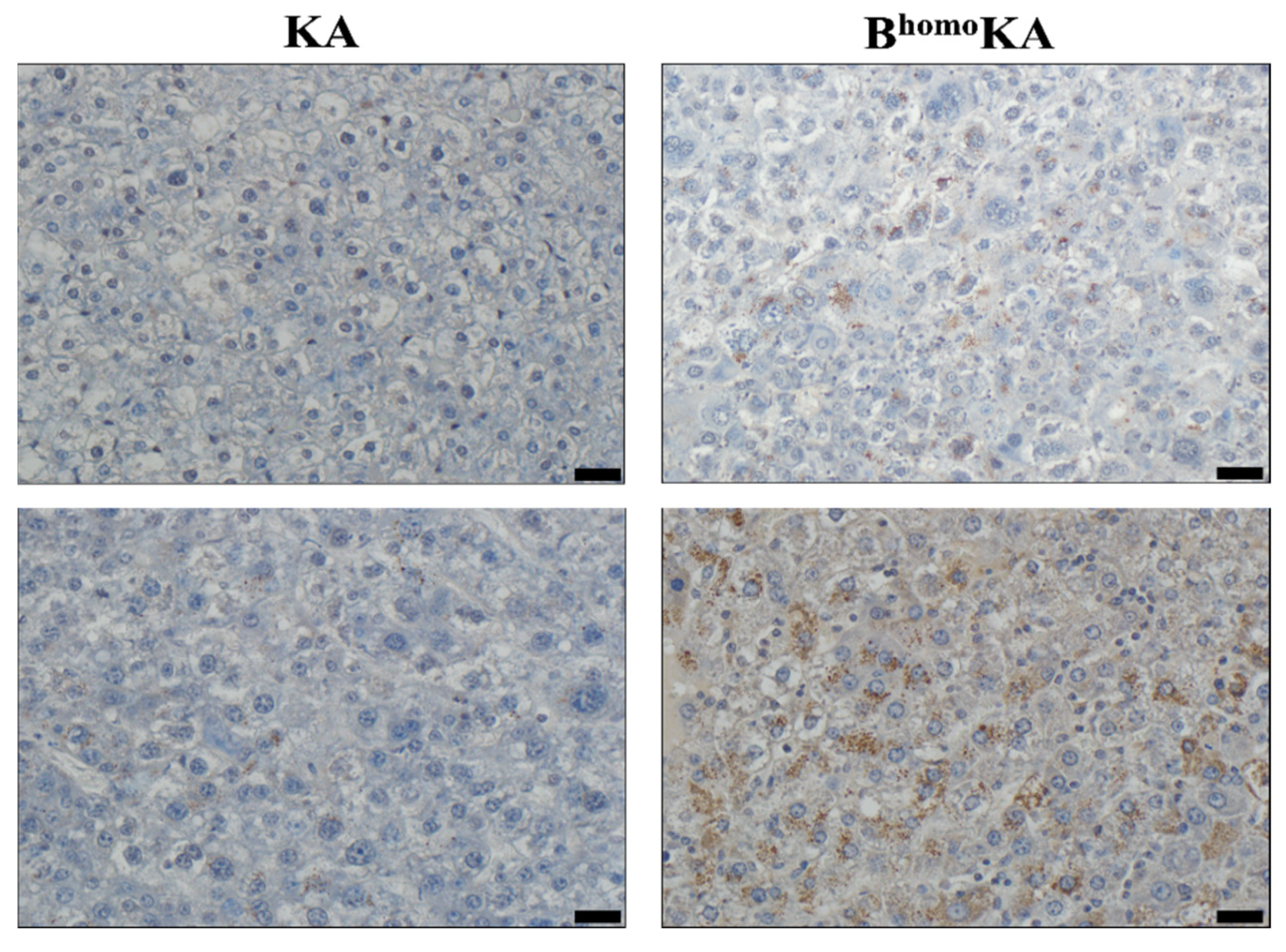
3.2. KrasG12D Activation in Combination with Homozygous Bap1 Deletion Results in Development of ICC
3.3. Transcriptomic Profiling of BhomoKA Mouse Tumors Identifies Potential Effector Pathways of Tumor Promotion Caused by Loss of BAP1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Rizvi, S.; Gores, G.J. Pathogenesis, Diagnosis, and Management of Cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef] [Green Version]
- Dodson, R.M.; Weiss, M.J.; Cosgrove, D.; Herman, J.M.; Kamel, I.; Anders, R.; Geschwind, J.-F.H.; Pawlik, T.M. Intrahepatic cholangiocarcinoma: Management options and emerging therapies. J. Am. Coll. Surg. 2013, 217, 736–750.e4. [Google Scholar] [CrossRef]
- Shaib, Y.; El-Serag, H.B. The epidemiology of cholangiocarcinoma. Semin. Liver Dis. 2004, 24, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Kirstein, M.M.; Vogel, A. Epidemiology and Risk Factors of Cholangiocarcinoma. Visc. Med. 2016, 32, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Shaib, Y.H.; Davila, J.A.; McGlynn, K.; El-Serag, H.B. Rising incidence of intrahepatic cholangiocarcinoma in the United States: A true increase? J. Hepatol. 2004, 40, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Zhu, A.X.; Fuchs, C.S.; Brooks, G.A. Forty-Year Trends in Cholangiocarcinoma Incidence in the U.S.: Intrahepatic Disease on the Rise. Oncologist 2016, 21, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, A.; Brandi, G. BILCAP trial and adjuvant capecitabine in resectable biliary tract cancer: Reflections on a standard of care. Expert Rev. Gastroenterol. Hepatol. 2020, 18, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Utuama, O.; Permuth, J.B.; Dagne, G.; Sanchez-Anguiano, A.; Alman, A.; Kumar, A.; Denbo, J.; Kim, R.; Fleming, J.B.; Anaya, D.A. Neoadjuvant Chemotherapy for Intrahepatic Cholangiocarcinoma: A Propensity Score Survival Analysis Supporting Use in Patients with High-Risk Disease. Ann. Surg. Oncol. 2021, 28, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Buettner, S.; Van Vugt, J.L.; IJzermans, J.N.; Groot Koerkamp, B. Intrahepatic cholangiocarcinoma: Current perspectives. OncoTargets Ther. 2017, 10, 1131–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef] [Green Version]
- Goere, D.; Wagholikar, G.D.; Pessaux, P.; Carrère, N.; Sibert, A.; Vilgrain, V.; Sauvanet, A.; Belghiti, J. Utility of staging laparoscopy in subsets of biliary cancers: Laparoscopy is a powerful diagnostic tool in patients with intrahepatic and gallbladder carcinoma. Surg. Endosc. 2006, 20, 721–725. [Google Scholar] [CrossRef]
- Franken, L.C.; Coelen, R.J.S.; Roos, E.; Verheij, J.; Phoa, S.S.; Besselink, M.G.; Busch, O.R.C.; van Gulik, T.M. Staging Laparoscopy in Patients with Intrahepatic Cholangiocarcinoma: Is It Still Useful? Visc. Med. 2020, 36, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Ross, P.; Wasan, H.S.; Hubner, R.A.; McNamara, M.G.; Lopes, A.; Manoharan, P.; Palmer, D.; Bridgewater, J.; Valle, J.W. Advanced Intrahepatic Cholangiocarcinoma: Post Hoc Analysis of the ABC-01, -02, and -03 Clinical Trials. J. Natl. Cancer Inst. 2020, 112, 200–210. [Google Scholar] [CrossRef]
- Kish, M.; Chan, K.; Perry, K.; Ko, Y.J. A systematic review and network meta-analysis of adjuvant therapy for curatively resected biliary tract cancers. Curr. Oncol. Tor. Ont. 2020, 27, e20–e26. [Google Scholar] [CrossRef] [Green Version]
- Lamarca, A.; Edeline, J.; McNamara, M.G.; Hubner, R.A.; Nagino, M.; Bridgewater, J.; Primrose, J.; Valle, J.W. Current standards and future perspectives in adjuvant treatment for biliary tract cancers. Cancer Treat. Rev. 2020, 84, 101936. [Google Scholar] [CrossRef]
- Ebata, T.; Hirano, S.; Konishi, M.; Uesaka, K.; Tsuchiya, Y.; Ohtsuka, M.; Kaneoka, Y.; Yamamoto, M.; Ambo, Y.; Shimizu, Y.; et al. Randomized clinical trial of adjuvant gemcitabine chemotherapy versus observation in resected bile duct cancer. Br. J. Surg. 2018, 105, 192–202. [Google Scholar] [CrossRef]
- Edeline, J.; Benabdelghani, M.; Bertaut, A.; Watelet, J.; Hammel, P.; Joly, J.-P.; Boudjema, K.; Fartoux, L.; Bouhier-Leporrier, K.; Jouve, J.-L.; et al. Gemcitabine and Oxaliplatin Chemotherapy or Surveillance in Resected Biliary Tract Cancer (PRODIGE 12-ACCORD 18-UNICANCER GI): A Randomized Phase III Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 658–667. [Google Scholar] [CrossRef]
- Rizzo, A.; Frega, G.; Ricci, A.D.; Palloni, A.; Abbati, F.; De Lorenzo, S.; Deserti, M.; Tavolari, S.; Brandi, G. Anti-EGFR Monoclonal Antibodies in Advanced Biliary Tract Cancer: A Systematic Review and Meta-analysis. In Vivo 2020, 34, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Malka, D.; Edeline, J. Adjuvant capecitabine in biliary tract cancer: A standard option? Lancet Oncol. 2019, 20, 606–608. [Google Scholar] [CrossRef]
- Primrose, J.N.; Fox, R.P.; Palmer, D.H.; Malik, H.Z.; Prasad, R.; Mirza, D.; Anthony, A.; Corrie, P.; Falk, S.; Finch-Jones, M.; et al. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): A randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 2019, 20, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.T.; Kennedy, E.B.; Bachini, M.; Bekaii-Saab, T.; Crane, C.; Edeline, J.; El-Khoueiry, A.; Feng, M.; Katz, M.H.G.; Primrose, J.; et al. Adjuvant Therapy for Resected Biliary Tract Cancer: ASCO Clinical Practice Guideline. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 1015–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Thornblade, L.W.; Wong, P.; Li, D.; Warner, S.G.; Chang, S.; Raoof, M.; Kessler, J.; Amini, A.; Lin, J.; Chung, V.; et al. Patterns of Whole Exome Sequencing in Resected Cholangiocarcinoma. Cancers 2021, 13, 4062. [Google Scholar] [CrossRef]
- Hu, L.-S.; Zhang, X.-F.; Weiss, M.; Popescu, I.; Marques, H.P.; Aldrighetti, L.; Maithel, S.K.; Pulitano, C.; Bauer, T.W.; Shen, F.; et al. Recurrence Patterns and Timing Courses Following Curative-Intent Resection for Intrahepatic Cholangiocarcinoma. Ann. Surg. Oncol. 2019, 26, 2549–2557. [Google Scholar] [CrossRef] [PubMed]
- Bagante, F.; Spolverato, G.; Weiss, M.; Alexandrescu, S.; Marques, H.P.; Aldrighetti, L.; Maithel, S.K.; Pulitano, C.; Bauer, T.W.; Shen, F.; et al. Defining Long-Term Survivors Following Resection of Intrahepatic Cholangiocarcinoma. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 2017, 21, 1888–1897. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.-C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef]
- Chan-on, W.; Nairismägi, M.-L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke–related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef]
- Johnson, L.; Mercer, K.; Greenbaum, D.; Bronson, R.T.; Crowley, D.; Tuveson, D.A.; Jacks, T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001, 410, 1111–1116. [Google Scholar] [CrossRef]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Ventii, K.H.; Devi, N.S.; Friedrich, K.L.; Chernova, T.A.; Tighiouart, M.; Van Meir, E.G.; Wilkinson, K.D. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res. 2008, 68, 6953–6962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchini, C.; Veronese, N.; Yachida, S.; Cheng, L.; Nottegar, A.; Stubbs, B.; Solmi, M.; Capelli, P.; Pea, A.; Barbareschi, M.; et al. Different prognostic roles of tumor suppressor gene BAP1 in cancer: A systematic review with meta-analysis. Genes Chromosomes Cancer 2016, 55, 741–749. [Google Scholar] [CrossRef]
- Peña-Llopis, S.; Vega-Rubín-de-Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef]
- Qin, J.; Zhou, Z.; Chen, W.; Wang, C.; Zhang, H.; Ge, G.; Shao, M.; You, D.; Fan, Z.; Xia, H.; et al. BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nat. Commun. 2015, 6, 8471. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 links metabolic regulation of ferroptosis to tumor suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef]
- Zhang, Y.; Koppula, P.; Gan, B. Regulation of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1. Cell Cycle 2019, 18, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dell, M.R.; Li Huang, J.; Whitney-Miller, C.L.; Deshpande, V.; Rothberg, P.; Grose, V.; Rossi, R.M.; Zhu, A.X.; Land, H.; Bardeesy, N.; et al. KrasG12D and p53 Mutation Cause Primary Intrahepatic Cholangiocarcinoma. Cancer Res. 2012, 72, 1557–1567. [Google Scholar] [CrossRef] [Green Version]
- Postic, C.; Magnuson, M.A. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 2000, 26, 149–150. [Google Scholar] [CrossRef]
- Gu, Y.-F.; Cohn, S.; Christie, A.; McKenzie, T.; Wolff, N.; Do, Q.N.; Madhuranthakam, A.J.; Pedrosa, I.; Wang, T.; Dey, A.; et al. Modeling Renal Cell Carcinoma in Mice: Bap1 and Pbrm1 Inactivation Drive Tumor Grade. Cancer Discov. 2017, 7, 900–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-S.; Gu, Y.-F.; Wolff, N.; Stefanius, K.; Christie, A.; Dey, A.; Hammer, R.E.; Xie, X.-J.; Rakheja, D.; Pedrosa, I.; et al. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 16538–16543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Pramanik, D.; Mukherjee, R.; Campbell, N.R.; Elumalai, S.; De Wilde, R.F.; Hong, S.-M.; Goggins, M.G.; De Jesus-Acosta, A.; Laheru, D.; et al. Molecular determinants of retinoic acid sensitivity in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinform. Oxf. Engl. 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Kühn, R.; Schwenk, F.; Aguet, M.; Rajewsky, K. Inducible Gene Targeting in Mice. Sci. New Ser. 1995, 269, 1427–1429. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Kobayashi, S.; Qiao, W.; Li, C.; Xiao, C.; Radaeva, S.; Stiles, B.; Wang, R.-H.; Ohara, N.; Yoshino, T.; et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption ofSmad4 and Pten in mice. J. Clin. Invest. 2006, 116, 1843–1852. [Google Scholar] [CrossRef]
- Kenerson, H.L.; Yeh, M.M.; Kazami, M.; Jiang, X.; Riehle, K.J.; McIntyre, R.L.; Park, J.O.; Kwon, S.; Campbell, J.S.; Yeung, R.S. Akt and mTORC1 have different roles during liver tumorigenesis in mice. Gastroenterology 2013, 144, 1055–1065. [Google Scholar] [CrossRef] [Green Version]
- Saborowski, A.; Saborowski, M.; Davare, M.A.; Druker, B.J.; Klimstra, D.S.; Lowe, S.W. Mouse model of intrahepatic cholangiocarcinoma validates FIG-ROS as a potent fusion oncogene and therapeutic target. Proc. Natl. Acad. Sci. USA 2013, 110, 19513–19518. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Seshasayee, D.; Noubade, R.; French, D.M.; Liu, J.; Chaurushiya, M.S.; Kirpatrick, D.S.; Pham, V.C.; Lill, J.R.; Bakalarski, C.E.; et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science 2012, 337, 1541–1546. [Google Scholar] [CrossRef] [Green Version]
- Balaton, A.J.; Nehama-Sibony, M.; Gotheil, C.; Callard, P.; Baviera, E.E. Distinction between hepatocellular carcinoma, cholangiocarcinoma, and metastatic carcinoma based on immunohistochemical staining for carcinoembryonic antigen and for cytokeratin 19 on paraffin sections. J. Pathol. 1988, 156, 305–310. [Google Scholar] [CrossRef]
- Johnson, D.E.; Herndier, B.G.; Medeiros, L.J.; Warnke, R.A.; Rouse, R.V. The diagnostic utility of the keratin profiles of hepatocellular carcinoma and cholangiocarcinoma. Am. J. Surg. Pathol. 1988, 12, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.G.; Ishizawa, S.; Wu, E.; Weiss, L.M. Hepatocyte antigen as a marker of hepatocellular carcinoma: An immunohistochemical comparison to carcinoembryonic antigen, CD10, and alpha-fetoprotein. Am. J. Surg. Pathol. 2002, 26, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Prakash, S.; Geller, S.A.; Alsabeh, R. Comparative immunohistochemical profile of hepatocellular carcinoma, cholangiocarcinoma, and metastatic adenocarcinoma. Hum. Pathol. 2002, 33, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Van de Rijn, M.; Montgomery, K.; Rouse, R.V. Hep par 1 antibody stain for the differential diagnosis of hepatocellular carcinoma: 676 tumors tested using tissue microarrays and conventional tissue sections. Mod. Pathol. 2003, 16, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.S.; Lee, K.; Shin, E.; Kim, S.H.; Jing, J.; Jung, H.Y.; Lee, H.; Jang, J.-J. Comparative Analysis of Immunohistochemical Markers for Differential Diagnosis of Hepatocelluar Carcinoma and Cholangiocarcinoma. Tumori J. 2012, 98, 478–484. [Google Scholar] [CrossRef]
- Gagliano, E.F. Reticulin stain in the fine needle aspiration differential diagnosis of liver nodules. Acta Cytol. 1995, 39, 596–598. [Google Scholar] [PubMed]
- Bergman, S.; Graeme-Cook, F.; Pitman, M.B. The usefulness of the reticulin stain in the differential diagnosis of liver nodules on fine-needle aspiration biopsy cell block preparations. Mod. Pathol. 1997, 10, 1258–1264. [Google Scholar]
- Wee, A. Fine needle aspiration biopsy of the liver: Algorithmic approach and current issues in the diagnosis of hepatocellular carcinoma. CytoJournal 2005, 2, 7. [Google Scholar] [CrossRef]
- Nahon, J.L.; Tratner, I.; Poliard, A.; Presse, F.; Poiret, M.; Gal, A.; Sala-Trepat, J.M.; Legrès, L.; Feldmann, G.; Bernuau, D. Albumin and alpha-fetoprotein gene expression in various nonhepatic rat tissues. J. Biol. Chem. 1988, 263, 11436–11442. [Google Scholar] [CrossRef]
- Ikenoue, T.; Terakado, Y.; Nakagawa, H.; Hikiba, Y.; Fujii, T.; Matsubara, D.; Noguchi, R.; Zhu, C.; Yamamoto, K.; Kudo, Y.; et al. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci. Rep. 2016, 6, 23899. [Google Scholar] [CrossRef]
- Lee, K.-P.; Lee, J.-H.; Kim, T.-S.; Kim, T.-H.; Park, H.-D.; Byun, J.-S.; Kim, M.-C.; Jeong, W.-I.; Calvisi, D.F.; Kim, J.-M.; et al. The Hippo–Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 8248–8253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sörensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Bektas, H.; Manns, M.P.; et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Xin, B.; Watanabe, K.; Ooshio, T.; Fujii, K.; Chen, X.; Okada, Y.; Abe, H.; Taguchi, Y.; Miyokawa, N.; et al. Oncogenic Determination of a Broad Spectrum of Phenotypes of Hepatocyte-Derived Mouse Liver Tumors. Am. J. Pathol. 2017, 187, 2711–2725. [Google Scholar] [CrossRef] [Green Version]
- Fausto, N. Liver regeneration and repair: Hepatocytes, progenitor cells, and stem cells. Hepatology 2004, 39, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T. Cancer Stem cells and their cellular origins in primary liver and biliary tract cancers. Hepatology 2016, 64, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Mean Survival, Range (wks) | Hepatic Histopathologic Findings | Mice With HCC, n (%) | Mice With ICC, n (%) |
|---|---|---|---|---|
| KA | 51, 34–83 | Fatty metamorphosis of hepatocytes, steatosis, parenchymal congestion, adenomas, HCC | 13 (87) | 0 (0) |
| BhomoA | 52, 28–76 | Fatty metamorphosis of hepatocytes, parenchymal congestion, adenomas, HCC | 15 (60) | 0 (0) |
| BhetKA | 45, 40–53 | Dilated blood vessels, adenomas, HCC | 16 (94) | 0 (0) |
| BhomoKA | 24, 14–35 | Fatty metamorphosis of hepatocytes, steatosis, steatohepatitis, parenchymal congestion, biliary hyperplasia, adenomas, HCC, ICC | 33 (87) | 38 (100) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcus, R.; Ferri-Borgogno, S.; Hosein, A.; Foo, W.C.; Ghosh, B.; Zhao, J.; Rajapakshe, K.; Brugarolas, J.; Maitra, A.; Gupta, S. Oncogenic KRAS Requires Complete Loss of BAP1 Function for Development of Murine Intrahepatic Cholangiocarcinoma. Cancers 2021, 13, 5709. https://doi.org/10.3390/cancers13225709
Marcus R, Ferri-Borgogno S, Hosein A, Foo WC, Ghosh B, Zhao J, Rajapakshe K, Brugarolas J, Maitra A, Gupta S. Oncogenic KRAS Requires Complete Loss of BAP1 Function for Development of Murine Intrahepatic Cholangiocarcinoma. Cancers. 2021; 13(22):5709. https://doi.org/10.3390/cancers13225709
Chicago/Turabian StyleMarcus, Rebecca, Sammy Ferri-Borgogno, Abdel Hosein, Wai Chin Foo, Bidyut Ghosh, Jun Zhao, Kimal Rajapakshe, James Brugarolas, Anirban Maitra, and Sonal Gupta. 2021. "Oncogenic KRAS Requires Complete Loss of BAP1 Function for Development of Murine Intrahepatic Cholangiocarcinoma" Cancers 13, no. 22: 5709. https://doi.org/10.3390/cancers13225709
APA StyleMarcus, R., Ferri-Borgogno, S., Hosein, A., Foo, W. C., Ghosh, B., Zhao, J., Rajapakshe, K., Brugarolas, J., Maitra, A., & Gupta, S. (2021). Oncogenic KRAS Requires Complete Loss of BAP1 Function for Development of Murine Intrahepatic Cholangiocarcinoma. Cancers, 13(22), 5709. https://doi.org/10.3390/cancers13225709