
Stringent Base Specific and Optimization-Free Multiplex Mediator Probe ddPCR for the Quantification of Point Mutations in Circulating Tumor DNA

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
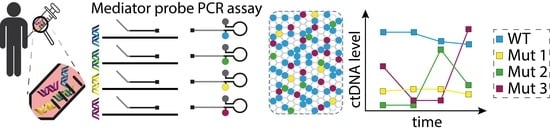
1. Introduction
2. Materials and Methods
2.1. Chemicals and Oligonucleotides for MP ddPCR Assay
2.2. Chemicals and Oligonucleotides for LNA PCR
2.3. MP ddPCR Assay Characterization
2.4. MP ddPCR Assay Validation with Clinical Samples
2.5. Principle for Point Mutation Detection by Mediator Probe PCR
2.6. Assay Design
2.7. Setup of the DOE for MP ddPCR Assay Development
- Annealing temperature (54 °C/60 °C)
- Annealing time (30 s/60 s)
- MP concentration (500 nM/1200 nM)
- MP:UR ratio (2:1/5:1)
- Forward primer (500 nM/1200 nM)
- Reverse primer (500 nM/1200 nM)
3. Results and Discussion
3.1. MP ddPCR Assay Development Using DOE
- Annealing temperature = 54 °C
- Annealing time = 60 s
- MP concentration = 1200 nM
- MP:UR ratio = 5:1 (1200 nM MP and 240 nM UR)
- Forward primer = 500 nM
- Reverse primer = 1200 nM
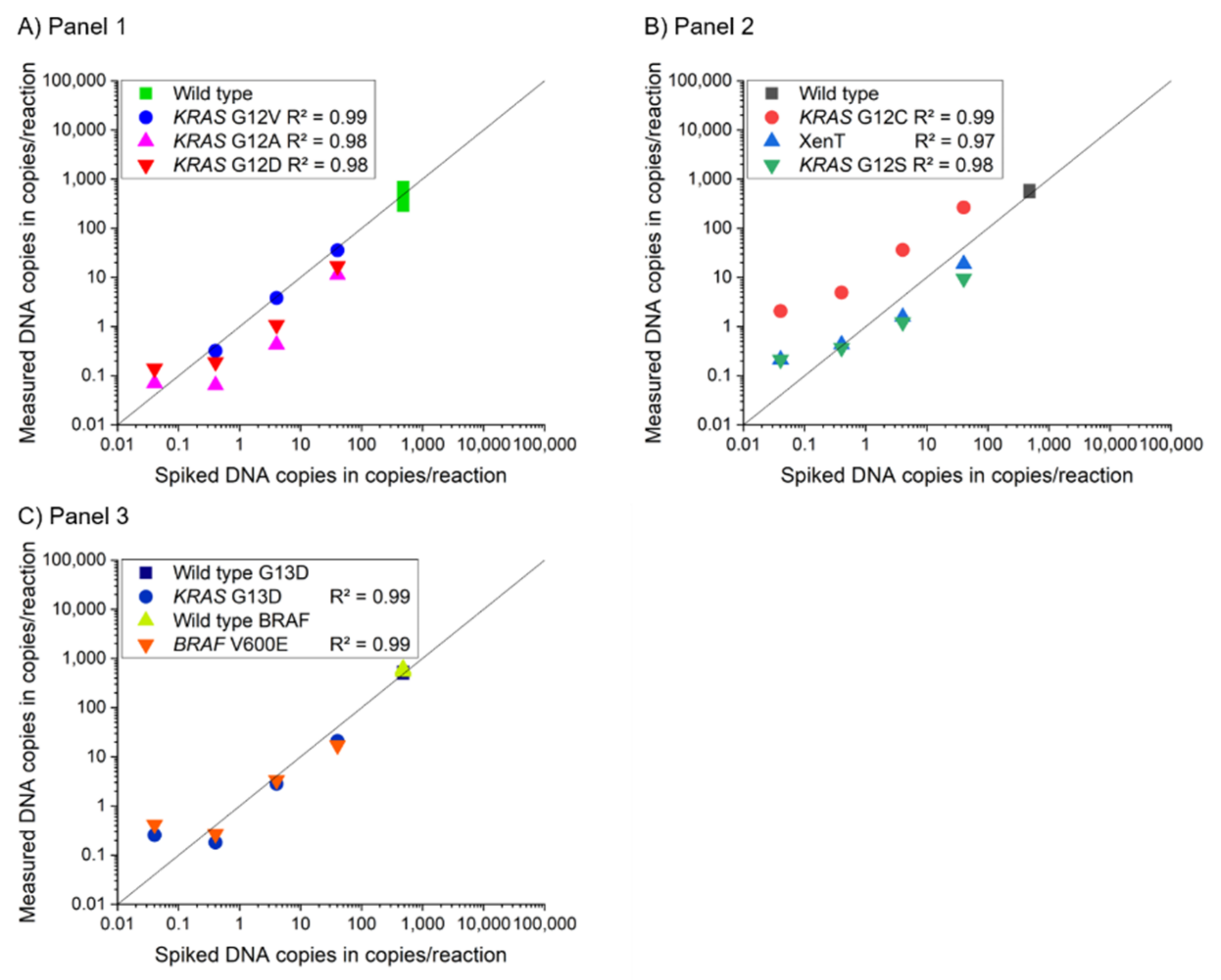
3.2. Assay Characterization of 2-Plex and 4-Plex MP ddPCR Assay
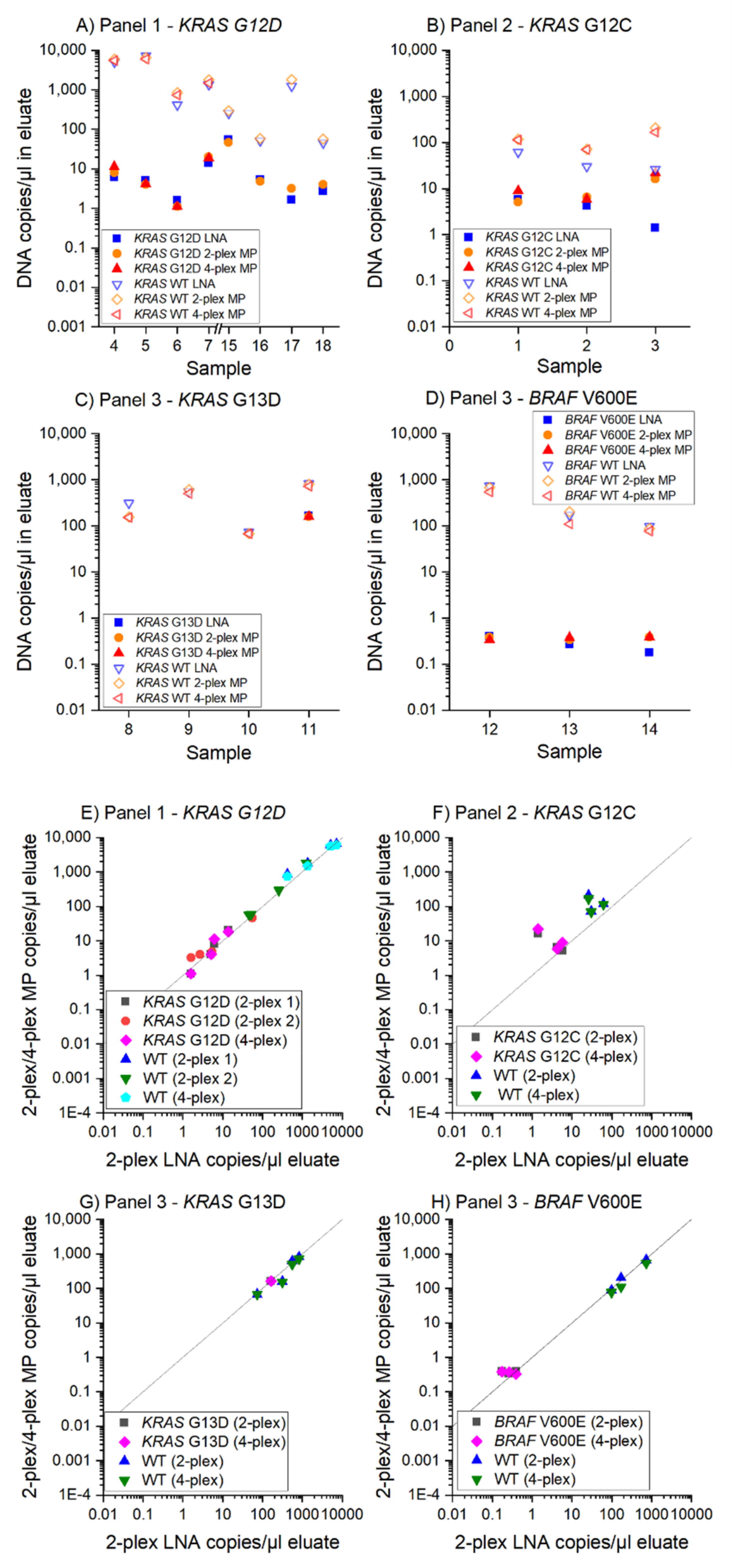
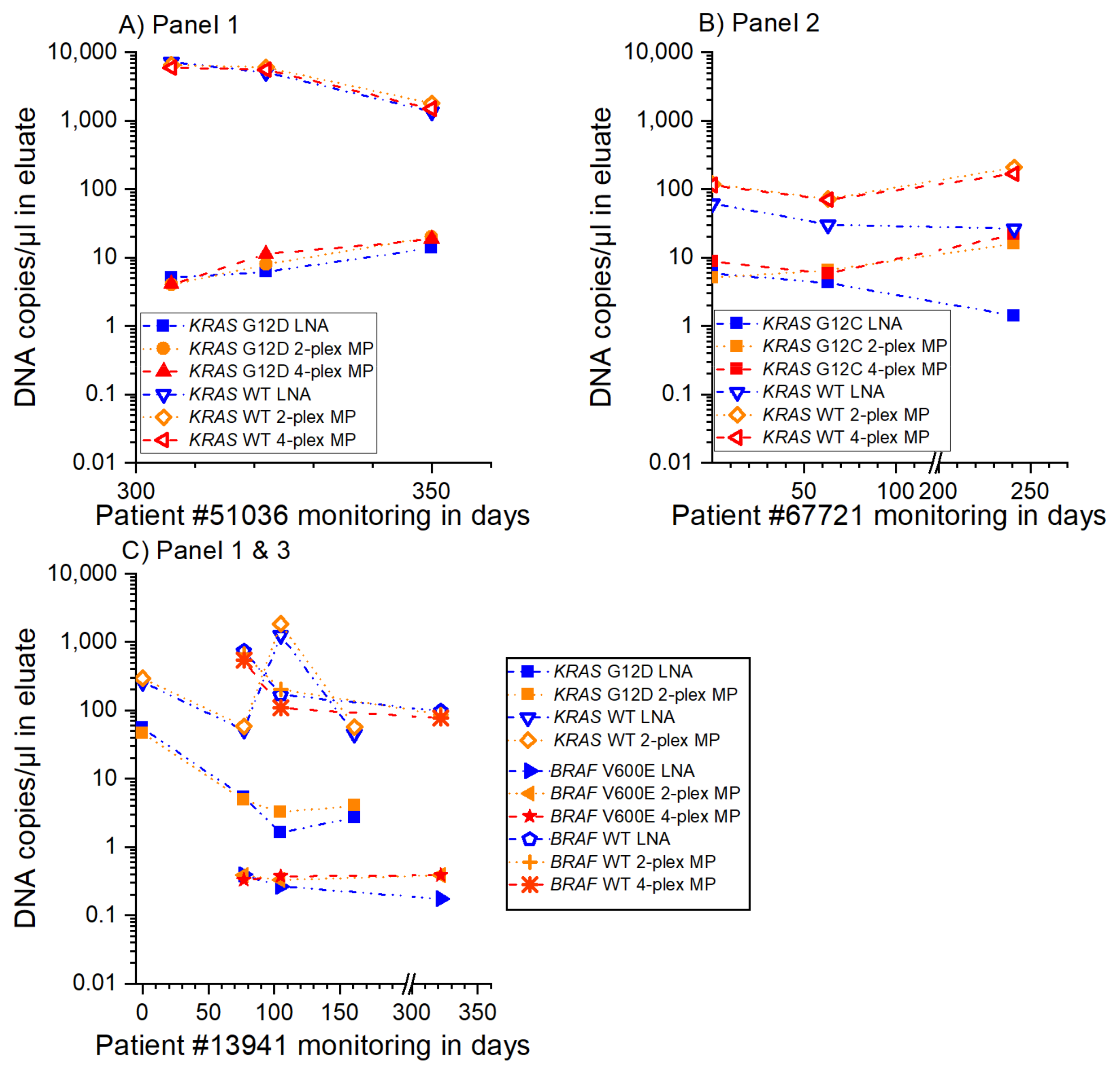
3.3. MP ddPCR Assay Validation with Clinical Samples
- 9/9 samples KRAS G12D positive
- 3/3 samples KRAS G12C positive
- 1/1 sample KRAS G13D positive
- 2/2 samples KRAS G13D negative and
- 3/3 samples BRAF V600E positive
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Madic, J.; Jovelet, C.; Lopez, J.; André, B.; Fatien, J.; Miran, I.; Honoré, A.; Lezquita, L.; Besse, B.; Lacroix, L.; et al. EGFR C797S, EGFR T790M, and EGFR sensitizing mutations in non-small chell lung cancer revealed by six-color crystal digital PCR. Oncotarget 2018, 9, 37393–37406. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, V.; Rutkowski, A.J.; Meuser, E.; Carr, T.H.; Harrington, E.A.; Barrett, J.C. Optimisation of robust singleplex and multiplex droplet digital PCR assays for high confidence mutation detection in circulating tumour DNA. Sci. Rep. 2019, 9, 12620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmedillas-López, S.; García-Arranz, M.; García-Olmo, D. Current and Emerging Applications of Droplet Digital PCR in Oncology. Mol. Diagn. Ther. 2017, 21, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Folprecht, G.; Reinacher-Schick, A.; Weitz, J.; Lugnier, C.; Kraeft, A.-L.; Wisser, S.; Aust, D.E.; Weiss, L.; Bubnoff, N.v.; Kramer, M.; et al. The CIRCULATE trial: Circulating tumour DNA based decision for adjuvant treatment in colon cancer stage II evaluation (AIO-KRK-0217). Clin. Colorectal Cancer 2021, 49, 1374. [Google Scholar] [CrossRef] [PubMed]
- Olsson, E.; Winter, C.; George, A.; Chen, Y.; Howlin, J.; Tang, M.-H.E.; Dahlgren, M.; Schulz, R.; Grabau, D.; Westen, D.; et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 2015, 7, 1034–1047. [Google Scholar] [CrossRef]
- Franczak, C.; Filhine-Tresarrieu, P.; Gilson, P.; Merlin, J.-L.; Au, L.; Harlé, A. Technical considerations for circulating tumor DNA detection in oncology. Expert Rev. Mol. Diagn. 2019, 19, 121–135. [Google Scholar] [CrossRef]
- Tan, C.R.C.; Zhou, L.; El-Deiry, W.S. Circulating Tumor Cells Versus Circulating Tumor DNA in Colorectal Cancer: Pros and Cons. Curr. Colorectal Cancer Rep. 2016, 12, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Taly, V.; Pekin, D.; Benhaim, L.; Kotsopoulos, S.K.; Le Corre, D.; Li, X.; Atochin, I.; Link, D.R.; Griffiths, A.D.; Pallier, K.; et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin. Chem. 2013, 59, 1722–1731. [Google Scholar] [CrossRef]
- Corné, J.; Le Du, F.; Quillien, V.; Godey, F.; Robert, L.; Bourien, H.; Brunot, A.; Crouzet, L.; Perrin, C.; Lefeuvre-Plesse, C.; et al. Development of multiplex digital PCR assays for the detection of PIK3CA mutations in the plasma of metastatic breast cancer patients. Sci. Rep. 2021, 11, 17316. [Google Scholar] [CrossRef]
- Diaz, L.A.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Hussung, S.; Follo, M.; Klar, R.F.U.; Michalczyk, S.; Fritsch, K.; Nollmann, F.; Hipp, J.; Duyster, J.; Scherer, F.; von Bubnoff, N.; et al. Development and Clinical Validation of Discriminatory Multitarget Digital Droplet PCR Assays for the Detection of Hot Spot KRAS and NRAS Mutations in Cell-Free DNA. J. Mol. Diagn. 2020, 22, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Madic, J.; Zocevic, A.; Senlis, V.; Fradet, E.; Andre, B.; Muller, S.; Dangla, R.; Droniou, M.E. Three-color crystal digital PCR. Biomol. Detect. Quantif. 2016, 10, 34–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussung, S.; Fritsch, R. Multi-target ddPCR Assays zur Detektion von KRAS- und NRAS-Mutationen. Biospektrum 2020, 26, 629–634. [Google Scholar] [CrossRef]
- Lehnert, M.; Kipf, E.; Schlenker, F.; Borst, N.; Zengerle, R.; von Stetten, F. Fluorescence signal-to-noise optimisation for real-time PCR using universal reporter oligonucleotides. Anal. Methods 2018, 10, 3444–3454. [Google Scholar] [CrossRef] [Green Version]
- Faltin, B.; Wadle, S.; Roth, G.; Zengerle, R.; von Stetten, F. Mediator probe PCR: A novel approach for detection of real-time PCR based on label-free primary probes and standardized secondary universal fluorogenic reporters. Clin. Chem. 2012, 58, 1546–1556. [Google Scholar] [CrossRef] [Green Version]
- Lyamichev, V.; Brow, M.A.; Dahlberg, J.E. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science 1993, 260, 778–783. [Google Scholar] [CrossRef]
- Wadle, S.; Lehnert, M.; Schuler, F.; Köppel, R.; Serr, A.; Zengerle, R.; von Stetten, F. Simplified development of multiplex real-time PCR through master mix augmented by universal fluorogenic reporters. Biotechniques 2016, 61, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Wadle, S.; Lehnert, M.; Rubenwolf, S.; Zengerle, R.; von Stetten, F. Real-time PCR probe optimization using design of experiments approach. Biomol. Detect. Quantif. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlenker, F.; Kipf, E.; Borst, N.; Hutzenlaub, T.; Zengerle, R.; von Stetten, F.; Juelg, P. Virtual Fluorescence Color Channels by Selective Photobleaching in Digital PCR Applied to the Quantification of KRAS Point Mutations. Anal. Chem. 2021, 93, 10538–10545. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Kim, D.; Lee, K.; Chun, J.-Y. Single-channel multiplexing without melting curve analysis in real-time PCR. Sci. Rep. 2014, 4, 7439. [Google Scholar] [CrossRef] [Green Version]
- Schlenker, F.; Kipf, E.; Borst, N.; Paust, N.; Zengerle, R.; von Stetten, F.; Juelg, P.; Hutzenlaub, T. Centrifugal Microfluidic Integration of 4-Plex ddPCR Demonstrated by the Quantification of Cancer-Associated Point Mutations. Processes 2021, 9, 97. [Google Scholar] [CrossRef]
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.M.; Schutten, M. The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5’ nuclease assay. J. Mol. Diagn. 2010, 12, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Robert, B.; Arnau Peyrotte, E.; Del Rio, M.; Ychou, M.; Molina, F.; Gongora, C.; Thierry, A.R. High fragmentation characterizes tumour-derived circulating DNA. PLoS ONE 2011, 6, e23418. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- SantaLucia, J. Physical principles and visual-OMP software for optimal PCR design. Methods Mol. Biol. 2007, 402, 3–34. [Google Scholar] [CrossRef] [PubMed]
- Kleppmann, W. Taschenbuch Versuchsplanung: Produkte Und Prozesse Optimieren; Carl Hanser Verlag GmbH & Co. KG: München, Germany, 2011; ISBN 9783446429420. [Google Scholar]
- Whale, A.S.; Cowen, S.; Foy, C.A.; Huggett, J.F. Methods for applying accurate digital PCR analysis on low copy DNA samples. PLoS ONE 2013, 8, e58177. [Google Scholar] [CrossRef] [Green Version]
- Wadle, S. Characterization and Optimization of Binding Energies in Mediator Probe PCR Enabling Multiplex Real-Time DNA and RNA Detection. Ph.D. Thesis, University Freiburg, Freiburg, Germany, 2016. [Google Scholar]
- Nikolaev, S.; Lemmens, L.; Koessler, T.; Blouin, J.-L.; Nouspikel, T. Circulating tumoral DNA: Preanalytical validation and quality control in a diagnostic laboratory. Anal. Biochem. 2018, 542, 34–39. [Google Scholar] [CrossRef]
- Devonshire, A.S.; Whale, A.S.; Gutteridge, A.; Jones, G.; Cowen, S.; Foy, C.A.; Huggett, J.F. Towards standardisation of cell-free DNA measurement in plasma: Controls for extraction efficiency, fragment size bias and quantification. Anal. Bioanal. Chem. 2014, 406, 6499–6512. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Panel | Gene | Mutation | Change of Nucleotide | Change of Amino Acid | 2-Plex Mediator Probe ddPCR | 4-Plex Mediator Probe ddPCR | 2-Plex Reference Assay | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LoB (Copies/mL Plasma) | LoD (% AF) | R2 2-Plex | LoB (Copies/mL Plasma) | LoD (% AF) | R2 4-Plex | LoB (Copies/mL Plasma) | LoD (% AF) | R2 2-Plex | |||||
| 1 | KRAS | Codon 12 | WT | ||||||||||
| KRAS | G12D | c.35G>A | p.Gly12Asp | 0.00 | 0.008 | 0.99 | 0.00 | 0.024 | 0.98 | 3.26 | 0.032 | 0.99 | |
| KRAS | G12A | c.35G>C | p.Gly12Ala | 0.00 | 0.004 | 0.99 | 0.00 | 0.009 | 0.98 | 0.00 | 0.074 | 0.99 | |
| KRAS | G12V | c.35G>T | p.Gly12Val | 0.00 | 0.011 | 0.99 | 0.00 | 0.047 | 0.99 | 2.21 | 0.011 | 0.99 | |
| 2 | KRAS | Codon 12 | WT | ||||||||||
| KRAS | G12S | c.34G>A | p.Gly12Ser | 0.00 | 0.019 | 0.99 | 0.00 | 0.040 | 0.98 | 1.43 | 0.097 | 0.99 | |
| KRAS | G12C | c.34G>T | p.Gly12Cys | 0.00 | 0.015 | 0.99 | 0.00 | 0.383 | 0.99 | 0.00 | 0.067 | 0.99 | |
| Xenopus tropicalis (XenT) extraction control | |||||||||||||
| 3 | KRAS | Codon 13 | WT | ||||||||||
| KRAS | G13D | c.38G>A | p.Gly13Asp | 0.00 | 0.050 | 0.99 | 9.82 | 0.034 | 0.99 | 1.68 | 0.170 | 0.99 | |
| BRAF | Codon 600 | WT | |||||||||||
| BRAF | V600E | c.1799T>A | p.Val600Glu | 0.00 | 0.039 | 0.99 | 16.29 | 0.050 | 0.99 | 5.90 | 0.047 | 0.99 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlenker, F.; Kipf, E.; Deuter, M.; Höffkes, I.; Lehnert, M.; Zengerle, R.; von Stetten, F.; Scherer, F.; Wehrle, J.; von Bubnoff, N.; et al. Stringent Base Specific and Optimization-Free Multiplex Mediator Probe ddPCR for the Quantification of Point Mutations in Circulating Tumor DNA. Cancers 2021, 13, 5742. https://doi.org/10.3390/cancers13225742
Schlenker F, Kipf E, Deuter M, Höffkes I, Lehnert M, Zengerle R, von Stetten F, Scherer F, Wehrle J, von Bubnoff N, et al. Stringent Base Specific and Optimization-Free Multiplex Mediator Probe ddPCR for the Quantification of Point Mutations in Circulating Tumor DNA. Cancers. 2021; 13(22):5742. https://doi.org/10.3390/cancers13225742
Chicago/Turabian StyleSchlenker, Franziska, Elena Kipf, Max Deuter, Inga Höffkes, Michael Lehnert, Roland Zengerle, Felix von Stetten, Florian Scherer, Julius Wehrle, Nikolas von Bubnoff, and et al. 2021. "Stringent Base Specific and Optimization-Free Multiplex Mediator Probe ddPCR for the Quantification of Point Mutations in Circulating Tumor DNA" Cancers 13, no. 22: 5742. https://doi.org/10.3390/cancers13225742
APA StyleSchlenker, F., Kipf, E., Deuter, M., Höffkes, I., Lehnert, M., Zengerle, R., von Stetten, F., Scherer, F., Wehrle, J., von Bubnoff, N., Juelg, P., Hutzenlaub, T., & Borst, N. (2021). Stringent Base Specific and Optimization-Free Multiplex Mediator Probe ddPCR for the Quantification of Point Mutations in Circulating Tumor DNA. Cancers, 13(22), 5742. https://doi.org/10.3390/cancers13225742