
Dual Targeting of CDK4/6 and cMET in Metastatic Uveal Melanoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
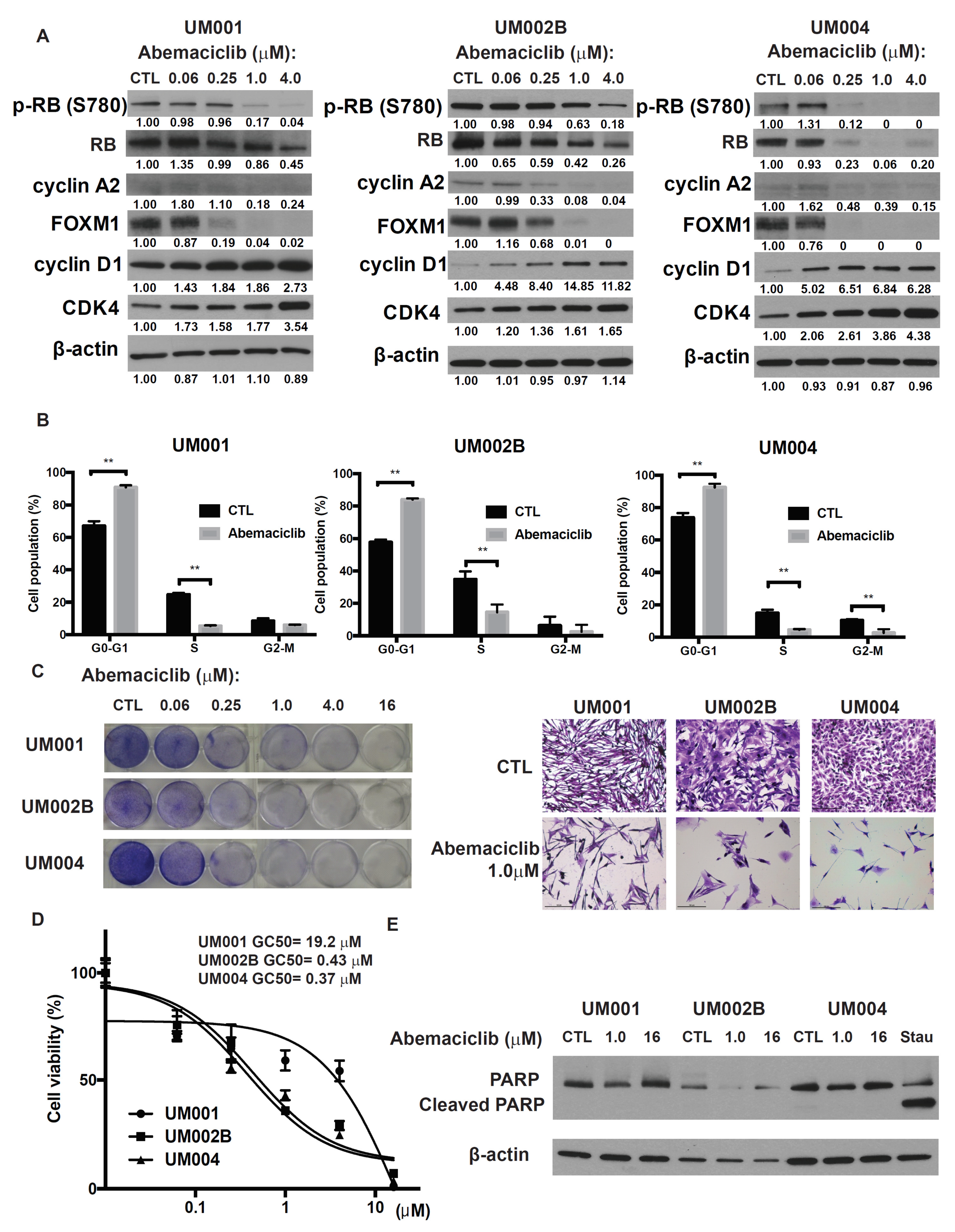
2.1. Abemaciclib Induces G1 Arrest and Decreases Cell Growth in Metastatic Uveal Melanoma Cells
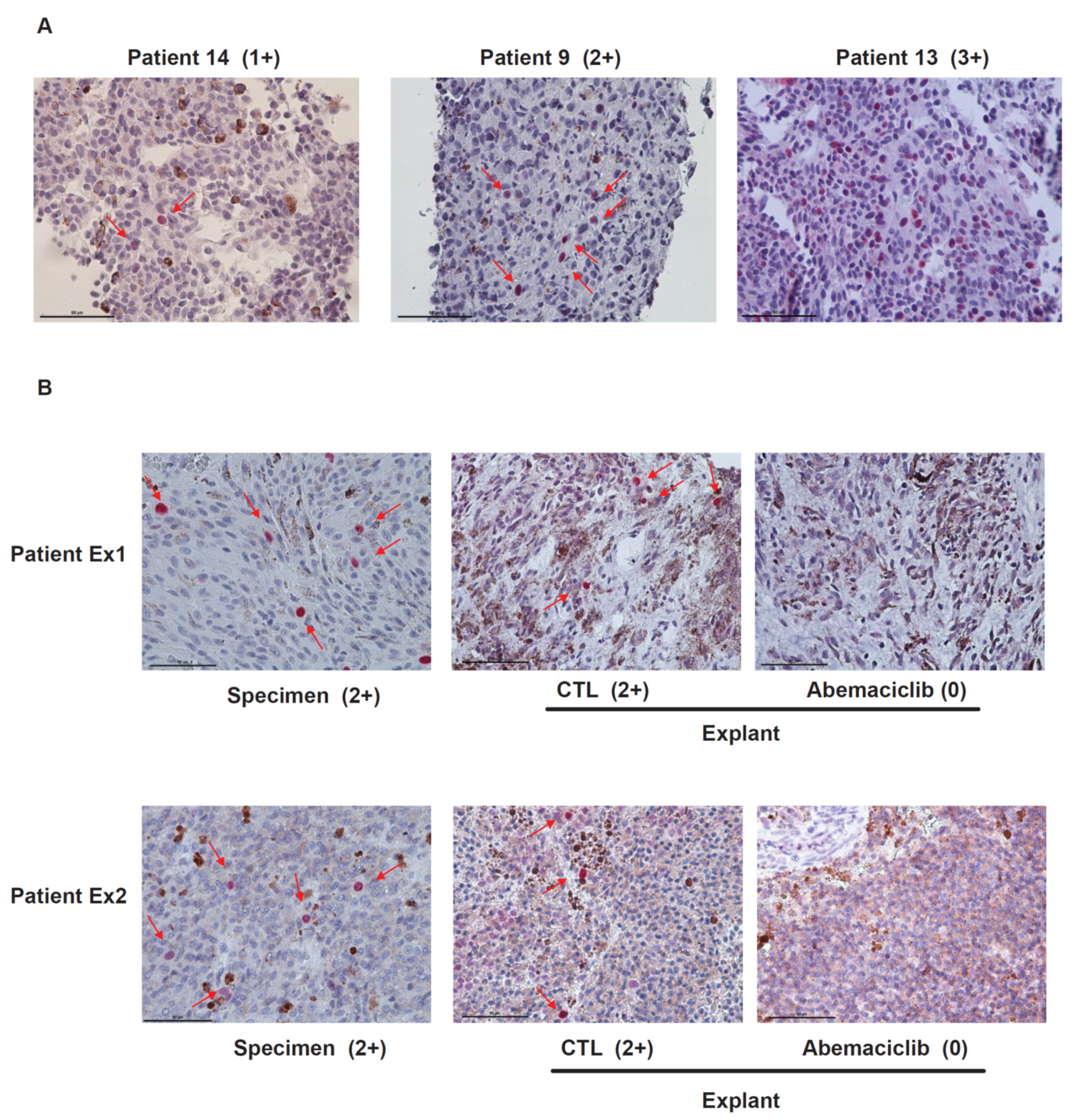
2.2. RB Protein Is Phosphorylated in Metastatic Uveal Melanoma Tissues and Abemaciclib Inhibits the Phosphorylation of RB in Metastatic UM Explants Obtained from Patients
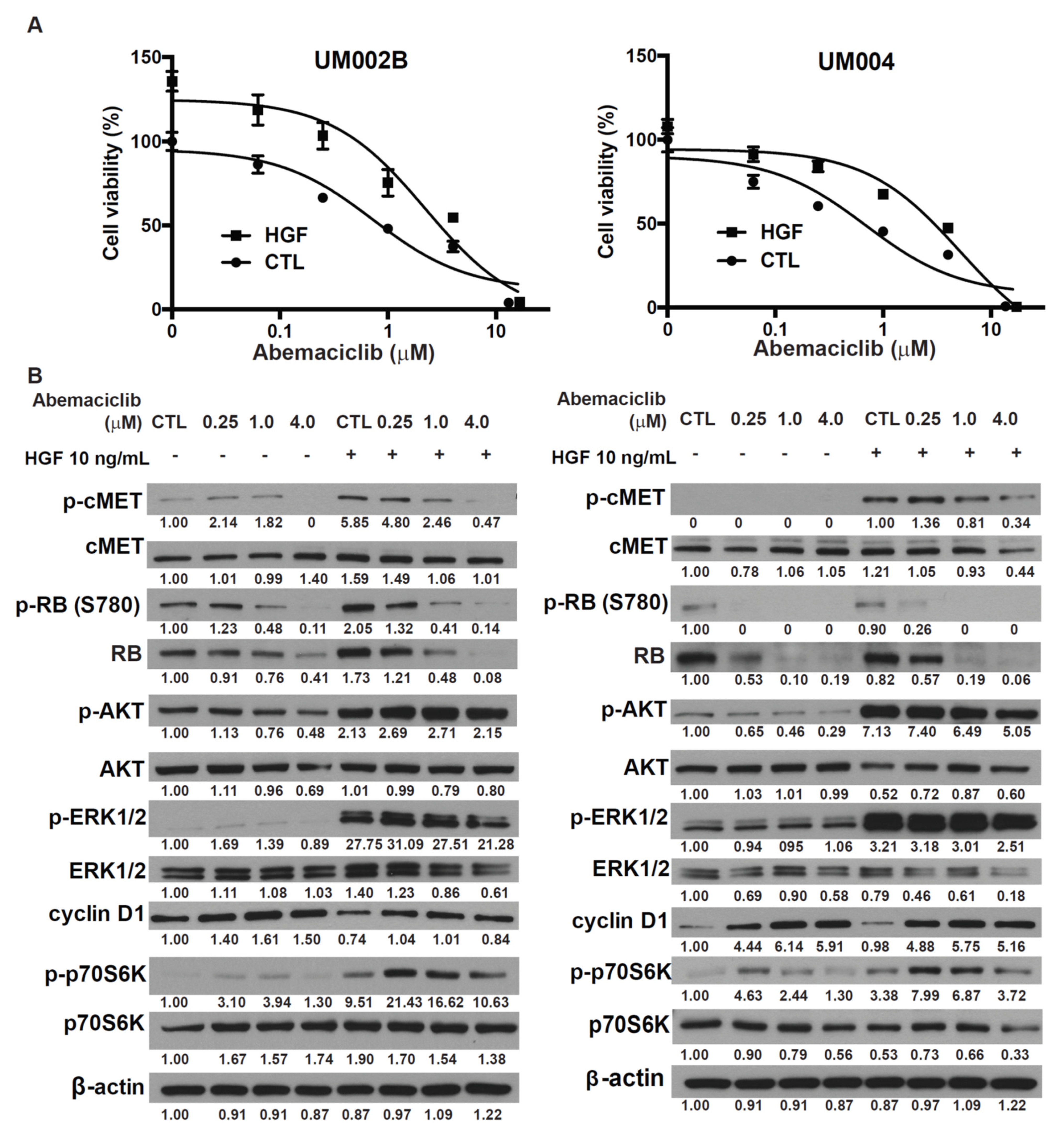
2.3. HGF Reduces the Growth-Inhibitory Effect of Abemaciclib in Metastatic Uveal Melanoma Cells
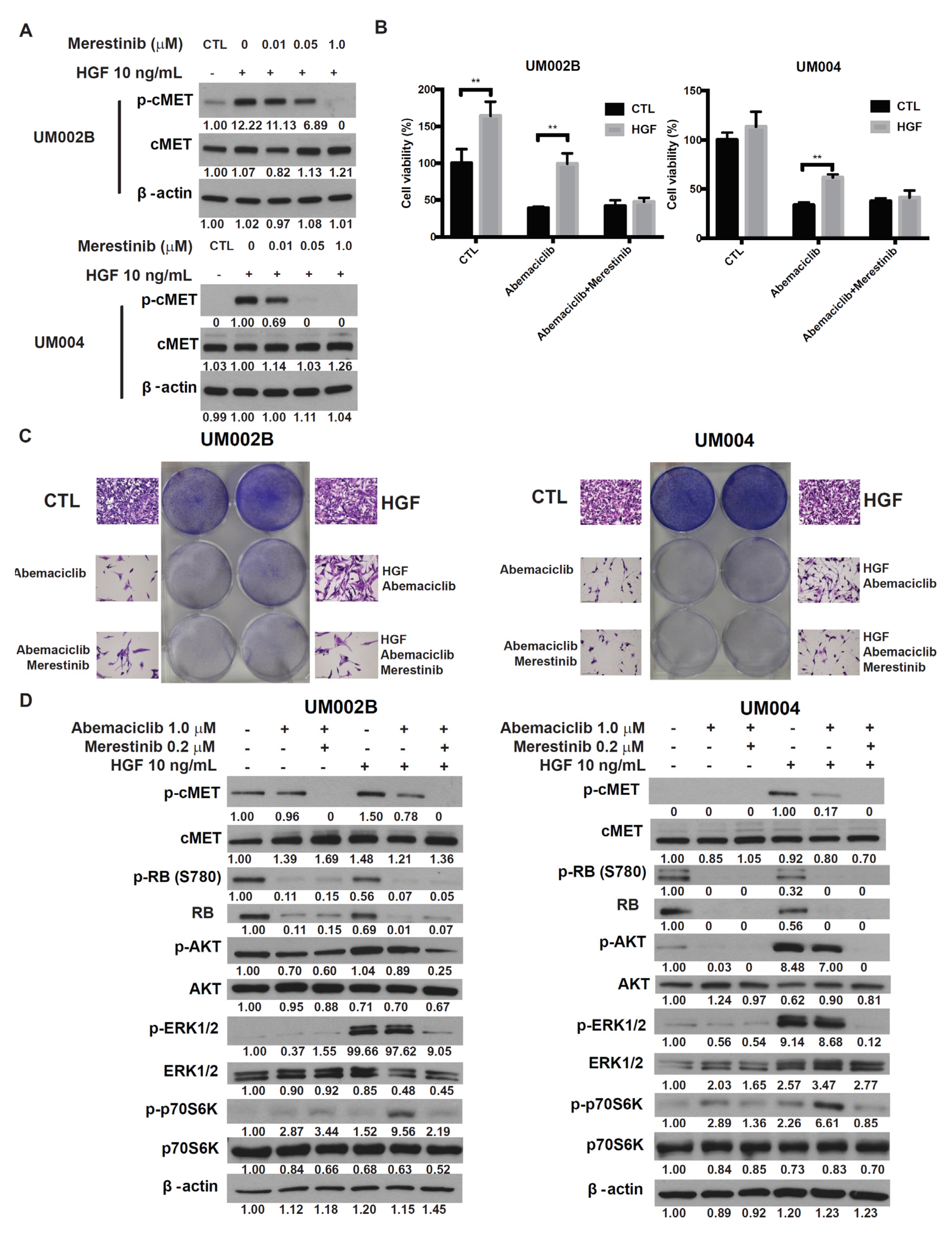
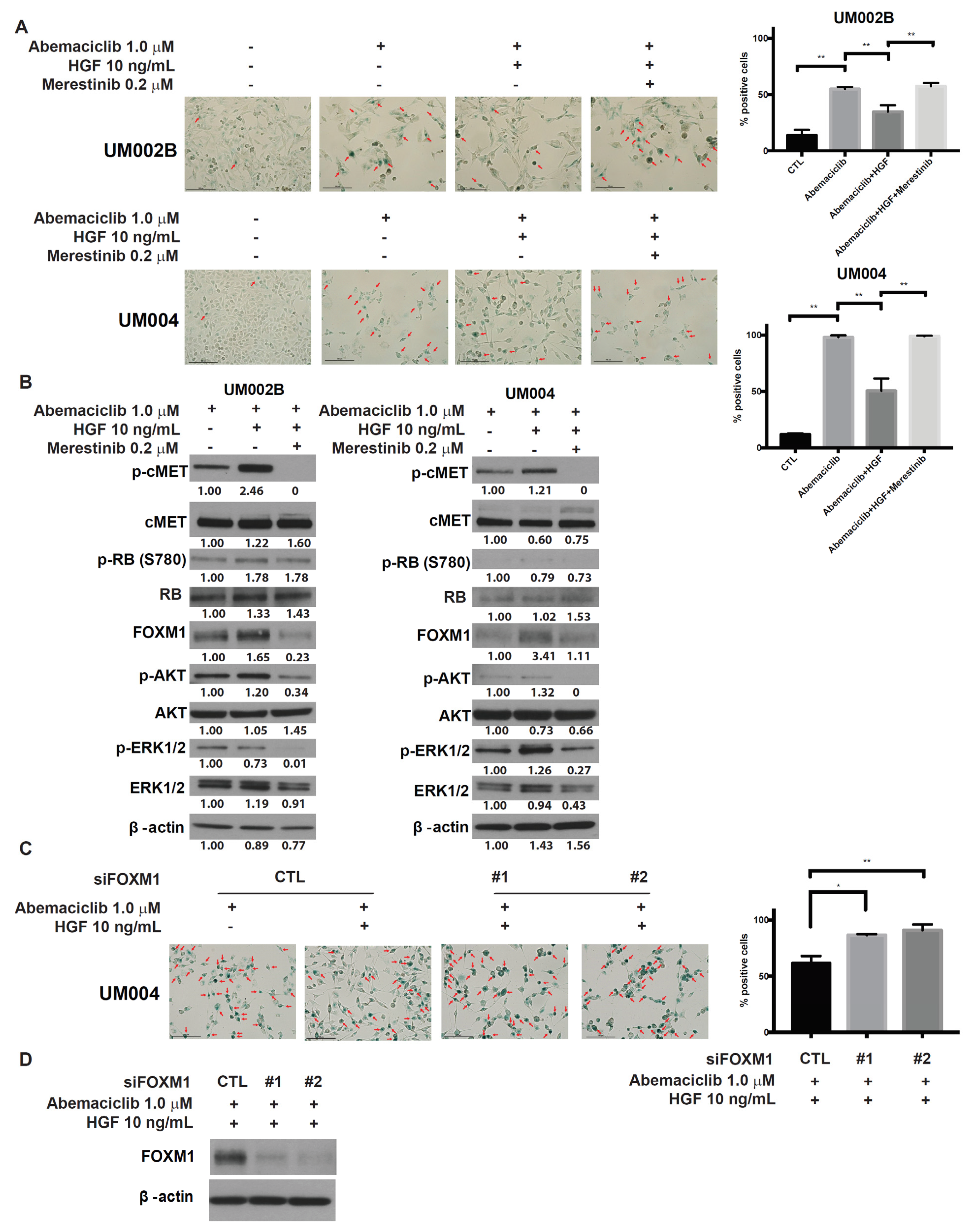
2.4. Merestinib Overcomes HGF-Mediated Resistance to Abemaciclib in Metastatic Uveal Melanoma Cells
2.5. Merestinib Inhibits the HGF-Mediated Protection from Cellular Senescence in Abemaciclib-Treated Uveal Melanoma Cells
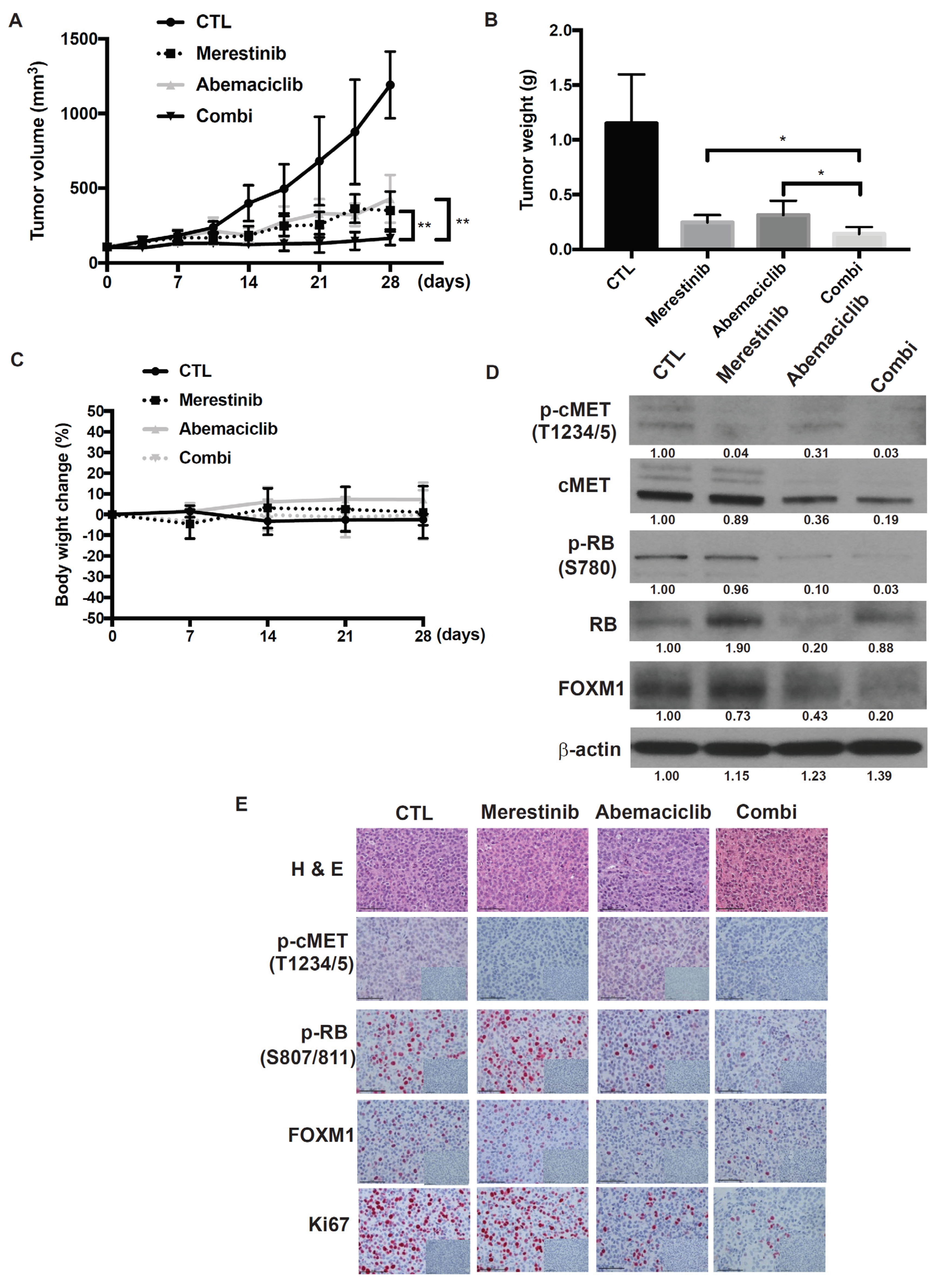
2.6. Abemaciclib in Combination with Merestinib Inhibits Metastatic Uveal Melanoma Tumor Growth in NSG-hHGFki Mice
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines
4.3. Short-Interfering RNA and Transfection
4.4. MTS Assays
4.5. Cell Growth Assays
4.6. Cell-Cycle Analysis
4.7. Protein Extraction and Western Immunoblotting
4.8. Senescence Assay
4.9. Xenograft Therapeutic Trials
4.10. Immunohistochemistry
4.11. Patient Tissue Samples
4.12. Ex Vivo UM Explants
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Rietschel, P.; Panageas, K.S.; Hanlon, C.; Patel, A.; Abramson, D.H.; Chapman, P.B. Variates of survival in metastatic uveal melanoma. J. Clin. Oncol. 2005, 23, 8076–8080. [Google Scholar] [CrossRef]
- Onken, M.D.; Worley, L.A.; Ehlers, J.P.; Harbour, J.W. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004, 64, 7205–7209. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W. A prognostic test to predict the risk of metastasis in uveal melanoma based on a 15-gene expression profile. Methods Mol. Biol. 2014, 1102, 427–440. [Google Scholar]
- Patel, M.; Smyth, E.; Chapman, P.B.; Wolchok, J.D.; Schwartz, G.K.; Abramson, D.H.; Carvajal, R.D. Therapeutic implications of the emerging molecular biology of uveal melanoma. Clin. Cancer Res. 2011, 17, 2087–2100. [Google Scholar] [CrossRef] [Green Version]
- Onken, M.D.; Worley, L.A.; Long, M.D.; Duan, S.; Council, M.L.; Bowcock, A.M.; Harbour, J.W. Oncogenic mutations in GNAQ occur early in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5230–5234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2008, 457, 599–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, V.; Lapadula, D.; Randolph, C.; Benovic, J.L.; Wedegaertner, P.B.; Aplin, A.E. Dysregulated GPCR signaling and therapeutic options in uveal melanoma. Mol. Cancer Res. 2017, 15, 501–506. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Babchia, N.; Calipel, A.; Mouriaux, F.; Faussat, A.M.; Mascarelli, F. The PI3K/Akt and mTOR/P70S6K signaling pathways in human uveal melanoma cells: Interaction with B-Raf/ERK. Investig. Ophthalmol. Vis. Sci. 2010, 51, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Von Euw, E.; Atefi, M.; Attar, N.; Chu, C.; Zachariah, S.; Burgess, B.L.; Mok, S.; Ng, C.; Wong, D.J.; Chmielowski, B.; et al. Antitumor effects of the investigational selective MEK inhibitor TAK733 against cutaneous and uveal melanoma cell lines. Mol. Cancer 2012, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wu, Q.; Tan, L.; Porter, D.; Jager, M.J.; Emery, C.; Bastian, B.C. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 2014, 33, 4724–4734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvajal, R.D.; Schwartz, G.K.; Tezel, T.; Marr, B.; Francis, J.H.; Nathan, P.D. Metastatic disease from uveal melanoma: Treatment options and future prospects. Br. J. Ophthalmol. 2017, 101, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zhou, J.; Rogers, A.M.; Jänne, P.A.; Benedettini, E.; Loda, M.; Hodi, F.S. c-Met, epidermal growth factor receptor, and insulin-like growth factor-1 receptor are important for growth in uveal melanoma and independently contribute to migration and metastatic potential. Melanoma Res. 2012, 22, 123–132. [Google Scholar] [CrossRef]
- Economou, M.A.; All-Ericsson, C.; Bykov, V.; Girnita, L.; Bartolazzi, A.; Larsson, O.; Seregard, S. Receptors for the liver synthesized growth factors IGF-1 and HGF/SF in uveal melanoma: Intercorrelation and prognostic implications. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4372–4375. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Terai, M.; Kageyama, K.; Ozaki, S.; McCue, P.A.; Sato, T.; Aplin, A.E. Paracrine Effect of NRG1 and HGF Drives Resistance to MEK Inhibitors in Metastatic Uveal Melanoma. Cancer Res. 2015, 75, 2737–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Chua, V.; Liao, C.; Purwin, T.J.; Terai, M.; Kageyama, K.; Davies, M.A.; Sato, T.; Aplin, A.E. Co-targeting HGF-cMET signaling with MEK inhibitors in metastatic uveal melanoma. Mol. Cancer Ther. 2017, 16, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Corso, S.; Comoglio, P.M.; Giordano, S. Cancer therapy: Can the challenge be MET? Trends Mol. Med. 2005, 11, 284–292. [Google Scholar] [CrossRef]
- Woodward, J.K.; Elshaw, S.R.; Murray, A.K.; Nichols, C.E.; Cross, N.; Laws, D.; Rennie, I.G.; Sisley, K. Stimulation and inhibition of uveal melanoma invasion by HGF, GRO, IL-1alpha and TGF-beta. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3144–3152. [Google Scholar]
- Chattopadhyay, C.; Grimm, E.A.; Woodman, S.E. Simultaneous inhibition of the HGF/MET and Erk1/2 pathways affect uveal melanoma cell growth and migration. PLoS ONE 2014, 9, e83957. [Google Scholar] [CrossRef] [Green Version]
- Gherardi, E.; Sharpe, M.; Lane, K.; Sirulnik, A.; Stoker, M. Hepatocyte growth factor/scatter factor (HGF/SF), the c-met receptor and the behaviour of epithelial cells. Symp. Soc. Exp. Biol. 1993, 47, 163–181. [Google Scholar]
- Mallikarjuna, K.; Pushparaj, V.; Biswas, J.; Krishnakumar, S. Expression of epidermal growth factor receptor, ezrin, hepatocyte growth factor, and c-Met in uveal melanoma: An immunohistochemical study. Curr. Eye Res. 2007, 32, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Topcu-Yilmaz, P.; Kiratli, H.; Saglam, A.; Söylemezoglu, F.; Hascelik, G. Correlation of clinicopathological parameters with HGF, c-Met, EGFR, and IGF-1R expression in uveal melanoma. Melanoma Res. 2010, 20, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Brantley, M.A., Jr.; Harbour, J.W. Inactivation of retinoblastoma protein in uveal melanoma by phosphorylation of sites in the COOH-terminal region. Cancer Res. 2000, 60, 4320–4323. [Google Scholar] [PubMed]
- Brantley, M.A., Jr.; Harbour, J.W. Deregulation of the Rb and p53 pathways in uveal melanoma. Am. J. Pathol. 2000, 157, 1795–1801. [Google Scholar] [CrossRef] [Green Version]
- Coupland, S.E.; Bechrakis, N.; Schuler, A.; Anagnostopoulos, I.; Hummel, M.; Bornfeld, N.; Stein, H. Expression patterns of cyclin D1 and related proteins regulating G1-S phase transition in uveal melanoma and retinoblastoma. Br. J. Ophthalmol. 1998, 82, 961–970. [Google Scholar] [CrossRef]
- Van der Velden, P.A.; Metzelaar-Blok, J.A.; Bergman, W.; Monique, H.; Hurks, H.; Frants, R.R.; Gruis, M.J.; Jager, M.J. Promoter hypermethylation: A common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res. 2001, 61, 5303–5306. [Google Scholar] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.B.; Peek, V.L.; Ajamie, R.; Buchanan, S.G.; Graff, J.R.; Heidler, S.A.; Hui, Y.-H.; Huss, K.L.; Konicek, B.W.; Manro, J.R.; et al. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Investig. New Drugs 2013, 31, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gypi, S.P.; Braun, P.; et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Xia, T.; Xie, D.; Gao, Y.; Jia, Z.; Wei, D.; Wang, L.; Huang, S.; Quan, M.; Xie, K. HGF/Met and FOXM1 form a positive feedback loop and render pancreatic cancer cells resistance to Met inhibition and aggressive phenotypes. Oncogene 2016, 35, 4708–4718. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, W.; Wen, L.; Yang, H.; Wen, M.; Yun, Y.; Zhao, L.; Zhu, X.; Tian, L.; Luo, E.; et al. FOXM1 confers resistance to gefitinib in lung adenocarcinoma via a MET/AKT-dependent positive feedback loop. Oncotarget 2016, 7, 59245–59259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrilllon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Franco, J.; Witkiewicz, A.K.; Knudsen, E.S. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 2014, 5, 6512–6525. [Google Scholar] [CrossRef] [Green Version]
- Teh, J.L.; Purwin, T.J.; Greenawalt, E.J.; Chervoneva, I.; Goldberg, A.; Davies, M.A.; Aplin, A.E. An In Vivo Reporter to Quantitatively and Temporally Analyze the Effects of CDK4/6 Inhibitor-Based Therapies in Melanoma. Cancer Res. 2016, 76, 5455–5466. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.; Burke, T.F.; Huber, L.; Van Horn, R.D.; Zhang, Y.; Buchanan, S.G.; Chan, E.M.; Starling, J.J.; Beckmann, R.P.; Peng, S.-B. The CDK4/6 Inhibitor LY2835219 Overcomes Vemurafenib Resistance Resulting from MAPK Reactivation and Cyclin D1 Upregulation. Mol. Cancer Ther. 2014, 13, 2253–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Zacharek, S.J.; Xiong, Y.; Shumway, S.D. Negative regulation of TSC1-TSC2 by mammalian D-type cyclins. Cancer Res. 2005, 65, 11354–11360. [Google Scholar] [CrossRef] [Green Version]
- Rivadeneira, D.B.; Mayhew, C.N.; Thangavel, C.; Sotillo, E.; Reed, C.A.; Grana, X.; Knudsen, E.S. Proliferative suppression by CDK4/6 inhibition: Complex function of the retinoblastoma pathway in liver tissue and hepatoma cells. Gastroenterology 2010, 138, 1920–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.L.; Thangavel, C.; McClendon, A.K.; Reed, C.A.; Knudsen, E.S. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 2010, 29, 4018–4032. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, A.; Panatta, E.; Lena, A.; Castiglia, D.; Di Daniele, N.; Melino, G.; Candi, E. FOXM1 regulates proliferation, senescence and oxidative stress in keratinocytes and cancer cells. Aging 2016, 8, 1384–1397. [Google Scholar] [CrossRef] [Green Version]
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 Inhibition Induces Cell-Cycle Arrest and Senescence in Neuroblastoma. Clin. Cancer Res. 2013, 19, 6173–6182. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, K.; Ohara, M.; Saito, K.; Ozaki, S.; Terai, M.; Mastrangelo, M.J.; Fortina, P.; Aplin, A.E.; Sato, T. Establishment of an orthotopic patient-derived xenograft mouse model using uveal melanoma hepatic metastasis. J. Transl. Med. 2017, 15, 145. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohara, M.; Saito, K.; Kageyama, K.; Terai, M.; Cheng, H.; Aplin, A.E.; Sato, T. Dual Targeting of CDK4/6 and cMET in Metastatic Uveal Melanoma. Cancers 2021, 13, 1104. https://doi.org/10.3390/cancers13051104
Ohara M, Saito K, Kageyama K, Terai M, Cheng H, Aplin AE, Sato T. Dual Targeting of CDK4/6 and cMET in Metastatic Uveal Melanoma. Cancers. 2021; 13(5):1104. https://doi.org/10.3390/cancers13051104
Chicago/Turabian StyleOhara, Masahiro, Kengo Saito, Ken Kageyama, Mizue Terai, Hanyin Cheng, Andrew E. Aplin, and Takami Sato. 2021. "Dual Targeting of CDK4/6 and cMET in Metastatic Uveal Melanoma" Cancers 13, no. 5: 1104. https://doi.org/10.3390/cancers13051104
APA StyleOhara, M., Saito, K., Kageyama, K., Terai, M., Cheng, H., Aplin, A. E., & Sato, T. (2021). Dual Targeting of CDK4/6 and cMET in Metastatic Uveal Melanoma. Cancers, 13(5), 1104. https://doi.org/10.3390/cancers13051104